
Understanding the Central Role of Citrate in the Metabolism of Cancer Cells and Tumors: An Update

 ,
, 

Abstract
:1. Introduction
2. The Central Role of Citrate in the Warburg Effect
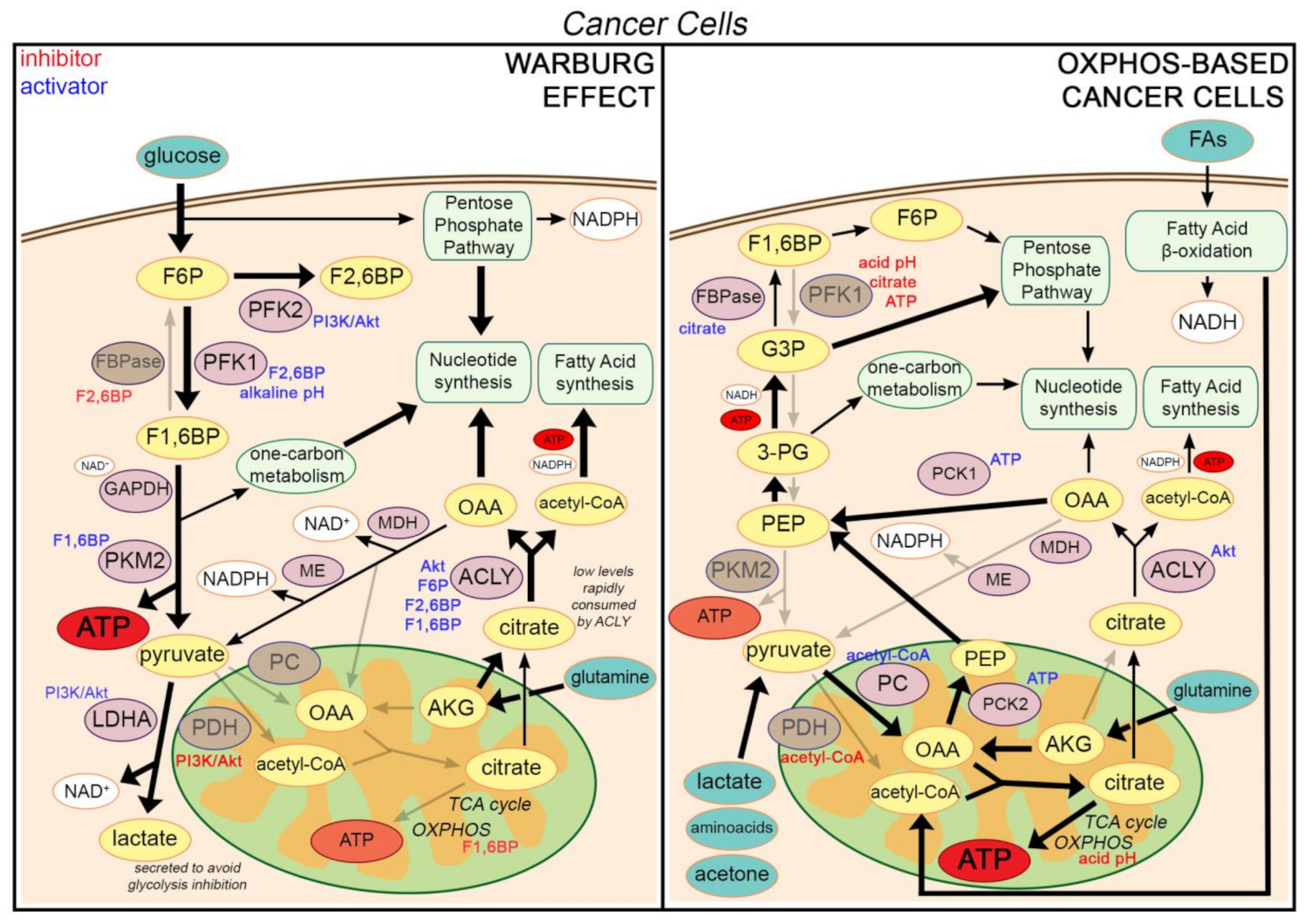
2.1. The Warburg Effect
2.2. A Low Citrate Level Is a Driving Force for the Warburg Effect
3. The Central Place of Citrate in Cancer Cells Relying on Oxidative Metabolism
3.1. Cancer Cells Are Not Inherently Glycolytic
3.2. Citrate Constitutes a Driving Force also in Cells Relying on Oxidative Metabolism
4. The Possible Role of Citrate in the Metabolism of the Microenvironment
5. The Anti-Cancer Effects of Citrate at High Dosages in Preclinical Experiments
5.1. In Vitro Studies
5.2. In Vivo Studies
6. Discussion and Therapeutic Perspectives
Author Contributions
Funding
Conflicts of Interest
References
- Icard, P.; Poulain, L.; Lincet, H. Understanding the central role of citrate in the metabolism of cancer cells. Biochim. Biophys. Acta Rev. Cancer 2012, 1825, 111–116. [Google Scholar] [CrossRef] [PubMed]
- Nelson, D.; Cox, M. Lehninger Principles of Biochemistry; W.H. Freeman & Co Ltd.: New York, NY, USA, 2021; ISBN 9781319228002. [Google Scholar]
- Nissler, K.; Petermann, H.; Wenz, I.; Brox, D. Fructose 2,6-bisphosphate metabolism in Ehrlich ascites tumour cells. J. Cancer Res. Clin. Oncol. 1995, 121, 739–745. [Google Scholar] [CrossRef]
- Icard, P.; Fournel, L.; Coquerel, A.; Gligorov, J.; Alifano, M.; Lincet, H. Citrate targets FBPase and constitutes an emerging novel approach for cancer therapy. Cancer Cell Int. 2018, 18, 175. [Google Scholar] [CrossRef]
- Cahill, G.F. Starvation in man. Clin. Endocrinol. Metab. 1976, 5, 397–415. [Google Scholar] [CrossRef]
- Rui, L. Energy Metabolism in the Liver. In Comprehensive Physiology; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2014; Volume 4, pp. 177–197. [Google Scholar]
- Hue, L.; Taegtmeyer, H. The Randle cycle revisited: A new head for an old hat. Am. J. Physiol. Metab. 2009, 297, E578–E591. [Google Scholar] [CrossRef] [Green Version]
- Berg, J.; Tymoczko, J.; Gatto, G.; Stryer, L. Biochemistry, 9th ed.; W.H. Freeman: New York, NY, USA, 2019; ISBN 978-1319114657. [Google Scholar]
- Brun, T.; Roche, E.; Assimacopoulos-Jeannet, F.; Corkey, B.E.; Kim, K.-H.; Prentki, M. Evidence for an Anaplerotic/Malonyl-CoA Pathway in Pancreatic-Cell Nutrient Signaling. Diabetes 1996, 45, 190–198. [Google Scholar] [CrossRef]
- Costello, L.C.; Franklin, R.B. Citrate metabolism of normal and malignant prostate epithelial cells. Urology 1997, 50, 3–12. [Google Scholar] [CrossRef]
- Costello, L.C.; Franklin, R.B.; Reynolds, M.A. The Important Role and Implications of Citrate in the Composition, Structure, and Function of Oral/Periodontal/Craniofacial Tissues. Madridge J. Dent. Oral Surg. 2018, 3, 85–90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costello, L.C.; Chellaiah, M.; Zou, J.; Franklin, R.B.; Reynolds, M.A. The status of citrate in the hydroxyapatite/collagen complex of bone; and Its role in bone formation. J. Regen. Med. Tissue Eng. 2014, 3, 4. [Google Scholar] [CrossRef] [Green Version]
- O’Neill, L.A.J.; Pearce, E.J. Immunometabolism governs dendritic cell and macrophage function. J. Exp. Med. 2016, 213, 15–23. [Google Scholar] [CrossRef]
- Williams, N.C.; O’Neill, L.A.J. A Role for the Krebs Cycle Intermediate Citrate in Metabolic Reprogramming in Innate Immunity and Inflammation. Front. Immunol. 2018, 9, 141. [Google Scholar] [CrossRef] [Green Version]
- Abdel-Salam, O.M.E.; Youness, E.R.; Mohammed, N.A.; Morsy, S.M.Y.; Omara, E.A.; Sleem, A.A. Citric Acid Effects on Brain and Liver Oxidative Stress in Lipopolysaccharide-Treated Mice. J. Med. Food 2014, 17, 588–598. [Google Scholar] [CrossRef] [Green Version]
- He, W.; Heinz, A.; Jahn, D.; Hiller, K. Complexity of macrophage metabolism in infection. Curr. Opin. Biotechnol. 2021, 68, 231–239. [Google Scholar] [CrossRef]
- Infantino, V.; Pierri, C.L.; Iacobazzi, V. Metabolic Routes in Inflammation: The Citrate Pathway and its Potential as Therapeutic Target. Curr. Med. Chem. 2020, 26, 7104–7116. [Google Scholar] [CrossRef] [PubMed]
- Mller, A.J.; Call, J.E.; Whiting, R.C. Comparison of Organic Acid Salts for Clostridium botulinum Control in an Uncured Turkey Product. J. Food Prot. 1993, 56, 958–962. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Cesario, T.; Owens, J.; Shanbrom, E.; Thrupp, L.D. Antibacterial activity of citrate and acetate. Nutrition 2002, 18, 665–666. [Google Scholar] [CrossRef]
- Lee, Y.-L.; Thrupp, L.; Owens, J.; Cesario, T.; Shanbrom, E. Bactericidal activity of citrate against Gram-positive cocci. Lett. Appl. Microbiol. 2001, 33, 349–351. [Google Scholar] [CrossRef]
- Icard, P.; Shulman, S.; Farhat, D.; Steyaert, J.-M.; Alifano, M.; Lincet, H. How the Warburg effect supports aggressiveness and drug resistance of cancer cells? Drug Resist. Update 2018, 38, 1–11. [Google Scholar] [CrossRef]
- Icard, P.; Wu, Z.; Alifano, M.; Fournel, L. Gluconeogenesis of Cancer Cells Is Disrupted by Citrate. Trends Cancer 2019, 5, 265–266. [Google Scholar] [CrossRef] [PubMed]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef]
- Salas, M.L.; Viñuela, E.; Salas, M.; Sols, A. Citrate inhibition of phosphofructokinase and the Pasteur effect. Biochem. Biophys. Res. Commun. 1965, 19, 371–376. [Google Scholar] [CrossRef]
- Zhang, X.; Varin, E.; Allouche, S.; Lu, Y.; Poulain, L.; Icard, P. Effect of Citrate on Malignant Pleural Mesothelioma Cells: A Synergistic Effect with Cisplatin. Anticancer Res. 2009, 29, 1249–1254. [Google Scholar] [PubMed]
- Lu, Y.; Zhang, X.; Zhang, H.; Lan, J.; Huang, G.; Varin, E.; Lincet, H.; Poulain, L.; Icard, P. Citrate induces apoptotic cell death: A promising way to treat gastric carcinoma? Anticancer Res. 2011, 31, 797–805. [Google Scholar] [PubMed]
- Lincet, H.; Kafara, P.; Giffard, F.; Abeilard-Lemoisson, E.; Duval, M.; Louis, M.-H.; Poulain, L.; Icard, P. Inhibition of Mcl-1 expression by citrate enhances the effect of Bcl-xL inhibitors on human ovarian carcinoma cells. J. Ovarian Res. 2013, 6, 72. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Chen, R.; Yu, Z.; Li, R.; Li, J.; Zhao, X.; Song, S.; Liu, J.; Huang, G. Dichloroacetate restores drug sensitivity in paclitaxel-resistant cells by inducing citric acid accumulation. Mol. Cancer 2015, 14, 63. [Google Scholar] [CrossRef] [Green Version]
- Tomasetti, M.; Nocchi, L.; Staffolani, S.; Manzella, N.; Amati, M.; Goodwin, J.; Kluckova, K.; Nguyen, M.; Strafella, E.; Bajzikova, M.; et al. MicroRNA-126 Suppresses Mesothelioma Malignancy by Targeting IRS1 and Interfering with the Mitochondrial Function. Antioxid. Redox Signal. 2014, 21, 2109–2125. [Google Scholar] [CrossRef] [Green Version]
- Zu, X.L.; Guppy, M. Cancer metabolism: Facts, fantasy, and fiction. Biochem. Biophys. Res. Commun. 2004, 313, 459–465. [Google Scholar] [CrossRef]
- Peiris-Pagès, M.; Martinez-Outschoorn, U.E.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer stem cell metabolism. Breast Cancer Res. 2016, 18, 55. [Google Scholar] [CrossRef] [PubMed]
- McGarry, J.D.; Takabayashi, Y.; Foster, D.W. The role of malonyl-CoA in the coordination of fatty acid synthesis and oxidation in isolated rat hepatocytes. J. Biol. Chem. 1978, 253, 8294–8300. [Google Scholar] [CrossRef]
- DeBerardinis, R.J.; Chandel, N.S. Fundamentals of cancer metabolism. Sci. Adv. 2016, 2, e1600200. [Google Scholar] [CrossRef] [Green Version]
- Martinez-Outschoorn, U.E.; Peiris-Pagés, M.; Pestell, R.G.; Sotgia, F.; Lisanti, M.P. Cancer metabolism: A therapeutic perspective. Nat. Rev. Clin. Oncol. 2017, 14, 11–31. [Google Scholar] [CrossRef]
- Heiden, M.G.V.; Cantley, L.C.; Thompson, C.B. Understanding the warburg effect: The metabolic requirements of cell proliferation. Science 2009, 324, 1029–1033. [Google Scholar] [CrossRef] [Green Version]
- Zhao, S.; Xu, W.; Jiang, W.; Yu, W.; Lin, Y.; Zhang, T.; Yao, J.; Zhou, L.; Zeng, Y.; Li, H.; et al. Regulation of Cellular Metabolism by Protein Lysine Acetylation. Science 2010, 327, 1000–1004. [Google Scholar] [CrossRef] [Green Version]
- Lv, L.; Li, D.; Zhao, D.; Lin, R.; Chu, Y.; Zhang, H.; Zha, Z.; Liu, Y.; Li, Z.; Xu, Y.; et al. Acetylation Targets the M2 Isoform of Pyruvate Kinase for Degradation through Chaperone-Mediated Autophagy and Promotes Tumor Growth. Mol. Cell 2011, 42, 719–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Icard, P.; Fournel, L.; Wu, Z.; Alifano, M.; Lincet, H. Interconnection between Metabolism and Cell Cycle in Cancer. Trends Biochem. Sci. 2019, 44, 490–501. [Google Scholar] [CrossRef]
- Mullen, A.R.; Wheaton, W.W.; Jin, E.S.; Chen, P.-H.; Sullivan, L.B.; Cheng, T.; Yang, Y.; Linehan, W.M.; Chandel, N.S.; DeBerardinis, R.J. Reductive carboxylation supports growth in tumour cells with defective mitochondria. Nature 2012, 481, 385–388. [Google Scholar] [CrossRef] [Green Version]
- Díaz-Ruiz, R.; Avéret, N.; Araiza, D.; Pinson, B.; Uribe-Carvajal, S.; Devin, A.; Rigoulet, M. Mitochondrial Oxidative Phosphorylation Is Regulated by Fructose 1,6-Bisphosphate. J. Biol. Chem. 2008, 283, 26948–26955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costello, L.C.; Liu, Y.; Zou, J.; Franklin, R.B. Mitochondrial aconitase gene expression is regulated by testosterone and prolactin in prostate epithelial cells. Prostate 2000, 42, 196–202. [Google Scholar] [CrossRef] [Green Version]
- Tong, W.-H.; Rouault, T.A. Metabolic regulation of citrate and iron by aconitases: Role of iron–sulfur cluster biogenesis. BioMetals 2007, 20, 549–564. [Google Scholar] [CrossRef] [PubMed]
- Lushchak, O.V.; Piroddi, M.; Galli, F.; Lushchak, V.I. Aconitase post-translational modification as a key in linkage between Krebs cycle, iron homeostasis, redox signaling, and metabolism of reactive oxygen species. Redox Rep. 2014, 19, 8–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, G.; Brownsey, R.W.; MacLeod, K.M. Regulation of mitochondrial aconitase by phosphorylation in diabetic rat heart. Cell. Mol. Life Sci. 2009, 66, 919–932. [Google Scholar] [CrossRef]
- Palmieri, E.M.; Gonzalez-Cotto, M.; Baseler, W.A.; Davies, L.C.; Ghesquière, B.; Maio, N.; Rice, C.M.; Rouault, T.A.; Cassel, T.; Higashi, R.M.; et al. Nitric oxide orchestrates metabolic rewiring in M1 macrophages by targeting aconitase 2 and pyruvate dehydrogenase. Nat. Commun. 2020, 11, 698. [Google Scholar] [CrossRef]
- Keohavong, P.; Kat, A.G.; Cariello, N.F.; Thilly, W.G. DNA Amplification In Vitro Using T4 DNA Polymerase. DNA 1988, 7, 63–70. [Google Scholar] [CrossRef]
- Marin-Hernandez, A.; Gallardo-Perez, J.; Ralph, S.; Rodriguez-Enriquez, S.; Moreno-Sanchez, R. HIF-1α Modulates Energy Metabolism in Cancer Cells by Inducing Over-Expression of Specific Glycolytic Isoforms. Mini Rev. Med. Chem. 2009, 9, 1084–1101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Tchernyshyov, I.; Semenza, G.L.; Dang, C.V. HIF-1-mediated expression of pyruvate dehydrogenase kinase: A metabolic switch required for cellular adaptation to hypoxia. Cell Metab. 2006, 3, 177–185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dhup, S.; Kumar Dadhich, R.; Ettore Porporato, P.; Sonveaux, P. Multiple Biological Activities of Lactic Acid in Cancer: Influences on Tumor Growth, Angiogenesis and Metastasis. Curr. Pharm. Des. 2012, 18, 1319–1330. [Google Scholar] [CrossRef] [Green Version]
- Husain, Z.; Huang, Y.; Seth, P.; Sukhatme, V.P. Tumor-Derived Lactate Modifies Antitumor Immune Response: Effect on Myeloid-Derived Suppressor Cells and NK Cells. J. Immunol. 2013, 191, 1486–1495. [Google Scholar] [CrossRef] [PubMed]
- Trivedi, B.; Danforth, W.H. Effect of pH on the kinetics of frog muscle phosphofructokinase. J. Biol. Chem. 1966, 241, 4110–4114. [Google Scholar] [CrossRef]
- Rodic, S.; Vincent, M.D. Reactive oxygen species (ROS) are a key determinant of cancer’s metabolic phenotype. Int. J. Cancer 2018, 142, 440–448. [Google Scholar] [CrossRef]
- Chen, J.-Q.Q.; Russo, J. Dysregulation of glucose transport, glycolysis, TCA cycle and glutaminolysis by oncogenes and tumor suppressors in cancer cells. Biochim. Biophys. Acta Rev. Cancer 2012, 1826, 370–384. [Google Scholar] [CrossRef]
- Cairns, R.A. Drivers of the Warburg Phenotype. Cancer J. 2015, 21, 56–61. [Google Scholar] [CrossRef]
- Wise, D.R.; Deberardinis, R.J.; Mancuso, A.; Sayed, N.; Zhang, X.Y.; Pfeiffer, H.K.; Nissim, I.; Daikhin, E.; Yudkoff, M.; McMahon, S.B.; et al. Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc. Natl. Acad. Sci. USA 2008, 105, 18782–18787. [Google Scholar] [CrossRef] [Green Version]
- Lin, C.C.; Cheng, T.L.; Tsai, W.H.; Tsai, H.J.; Hu, K.H.; Chang, H.C.; Yeh, C.W.; Chen, Y.C.; Liao, C.C.; Chang, W.T. Loss of the respiratory enzyme citrate synthase directly links the Warburg effect to tumor malignancy. Sci. Rep. 2012, 2, 785. [Google Scholar] [CrossRef]
- Na, F.; Wang, J.; Li, C.; Deng, L.; Xue, J.; Lu, Y. Primary Tumor Standardized Uptake Value Measured on F18-Fluorodeoxyglucose Positron Emission Tomography Is of Prediction Value for Survival and Local Control in Non–Small-Cell Lung Cancer Receiving Radiotherapy: Meta-Analysis. J. Thorac. Oncol. 2014, 9, 834–842. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smolle, E.; Leko, P.; Stacher-Priehse, E.; Brcic, L.; El-Heliebi, A.; Hofmann, L.; Quehenberger, F.; Hrzenjak, A.; Popper, H.H.; Olschewski, H.; et al. Distribution and prognostic significance of gluconeogenesis and glycolysis in lung cancer. Mol. Oncol. 2020, 14, 2853–2867. [Google Scholar] [CrossRef] [PubMed]
- Hudson, C.D.; Hagemann, T.; Mather, S.J.; Avril, N. Resistance to the tyrosine kinase inhibitor axitinib is associated with increased glucose metabolism in pancreatic adenocarcinoma. Cell Death Dis. 2014, 5, e1160. [Google Scholar] [CrossRef] [Green Version]
- Rotow, J.; Bivona, T.G. Understanding and targeting resistance mechanisms in NSCLC. Nat. Rev. Cancer 2017, 17, 637–658. [Google Scholar] [CrossRef]
- Crochet, R.B.; Kim, J.D.; Lee, H.; Yim, Y.S.; Kim, S.G.; Neau, D.; Lee, Y.H. Crystal structure of heart 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase (PFKFB2) and the inhibitory influence of citrate on substrate binding. Proteins Struct. Funct. Bioinform. 2017, 85, 117–124. [Google Scholar] [CrossRef] [Green Version]
- Yalcin, A.; Clem, B.F.; Imbert-Fernandez, Y.; Ozcan, S.C.; Peker, S.; O’Neal, J.; Klarer, A.C.; Clem, A.L.; Telang, S.; Chesney, J. 6-Phosphofructo-2-kinase (PFKFB3) promotes cell cycle progression and suppresses apoptosis via Cdk1-mediated phosphorylation of p27. Cell Death Dis. 2014, 5, e1337. [Google Scholar] [CrossRef] [PubMed]
- Houddane, A.; Bultot, L.; Novellasdemunt, L.; Johanns, M.; Gueuning, M.-A.; Vertommen, D.; Coulie, P.G.; Bartrons, R.; Hue, L.; Rider, M.H. Role of Akt/PKB and PFKFB isoenzymes in the control of glycolysis, cell proliferation and protein synthesis in mitogen-stimulated thymocytes. Cell Signal. 2017, 34, 23–37. [Google Scholar] [CrossRef] [PubMed]
- Jurica, M.S.; Mesecar, A.; Heath, P.J.; Shi, W.; Nowak, T.; Stoddard, B.L. The allosteric regulation of pyruvate kinase by fructose-1,6-bisphosphate. Structure 1998, 6, 195–210. [Google Scholar] [CrossRef] [Green Version]
- Ashizawa, K.; Willingham, M.C.; Liang, C.M.; Cheng, S.Y. In vivo regulation of monomer-tetramer conversion of pyruvate kinase subtype M2 by glucose is mediated via fructose 1,6-bisphosphate. J. Biol. Chem. 1991, 266, 16842–16846. [Google Scholar] [CrossRef]
- Macpherson, J.A.; Theisen, A.; Masino, L.; Fets, L.; Driscoll, P.C.; Encheva, V.; Snijders, A.P.; Martin, S.R.; Kleinjung, J.; Barran, P.E.; et al. Functional cross-talk between allosteric effects of activating and inhibiting ligands underlies PKM2 regulation. Elife 2019, 8, e45068. [Google Scholar] [CrossRef] [PubMed]
- Peeters, K.; Van Leemputte, F.; Fischer, B.; Bonini, B.M.; Quezada, H.; Tsytlonok, M.; Haesen, D.; Vanthienen, W.; Bernardes, N.; Gonzalez-Blas, C.B.; et al. Fructose-1,6-bisphosphate couples glycolytic flux to activation of Ras. Nat. Commun. 2017, 8, 922. [Google Scholar] [CrossRef] [PubMed]
- Das, J.; Ho, M.; Zikherman, J.; Govern, C.; Yang, M.; Weiss, A.; Chakraborty, A.K.; Roose, J.P. Digital Signaling and Hysteresis Characterize Ras Activation in Lymphoid Cells. Cell 2009, 136, 337–351. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.; Ramjaun, A.R.; Haiko, P.; Wang, Y.; Warne, P.H.; Nicke, B.; Nye, E.; Stamp, G.; Alitalo, K.; Downward, J. Binding of Ras to Phosphoinositide 3-Kinase p110α Is Required for Ras- Driven Tumorigenesis in Mice. Cell 2007, 129, 957–968. [Google Scholar] [CrossRef] [Green Version]
- Lim, S.-O.; Li, C.-W.; Xia, W.; Lee, H.-H.; Chang, S.-S.; Shen, J.; Hsu, J.L.; Raftery, D.; Djukovic, D.; Gu, H.; et al. EGFR Signaling Enhances Aerobic Glycolysis in Triple-Negative Breast Cancer Cells to Promote Tumor Growth and Immune Escape. Cancer Res. 2016, 76, 1284–1296. [Google Scholar] [CrossRef] [Green Version]
- Minchenko, O.; Opentanova, I.; Caro, J. Hypoxic regulation of the 6-phosphofructo-2-kinase/fructose-2,6-bisphosphatase gene family (PFKFB-1-4) expression in vivo. FEBS Lett. 2003, 554, 264–270. [Google Scholar] [CrossRef]
- Wang, Z.; Dong, C. Gluconeogenesis in Cancer: Function and Regulation of PEPCK, FBPase, and G6Pase. Trends Cancer 2019, 5, 30–45. [Google Scholar] [CrossRef]
- Chae, Y.C.; Vaira, V.; Caino, M.C.; Tang, H.-Y.Y.; Seo, J.H.; Kossenkov, A.V.; Ottobrini, L.; Martelli, C.; Lucignani, G.; Bertolini, I.; et al. Mitochondrial Akt Regulation of Hypoxic Tumor Reprogramming. Cancer Cell 2016, 30, 257–272. [Google Scholar] [CrossRef] [Green Version]
- Courtnay, R.; Ngo, D.C.; Malik, N.; Ververis, K.; Tortorella, S.M.; Karagiannis, T.C. Cancer metabolism and the Warburg effect: The role of HIF-1 and PI3K. Mol. Biol. Rep. 2015, 42, 841–851. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.-A.; Zhang, X.-D.; Guo, X.-Y.; Xian, S.-L.; Lu, Y.-F. 3-Bromopyruvate and sodium citrate target glycolysis, suppress survivin, and induce mitochondrial-mediated apoptosis in gastric cancer cells and inhibit gastric orthotopic transplantation tumor growth. Oncol. Rep. 2016, 35, 1287–1296. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Zhang, X.; Wang, T.; Xian, S.; Lu, Y. 3-Bromopyruvate and sodium citrate induce apoptosis in human gastric cancer cell line MGC-803 by inhibiting glycolysis and promoting mitochondria-regulated apoptosis pathway. Biochem. Biophys. Res. Commun. 2016, 475, 37–43. [Google Scholar] [CrossRef] [Green Version]
- Ren, J.-G.; Seth, P.; Ye, H.; Guo, K.; Hanai, J.; Husain, Z.; Sukhatme, V.P. Citrate Suppresses Tumor Growth in Multiple Models through Inhibition of Glycolysis, the Tricarboxylic Acid Cycle and the IGF-1R Pathway. Sci. Rep. 2017, 7, 4537. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hung, K.-C.; Wang, S.-G.; Lin, M.-L.; Chen, S.-S. Citrate-Induced p85α–PTEN Complex Formation Causes G2/M Phase Arrest in Human Pharyngeal Squamous Carcinoma Cell Lines. Int. J. Mol. Sci. 2019, 20, 2105. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, Z.; Levine, K.M.; Bahreini, A.; Wang, P.; Chu, D.; Park, B.H.; Oesterreich, S.; Lee, A.V. Upregulation of IRS1 Enhances IGF1 Response in Y537S and D538G ESR1 Mutant Breast Cancer Cells. Endocrinology 2018, 159, 285–296. [Google Scholar] [CrossRef]
- Icard, P.; Wu, Z.; Fournel, L.; Coquerel, A.; Lincet, H.; Alifano, M. ATP citrate lyase: A central metabolic enzyme in cancer. Cancer Lett. 2020, 471, 125–134. [Google Scholar] [CrossRef] [PubMed]
- Icard, P.; Lincet, H. Corrigendum to “The reduced concentration of citrate in cancer cells: An indicator of cancer aggressiveness and a possible therapeutic target”. Drug Resist. Update 2017, 30, 63. [Google Scholar] [CrossRef]
- Berwick, D.C.; Hers, I.; Heesom, K.J.; Moule, S.K.; Tavareá, J.M. The Identification of ATP-citrate Lyase as a Protein Kinase B (Akt) Substrate in Primary Adipocytes. J. Biol. Chem. 2002, 277, 33895–33900. [Google Scholar] [CrossRef] [Green Version]
- Bauer, D.E.; Hatzivassiliou, G.; Zhao, F.; Andreadis, C.; Thompson, C.B. ATP citrate lyase is an important component of cell growth and transformation. Oncogene 2005, 24, 6314–6322. [Google Scholar] [CrossRef] [Green Version]
- Potapova, I.A.; El-Maghrabi, M.R.; Doronin, S.V.; Benjamin, W.B. Phosphorylation of Recombinant Human ATP:Citrate Lyase by cAMP-Dependent Protein Kinase Abolishes Homotropic Allosteric Regulation of the Enzyme by Citrate and Increases the Enzyme Activity. Allosteric Activation of ATP:Citrate Lyase by Phosphorylated Sug. Biochemistry 2000, 39, 1169–1179. [Google Scholar] [CrossRef]
- Zhao, J.; Zhang, X.; Gao, T.; Wang, S.; Hou, Y.; Yuan, P.; Yang, Y.; Yang, T.; Xing, J.; Li, J.; et al. SIK2 enhances synthesis of fatty acid and cholesterol in ovarian cancer cells and tumor growth through PI3K/Akt signaling pathway. Cell Death Dis. 2020, 11, 25. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.V.; Carrer, A.; Shah, S.; Snyder, N.W.; Wei, S.; Venneti, S.; Worth, A.J.; Yuan, Z.F.; Lim, H.W.; Liu, S.; et al. Akt-dependent metabolic reprogramming regulates tumor cell Histone acetylation. Cell Metab. 2014, 20, 306–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hanai, J.I.; Doro, N.; Sasaki, A.T.; Kobayashi, S.; Cantley, L.C.; Seth, P.; Sukhatme, V.P. Inhibition of lung cancer growth: ATP citrate lyase knockdown and statin treatment leads to dual blockade of mitogen-activated protein Kinase (MAPK) and Phosphatidylinositol-3-kinase (PI3K)/AKT pathways. J. Cell. Physiol. 2012, 227, 1709–1720. [Google Scholar] [CrossRef] [Green Version]
- Hanai, J.; Doro, N.; Seth, P.; Sukhatme, V.P. ATP citrate lyase knockdown impacts cancer stem cells in vitro. Cell Death Dis. 2013, 4, e696. [Google Scholar] [CrossRef] [Green Version]
- Han, Q.; Chen, C.-A.; Yang, W.; Liang, D.; Lv, H.-W.; Lv, G.-S.; Zong, Q.-N.; Wang, H.-Y. ATP-citrate lyase regulates stemness and metastasis in hepatocellular carcinoma via the Wnt/β-catenin signaling pathway. Hepatobiliary Pancreat. Dis. Int. 2020, in press. [Google Scholar] [CrossRef]
- Lecarpentier, Y.; Schussler, O.; Hébert, J.-L.; Vallée, A. Multiple Targets of the Canonical WNT/β-Catenin Signaling in Cancers. Front. Oncol. 2019, 9, 1248. [Google Scholar] [CrossRef]
- Lee, S.; Choi, E.J.; Cho, E.J.; Lee, Y.B.; Lee, J.-H.; Yu, S.J.; Yoon, J.-H.; Kim, Y.J. Inhibition of PI3K/Akt signaling suppresses epithelial-to-mesenchymal transition in hepatocellular carcinoma through the Snail/GSK-3/beta-catenin pathway. Clin. Mol. Hepatol. 2020, 26, 529–539. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Settleman, J. EMT, cancer stem cells and drug resistance: An emerging axis of evil in the war on cancer. Oncogene 2010, 29, 4741–4751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lincet, H.; Icard, P. How do glycolytic enzymes favour cancer cell proliferation by nonmetabolic functions? Oncogene 2015, 34, 3751–3759. [Google Scholar] [CrossRef]
- Jacquin, M.A.; Chiche, J.; Zunino, B.; Bénéteau, M.; Meynet, O.; Pradelli, L.A.; Marchetti, S.; Cornille, A.; Carles, M.; Ricci, J.-E. GAPDH binds to active Akt, leading to Bcl-xL increase and escape from caspase-independent cell death. Cell Death Differ. 2013, 20, 1043–1054. [Google Scholar] [CrossRef] [Green Version]
- Carujo, S.; Estanyol, J.M.; Ejarque, A.; Agell, N.; Bachs, O.; Pujol, M.J. Glyceraldehyde 3-phosphate dehydrogenase is a SET-binding protein and regulates cyclin B-cdk1 activity. Oncogene 2006, 25, 4033–4042. [Google Scholar] [CrossRef] [Green Version]
- Rodrigues, L.M.; Uribe-Lewis, S.; Madhu, B.; Honess, D.J.; Stubbs, M.; Griffiths, J.R. The action of β-hydroxybutyrate on the growth, metabolism and global histone H3 acetylation of spontaneous mouse mammary tumours: Evidence of a β-hydroxybutyrate paradox. Cancer Metab. 2017, 5, 4. [Google Scholar] [CrossRef] [Green Version]
- Nieman, K.M.; Kenny, H.A.; Penicka, C.V.; Ladanyi, A.; Buell-Gutbrod, R.; Zillhardt, M.R.; Romero, I.L.; Carey, M.S.; Mills, G.B.; Hotamisligil, G.S.; et al. Adipocytes promote ovarian cancer metastasis and provide energy for rapid tumor growth. Nat. Med. 2011, 17, 1498–1503. [Google Scholar] [CrossRef] [Green Version]
- Visweswaran, M.; Arfuso, F.; Warrier, S.; Dharmarajan, A. Aberrant lipid metabolism as an emerging therapeutic strategy to target cancer stem cells. Stem Cells 2020, 38, 6–14. [Google Scholar] [CrossRef] [Green Version]
- Camarda, R.; Zhou, A.Y.; Kohnz, R.A.; Balakrishnan, S.; Mahieu, C.; Anderton, B.; Eyob, H.; Kajimura, S.; Tward, A.; Krings, G.; et al. Inhibition of fatty acid oxidation as a therapy for MYC-overexpressing triple-negative breast cancer. Nat. Med. 2016, 22, 427–432. [Google Scholar] [CrossRef]
- Ito, K.; Carracedo, A.; Weiss, D.; Arai, F.; Ala, U.; Avigan, D.E.; Schafer, Z.T.; Evans, R.M.; Suda, T.; Lee, C.-H.; et al. A PML–PPAR-δ pathway for fatty acid oxidation regulates hematopoietic stem cell maintenance. Nat. Med. 2012, 18, 1350–1358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carracedo, A.; Weiss, D.; Leliaert, A.K.; Bhasin, M.; de Boer, V.C.J.; Laurent, G.; Adams, A.C.; Sundvall, M.; Song, S.J.; Ito, K.; et al. A metabolic prosurvival role for PML in breast cancer. J. Clin. Investig. 2012, 122, 3088–3100. [Google Scholar] [CrossRef] [PubMed]
- Mihaylova, M.M.; Shaw, R.J. The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nat. Cell Biol. 2011, 13, 1016–1023. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Liu, T.; Zhou, J.; Wang, Y.; Wang, X.; Di, W.; Zhang, S. Citrate synthase expression affects tumor phenotype and drug resistance in human ovarian carcinoma. PLoS ONE 2014, 9, e115708. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schlichtholz, B.; Turyn, J.; Goyke, E.; Biernacki, M.; Jaskiewicz, K.; Sledzinski, Z.; Swierczynski, J. Enhanced citrate synthase activity in human pancreatic cancer. Pancreas 2005, 30, 99–104. [Google Scholar] [CrossRef]
- Aleskandarany, M.A.; Negm, O.H.N.; Rakha, E.A.; Ahmed, M.A.H.; Nolan, C.C.; Ball, G.R.; Caldas, C.; Green, A.R.; Tighe, P.J.; Ellis, I.O. TOMM34 expression in early invasive breast cancer: A biomarker associated with poor outcome. Breast Cancer Res. Treat. 2012, 136, 419–427. [Google Scholar] [CrossRef]
- Icard, P.; Kafara, P.; Steyaert, J.-M.; Schwartz, L.; Lincet, H. The metabolic cooperation between cells in solid cancer tumors. Biochim. Biophys. Acta Rev. Cancer 2014, 1846, 216–225. [Google Scholar] [CrossRef] [PubMed]
- Curry, J.M.; Tuluc, M.; Whitaker-Menezes, D.; Ames, J.A.; Anantharaman, A.; Butera, A.; Leiby, B.; Cognetti, D.; Sotgia, F.; Lisanti, M.P.; et al. Cancer metabolism, stemness and tumor recurrence. Cell Cycle 2013, 12, 1371–1384. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bonuccelli, G.; Tsirigos, A.; Whitaker-Menezes, D.; Pavlides, S.; Pestell, R.G.; Chiavarina, B.; Frank, P.G.; Flomenberg, N.; Howell, A.; Martinez-Outschoorn, U.E.; et al. Ketones and lactate “fuel” tumor growth and metastasis. Cell Cycle 2010, 9, 3506–3514. [Google Scholar] [CrossRef]
- Corbet, C.; Pinto, A.; Martherus, R.; Santiago de Jesus, J.P.; Polet, F.; Feron, O. Acidosis Drives the Reprogramming of Fatty Acid Metabolism in Cancer Cells through Changes in Mitochondrial and Histone Acetylation. Cell Metab. 2016, 24, 311–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hillar, M.; Lott, V.; Lennox, B. Correlation of the effects of citric acid cycle metabolites on succinate oxidation by rat liver mitochondria and submitochondrial particles. J. Bioenerg. 1975, 7, 1–15. [Google Scholar] [CrossRef]
- Fink, B.D.; Bai, F.; Yu, L.; Sheldon, R.D.; Sharma, A.; Taylor, E.B.; Sivitz, W.I. Oxaloacetic acid mediates ADP-dependent inhibition of mitochondrial complex II-driven respiration. J. Biol. Chem. 2018, 293, 19932–19941. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bae, J.; Salamon, R.J.; Brandt, E.B.; Paltzer, W.G.; Zhang, Z.; Britt, E.C.; Hacker, T.A.; Fan, J.; Mahmoud, A.I. Malonate Promotes Adult Cardiomyocyte Proliferation and Heart Regeneration. Circulation 2021, 143, 1973–1986. [Google Scholar] [CrossRef]
- Eidelman, E.; Twum-Ampofo, J.; Ansari, J.; Siddiqui, M.M. The Metabolic Phenotype of Prostate Cancer. Front. Oncol. 2017, 7, 131. [Google Scholar] [CrossRef]
- Liu, Y. Fatty acid oxidation is a dominant bioenergetic pathway in prostate cancer. Prostate Cancer Prostatic Dis. 2006, 9, 230–234. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costello, L.C.; Franklin, R.B. A comprehensive review of the role of zinc in normal prostate function and metabolism; and its implications in prostate cancer. Arch. Biochem. Biophys. 2016, 611, 100–112. [Google Scholar] [CrossRef] [Green Version]
- Andersen, M.K.; Høiem, T.S.; Claes, B.S.R.; Balluff, B.; Martin-Lorenzo, M.; Richardsen, E.; Krossa, S.; Bertilsson, H.; Heeren, R.M.A.; Rye, M.B.; et al. Spatial differentiation of metabolism in prostate cancer tissue by MALDI-TOF MSI. Cancer Metab. 2021, 9, 9. [Google Scholar] [CrossRef] [PubMed]
- Giskeødegård, G.F.; Bertilsson, H.; Selnæs, K.M.; Wright, A.J.; Bathen, T.F.; Viset, T.; Halgunset, J.; Angelsen, A.; Gribbestad, I.S.; Tessem, M.-B. Spermine and Citrate as Metabolic Biomarkers for Assessing Prostate Cancer Aggressiveness. PLoS ONE 2013, 8, e62375. [Google Scholar] [CrossRef] [Green Version]
- Campbell, P.N.; Smith, A.D.; Peters, T.J. Biochemistry Illustrated; Churchill Livingstone: London, UK, 2005; ISBN 978-0443100345. [Google Scholar]
- Caruana, I.; Simula, L.; Locatelli, F.; Campello, S. T lymphocytes against solid malignancies: Winning ways to defeat tumours. Cell Stress 2018, 2, 200–212. [Google Scholar] [CrossRef] [Green Version]
- Ho, P.-C.; Liu, P.-S. Metabolic communication in tumors: A new layer of immunoregulation for immune evasion. J. Immunother. Cancer 2016, 4, 4. [Google Scholar] [CrossRef] [Green Version]
- Chang, C.H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.W.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sukumar, M.; Roychoudhuri, R.; Restifo, N.P. Nutrient Competition: A New Axis of Tumor Immunosuppression. Cell 2015, 162, 1206–1208. [Google Scholar] [CrossRef] [Green Version]
- Michalek, R.D.; Gerriets, V.A.; Jacobs, S.R.; Macintyre, A.N.; MacIver, N.J.; Mason, E.F.; Sullivan, S.A.; Nichols, A.G.; Rathmell, J.C. Cutting edge: Distinct glycolytic and lipid oxidative metabolic programs are essential for effector and regulatory CD4+ T cell subsets. J. Immunol. 2011, 186, 3299–3303. [Google Scholar] [CrossRef] [Green Version]
- Rueda, C.M.; Rodríguez-Perea, A.L.; Moreno-Fernandez, M.; Jackson, C.M.; Melchior, J.T.; Davidson, W.S.; Chougnet, C.A. High density lipoproteins selectively promote the survival of human regulatory T cells. J. Lipid Res. 2017, 58, 1514–1523. [Google Scholar] [CrossRef] [Green Version]
- Cho, Y.; Cho, E.J.; Lee, J.-H.; Yu, S.J.; Kim, Y.J.; Kim, C.Y.; Yoon, J.-H. Hypoxia Enhances Tumor-Stroma Crosstalk that Drives the Progression of Hepatocellular Carcinoma. Dig. Dis. Sci. 2016, 61, 2568–2577. [Google Scholar] [CrossRef]
- Pavlides, S.; Whitaker-Menezes, D.; Castello-Cros, R.; Flomenberg, N.; Witkiewicz, A.K.; Frank, P.G.; Casimiro, M.C.; Wang, C.; Fortina, P.; Addya, S.; et al. The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009, 8, 3984–4001. [Google Scholar] [CrossRef] [Green Version]
- Icard, P.; Loi, M.; Wu, Z.; Ginguay, A.; Lincet, H.; Robin, E.; Coquerel, A.; Berzan, D.; Fournel, L.; Alifano, M. Metabolic Strategies for Inhibiting Cancer Development. Adv. Nutr. 2021, nmaa174. [Google Scholar] [CrossRef] [PubMed]
- Weiss, J.M.; Davies, L.C.; Karwan, M.; Ileva, L.; Ozaki, M.K.; Cheng, R.Y.S.; Ridnour, L.A.; Annunziata, C.M.; Wink, D.A.; McVicar, D.W. Itaconic acid mediates crosstalk between macrophage metabolism and peritoneal tumors. J. Clin. Investig. 2018, 128, 3794–3805. [Google Scholar] [CrossRef]
- O’Neill, L.A.J.; Artyomov, M.N. Itaconate: The poster child of metabolic reprogramming in macrophage function. Nat. Rev. Immunol. 2019, 19, 273–281. [Google Scholar] [CrossRef] [PubMed]
- Mycielska, M.E.; Dettmer, K.; Rümmele, P.; Schmidt, K.; Prehn, C.; Milenkovic, V.M.; Jagla, W.; Madej, G.M.; Lantow, M.; Schladt, M.; et al. Extracellular Citrate Affects Critical Elements of Cancer Cell Metabolism and Supports Cancer Development In Vivo. Cancer Res. 2018, 78, 2513–2523. [Google Scholar] [CrossRef] [Green Version]
- Haferkamp, S.; Drexler, K.; Federlin, M.; Schlitt, H.J.; Berneburg, M.; Adamski, J.; Gaumann, A.; Geissler, E.K.; Ganapathy, V.; Parkinson, E.K.; et al. Extracellular Citrate Fuels Cancer Cell Metabolism and Growth. Front. Cell Dev. Biol. 2020, 8, 1518. [Google Scholar] [CrossRef]
- Petillo, A.; Abruzzese, V.; Koshal, P.; Ostuni, A.; Bisaccia, F. Extracellular Citrate Is a Trojan Horse for Cancer Cells. Front. Mol. Biosci. 2020, 7, 593866. [Google Scholar] [CrossRef] [PubMed]
- Costello, L.C.; Franklin, R.B. The implications of the hypocitricemic response to surgery and the role of liver function and hepatocyte metabolism: An important, but neglected, clinical relationship. Liver Res. Disord. Ther. 2018, 4, 114–119. [Google Scholar] [CrossRef] [PubMed]
- Granchi, D.; Baldini, N.; Ulivieri, F.M.; Caudarella, R. Role of Citrate in Pathophysiology and Medical Management of Bone Diseases. Nutrients 2019, 11, 2576. [Google Scholar] [CrossRef] [Green Version]
- Yi, C.H.; Pan, H.; Seebacher, J.; Jang, I.-H.; Hyberts, S.G.; Heffron, G.J.; Vander Heiden, M.G.; Yang, R.; Li, F.; Locasale, J.W.; et al. Metabolic Regulation of Protein N-Alpha-Acetylation by Bcl-xL Promotes Cell Survival. Cell 2011, 146, 607–620. [Google Scholar] [CrossRef] [Green Version]
- Jeong, Y.-M.M.; Lee, J.E.; Kim, S.Y.; Yun, H.-Y.Y.; Baek, K.J.; Kwon, N.S.; Kim, D.-S.S. Enhanced effects of citrate on UVB-induced apoptosis of B16 melanoma cells. Pharmazie 2009, 64, 829–833. [Google Scholar] [CrossRef]
- Kruspig, B.; Nilchian, A.; Orrenius, S.; Zhivotovsky, B.; Gogvadze, V. Citrate kills tumor cells through activation of apical caspases. Cell. Mol. Life Sci. 2012, 69, 4229–4237. [Google Scholar] [CrossRef] [Green Version]
- El Sayed, S.M.; El-Magd, R.M.A.; Shishido, Y.; Yorita, K.; Chung, S.P.; Tran, D.H.; Sakai, T.; Watanabe, H.; Kagami, S.; Fukui, K. D-Amino acid oxidase-induced oxidative stress, 3-bromopyruvate and citrate inhibit angiogenesis, exhibiting potent anticancer effects. J. Bioenerg. Biomembr. 2012, 44, 513–523. [Google Scholar] [CrossRef] [PubMed]
- Xu, X.; LI, B.; Huang, P.; Wan, X.; Qin, Y.; Zhou, L.; Liu, H.; Bai, H.; Gao, Y.; Wang, C.; et al. Citrate induces apoptosis of the acute monocytic leukemia U937 cell line through regulation of HIF-1α signaling. Mol. Med. Rep. 2013, 8, 1379–1384. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Lv, Q.; Liu, Y.; Deng, W. Effect of food additive citric acid on the growth of human esophageal carcinoma cell line EC109. Cell J. 2016, 18, 493–502. [Google Scholar] [CrossRef] [PubMed]
- Xia, Y.; Zhang, X.; Bo, A.; Sun, J.; Li, M. Sodium citrate inhibits the proliferation of human gastric adenocarcinoma epithelia cells. Oncol. Lett. 2018, 15, 6622–6628. [Google Scholar] [CrossRef] [PubMed]
- Abe, K.; Yamamoto, N.; Hayashi, K.; Takeuchi, A.; Tsuchiya, H. Caffeine citrate enhanced cisplatin antitumor effects in osteosarcoma and fibrosarcoma in vitro and in vivo. BMC Cancer 2019, 19, 689. [Google Scholar] [CrossRef] [Green Version]
- Fagundes, D.A.; Leonel, L.V.; Fernandez-Outon, L.E.; Ardisson, J.D.; dos Santos, R.G. Radiosensitizing effects of citrate-coated cobalt and nickel ferrite nanoparticles on breast cancer cells. Nanomedicine 2020, 15, 2823–2836. [Google Scholar] [CrossRef] [PubMed]
- Zhou, S.; Sakamoto, K. Citric acid promoted melanin synthesis in B16F10 mouse melanoma cells, but inhibited it in human epidermal melanocytes and HMVII melanoma cells via the GSK3β/β-catenin signaling pathway. PLoS ONE 2020, 15, e0243565. [Google Scholar] [CrossRef]
- Fan, X.; Zhou, J.; Yan, X.; Bi, X.; Liang, J.; Lu, S.; Luo, L.; Zhou, D.; Yin, Z. Citrate activates autophagic death of prostate cancer cells via downregulation CaMKII/AKT/mTOR pathway. Life Sci. 2021, 275, 119355. [Google Scholar] [CrossRef] [PubMed]
- Ando, H.; Eshima, K.; Ishida, T. Neutralization of Acidic Tumor Microenvironment (TME) with Daily Oral Dosing of Sodium Potassium Citrate (K/Na Citrate) Increases Therapeutic Effect of Anti-cancer Agent in Pancreatic Cancer Xenograft Mice Model. Biol. Pharm. Bull. 2021, 44, 266–270. [Google Scholar] [CrossRef]
- Iacobazzi, V.; Infantino, V. Citrate-new functions for an old metabolite. Biol. Chem. 2014, 395, 387–399. [Google Scholar] [CrossRef]
- Huang, L.; Wang, C.; Xu, H.; Peng, G. Targeting citrate as a novel therapeutic strategy in cancer treatment. Biochim. Biophys. Acta Rev. Cancer 2020, 1873, 188332. [Google Scholar] [CrossRef] [PubMed]
- Philippe, I.; Lincet, H. The reduced concentration of citrate in cancer cells: An indicator of cancer aggressiveness and a possible therapeutic target. Drug Resist. Update 2016, 29, 47–53. [Google Scholar] [CrossRef]
- Simon-Molas, H.; Arnedo-Pac, C.; Fontova, P.; Vidal-Alabró, A.; Castaño, E.; Rodríguez-García, A.; Navarro-Sabaté, À.; Lloberas, N.; Manzano, A.; Bartrons, R. PI3K–Akt signaling controls PFKFB3 expression during human T-lymphocyte activation. Mol. Cell. Biochem. 2018, 448, 187–197. [Google Scholar] [CrossRef]
- Mottet, D.; Dumont, V.; Deccache, Y.; Demazy, C.; Ninane, N.; Raes, M.; Michiels, C. Regulation of hypoxia-inducible factor-1α protein level during hypoxic conditions by the phosphatidylinositol 3-kinase/Akt/glycogen synthase kinase 3β pathway in HepG2 cells. J. Biol. Chem. 2003, 278, 31277–31285. [Google Scholar] [CrossRef] [Green Version]
- Raghunand, N.; He, X.; van Sluis, R.; Mahoney, B.; Baggett, B.; Taylor, C.W.; Paine-Murrieta, G.; Roe, D.; Bhujwalla, Z.M.; Gillies, R.J.; et al. Enhancement of chemotherapy by manipulation of tumour pH. Br. J. Cancer 1999, 80, 1005–1011. [Google Scholar] [CrossRef] [Green Version]
- Faes, S.; Duval, A.P.; Planche, A.; Uldry, E.; Santoro, T.; Pythoud, C.; Stehle, J.-C.C.; Horlbeck, J.; Letovanec, I.; Riggi, N.; et al. Acidic tumor microenvironment abrogates the efficacy of mTORC1 inhibitors. Mol. Cancer 2016, 15, 78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pilon-Thomas, S.; Kodumudi, K.N.; El-Kenawi, A.E.; Russell, S.; Weber, A.M.; Luddy, K.; Damaghi, M.; Wojtkowiak, J.W.; Mulé, J.J.; Ibrahim-Hashim, A.; et al. Neutralization of Tumor Acidity Improves Antitumor Responses to Immunotherapy. Cancer Res. 2016, 76, 1381–1390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mycielska, M.E.; Geissler, E.K. Extracellular Citrate and Cancer Metabolism—Response. Cancer Res. 2018, 78, 5177. [Google Scholar] [CrossRef] [Green Version]
- Caiazza, C.; D’Agostino, M.; Passaro, F.; Faicchia, D.; Mallardo, M.; Paladino, S.; Pierantoni, G.M.; Tramontano, D. Effects of Long-Term Citrate Treatment in the PC3 Prostate Cancer Cell Line. Int. J. Mol. Sci. 2019, 20, 2613. [Google Scholar] [CrossRef] [Green Version]
- Guo, J.Y.; White, E. Autophagy is required for mitochondrial function, lipid metabolism, growth, and fate of KRAS G12D -driven lung tumors. Autophagy 2013, 9, 1636–1638. [Google Scholar] [CrossRef] [Green Version]
- Schug, Z.T.; Vande Voorde, J.; Gottlieb, E. The metabolic fate of acetate in cancer. Nat. Rev. Cancer 2016, 16, 708–717. [Google Scholar] [CrossRef] [PubMed]
- Diaz-Moralli, S.; Aguilar, E.; Marin, S.; Coy, J.F.; Dewerchin, M.; Antoniewicz, M.R.; Meca-Cortés, O.; Notebaert, L.; Ghesquière, B.; Eelen, G.; et al. A key role for transketolase-like 1 in tumor metabolic reprogramming. Oncotarget 2016, 7, 51875–51897. [Google Scholar] [CrossRef] [PubMed]
- Singh, S. Preclinical Pharmacokinetics: An Approach Towards Safer and Efficacious Drugs. Curr. Drug Metab. 2006, 7, 165–182. [Google Scholar] [CrossRef] [PubMed]
- D’Angelo, A.; Sobhani, N.; Chapman, R.; Bagby, S.; Bortoletti, C.; Traversini, M.; Ferrari, K.; Voltolini, L.; Darlow, J.; Roviello, G. Focus on ROS1-Positive Non-Small Cell Lung Cancer (NSCLC): Crizotinib, Resistance Mechanisms and the Newer Generation of Targeted Therapies. Cancers 2020, 12, 3293. [Google Scholar] [CrossRef] [PubMed]
- Romaniello, D.; Marrocco, I.; Belugali Nataraj, N.; Ferrer, I.; Drago-Garcia, D.; Vaknin, I.; Oren, R.; Lindzen, M.; Ghosh, S.; Kreitman, M.; et al. Targeting HER3, a Catalytically Defective Receptor Tyrosine Kinase, Prevents Resistance of Lung Cancer to a Third-Generation EGFR Kinase Inhibitor. Cancers 2020, 12, 2394. [Google Scholar] [CrossRef]


{kind=link}
| Cells | Cancer Type | In Vivo/In Vitro | Citrate Targets/Citrate Effects on Cells | Year | Ref. |
|---|---|---|---|---|---|
| MSTO-211H cells | Mesothelioma cells | In vitro | Decreased expression of anti-apoptotic protein Mcl-1 and Bcl-xL. Depletion of ATP. Increase in the cisplatin antitumor effect. | 2009 | [25] |
| B16 cells | Murine melanoma | In vitro | Combined treatment with UVB increases c-Jun and p38 phosphorylation and the activity of caspase-3 and -9. | 2009 | [136] |
| HeLa cells | Adenocarcinoma | In vitro | Increase in the protein N-alpha-acetylation. Sensitivity to apoptotic stimuli. | 2011 | [135] |
| BGC-823 cells SGC-7901 cells | Gastric cancer | In vitro | Decreased expression of anti-apoptotic protein Mcl-1. | 2011 | [26] |
| Tet21N cells SKNAS cells SKNSH cells SK-N-BE(2) cells U1810 cells | Neuroblastoma Neuroblastoma Neuroblastoma Neuroblastoma Lung cancer | In vitro | Activation of the apical caspases-8 and -2. Increased release of the cytochrome C. | 2012 | [137] |
| C6 cells | Glioblastoma | In vitro | Inhibition of the number of vascular branching points. Inhibition of the length of vascular tubules. Inhibition of angiogenesis. | 2012 | [138] |
| SKOV3 cells IGROV1-R10 cells | Ovarian cancer | In vitro | Decreased expression of the anti-apoptotic protein Mcl-1. Increase in the efficacy of ABT 737 treatment. | 2013 | [27] |
| U937 cells | Acute Monocytic Leukemia | In vitro | Increase in caspase-3 and -9 activities. Decreased expression of anti-apoptotic protein Bcl-2. | 2013 | [139] |
| MGC-803 cells | Gastric cancer | In vitro | Increased expression of the pro-apoptotic protein Bax. Inhibition of the PFK activity. Decrease in lactate and ATP production. | 2016 | [76] |
| SGC-7901 cells | Gastric cancer | In vitro and in vivo | Inhibition of the PFK1 activity. Decreased tumor growth and increased apoptosis (increased cytC release and Bax expression, reduced Bcl-2). | 2016 | [75] |
| A549 cells Pan02 cells Her2/Neu model | Lung cancer Pancreatic cancer Breast cancer model | In vitro and in vivo | Inhibition of IGF-1R/AKT pathway via PTEN. Inhibition of tumor growth in Ras-driven lung cancer, Pan02 xenograft and Her2/Neu models. | 2017 | [77] |
| EC109 cells | Oesophageal cancer | In vitro | Increase in apoptosis. | 2017 | [140] |
| AGS cells | Gastric cancer | In vitro | Increase in the levels of interleukin-1β, IL-8 and TNF. Increased activity of caspase-3 and -9. | 2018 | [141] |
| HOS & LM8 cells HT1080 cells | Osteosarcoma Fibrosarcoma | In vitro and in vivo | Enhancement of cisplatin anti-tumor effect. | 2019 | [142] |
| PSC cells | Pharyngeal carcinoma | In vitro | Cell cycle arrest at the G2/M phase. Stabilization of cyclinB1-CDK1 through p85α-PTEN. | 2019 | [78] |
| MCF-7 cells | Breast cancer | In vitro | Increase in the efficacy of radiation therapy. | 2020 | [143] |
| HMV-II cells | Melanoma | In vitro | Inhibition of cancer cell proliferation. Reduction in β-catenin levels. | 2020 | [144] |
| HepG2 cells | Hepatoma | In vitro | Inhibition of the ACLY-mediated H4 acetylation and lipid deposition. | 2020 | [132] |
| PCa cells | Prostate cancer | In vitro and in vivo | Induction of cell death through autophagy modulation. Inhibition of tumor growth in a xenograft model. | 2021 | [145] |
| Panc-1 cells | Pancreatic cancer | In vivo | Neutralization of TME acidity. Potentiation of anti-tumor effect of 5-FU derivative. Reduction in tumor growth (xenograft model). | 2021 | [146] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Icard, P.; Coquerel, A.; Wu, Z.; Gligorov, J.; Fuks, D.; Fournel, L.; Lincet, H.; Simula, L. Understanding the Central Role of Citrate in the Metabolism of Cancer Cells and Tumors: An Update. Int. J. Mol. Sci. 2021, 22, 6587. https://doi.org/10.3390/ijms22126587
Icard P, Coquerel A, Wu Z, Gligorov J, Fuks D, Fournel L, Lincet H, Simula L. Understanding the Central Role of Citrate in the Metabolism of Cancer Cells and Tumors: An Update. International Journal of Molecular Sciences. 2021; 22(12):6587. https://doi.org/10.3390/ijms22126587
Chicago/Turabian StyleIcard, Philippe, Antoine Coquerel, Zherui Wu, Joseph Gligorov, David Fuks, Ludovic Fournel, Hubert Lincet, and Luca Simula. 2021. "Understanding the Central Role of Citrate in the Metabolism of Cancer Cells and Tumors: An Update" International Journal of Molecular Sciences 22, no. 12: 6587. https://doi.org/10.3390/ijms22126587
APA StyleIcard, P., Coquerel, A., Wu, Z., Gligorov, J., Fuks, D., Fournel, L., Lincet, H., & Simula, L. (2021). Understanding the Central Role of Citrate in the Metabolism of Cancer Cells and Tumors: An Update. International Journal of Molecular Sciences, 22(12), 6587. https://doi.org/10.3390/ijms22126587