
Interleukin-1β Induces Tissue Factor Expression in A549 Cells via EGFR-Dependent and -Independent Mechanisms

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
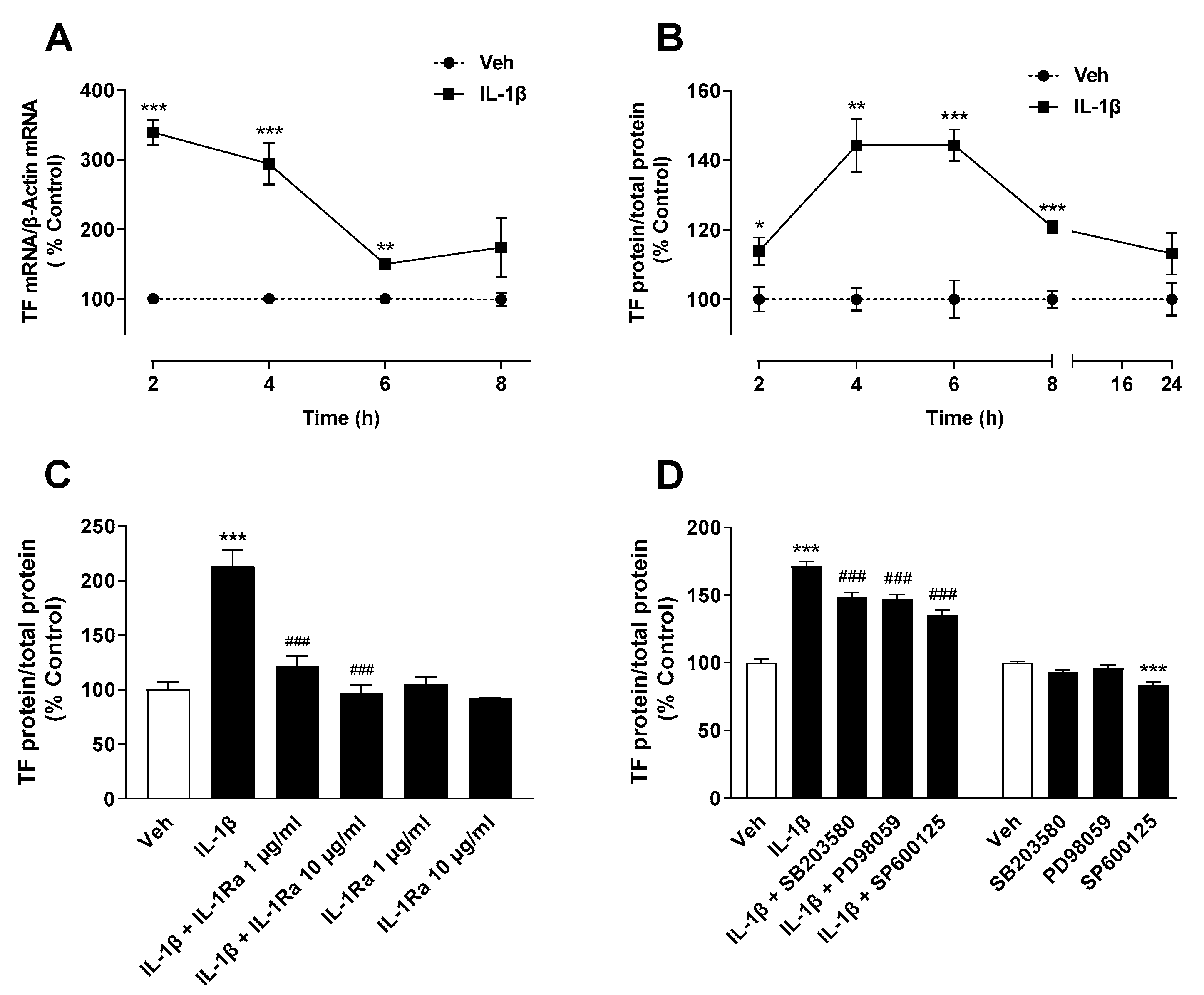
2.1. IL-1β Induces TF Expression in A549 Lung Cancer Cells
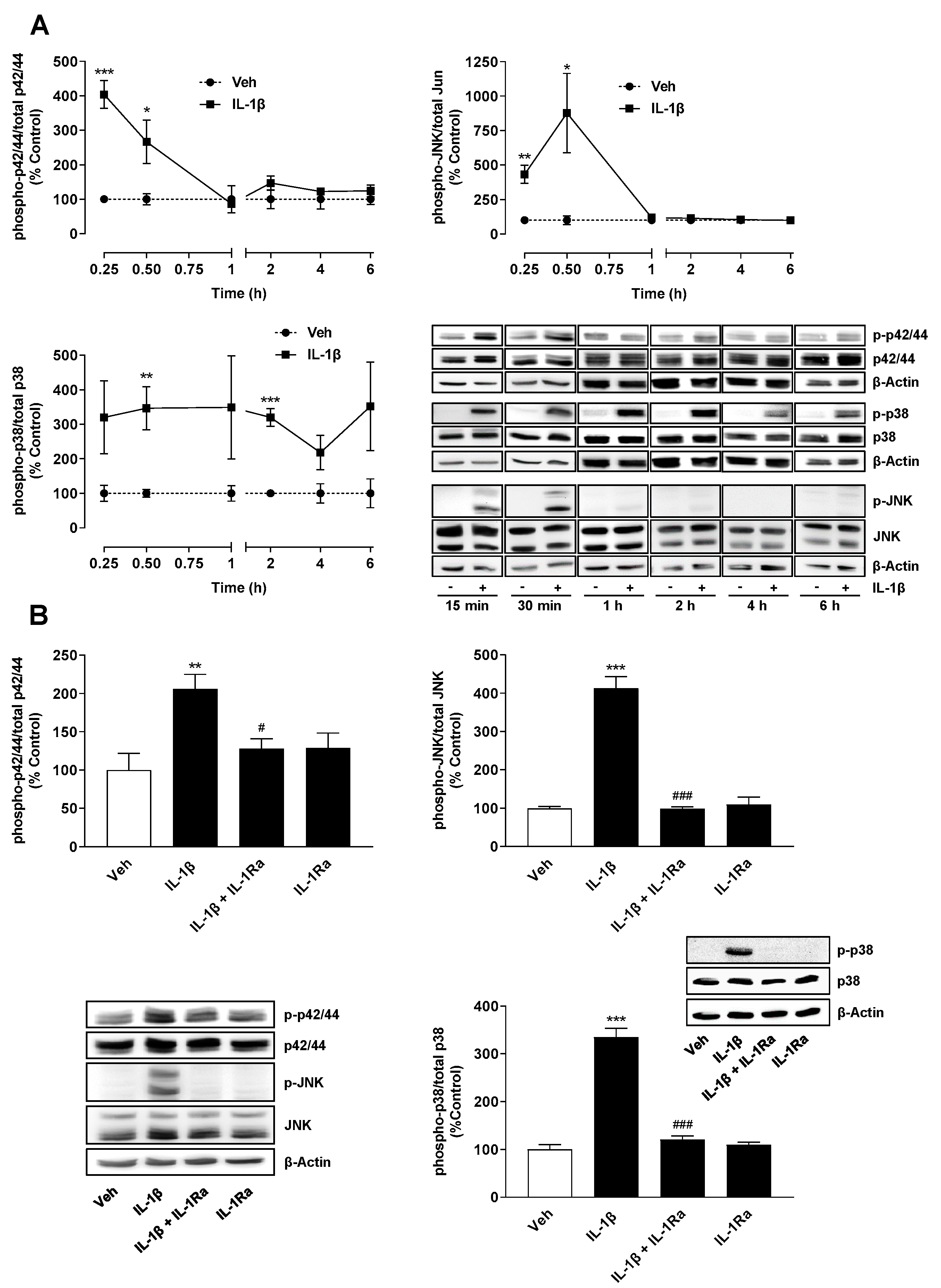
2.2. TF Expression Induced by IL-1β Is Mediated via IL-1 Receptor-Dependent Activation of MAP Kinases p38, p42/44 and JNK
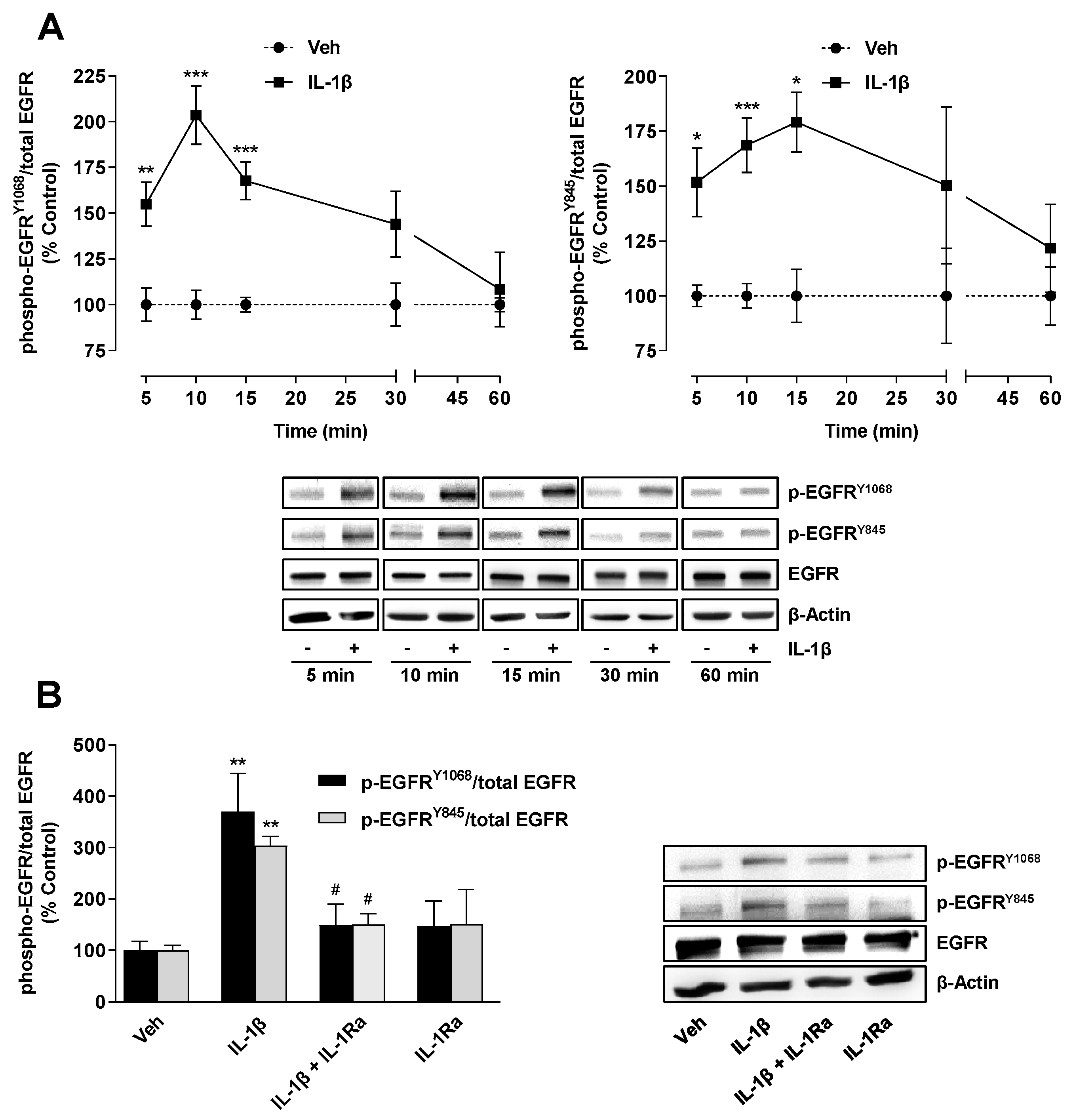
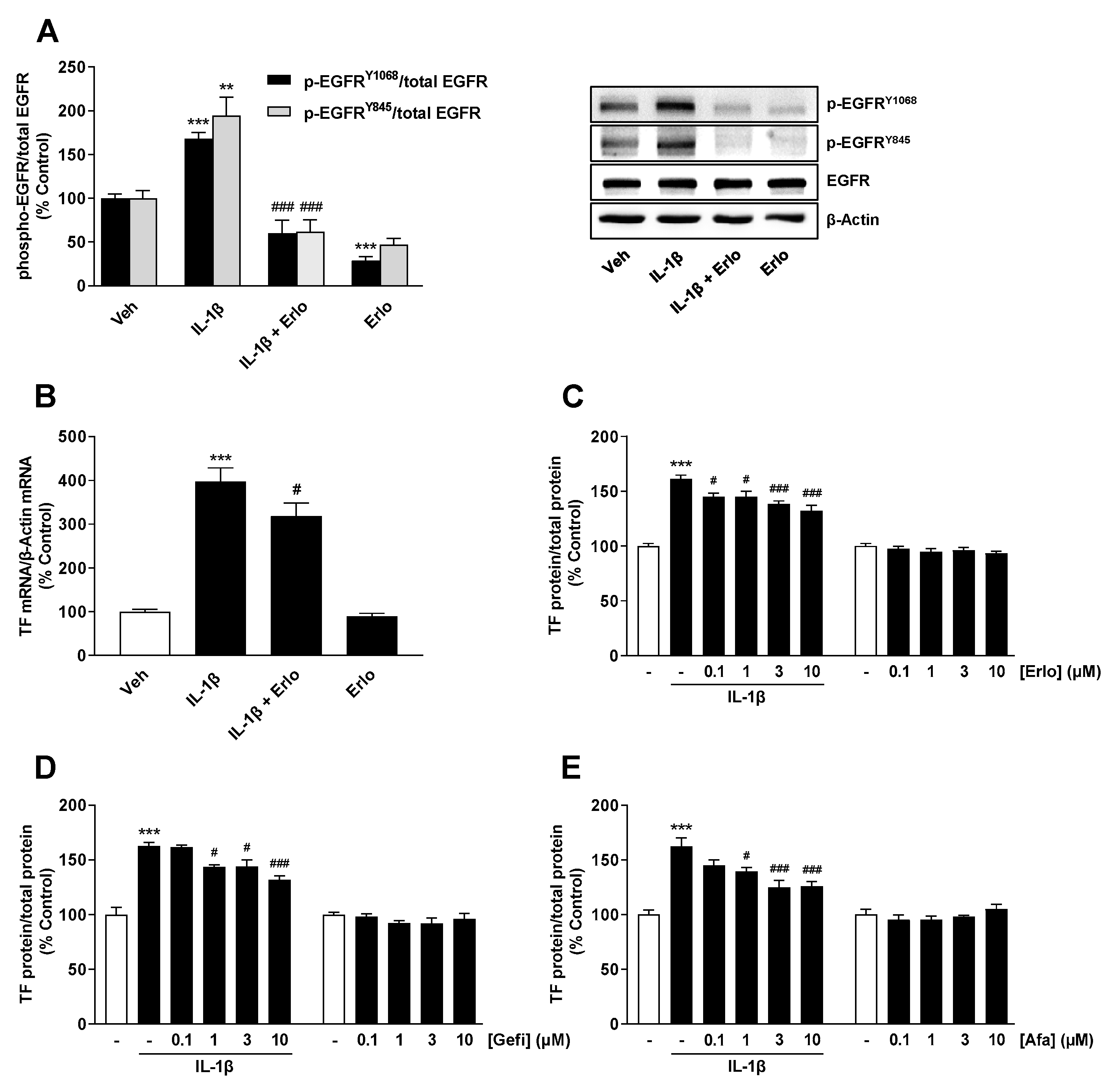
2.3. TF Expression Induced by IL-1β Is Mediated by an IL-1 Receptor-Dependent Activation of EGFR
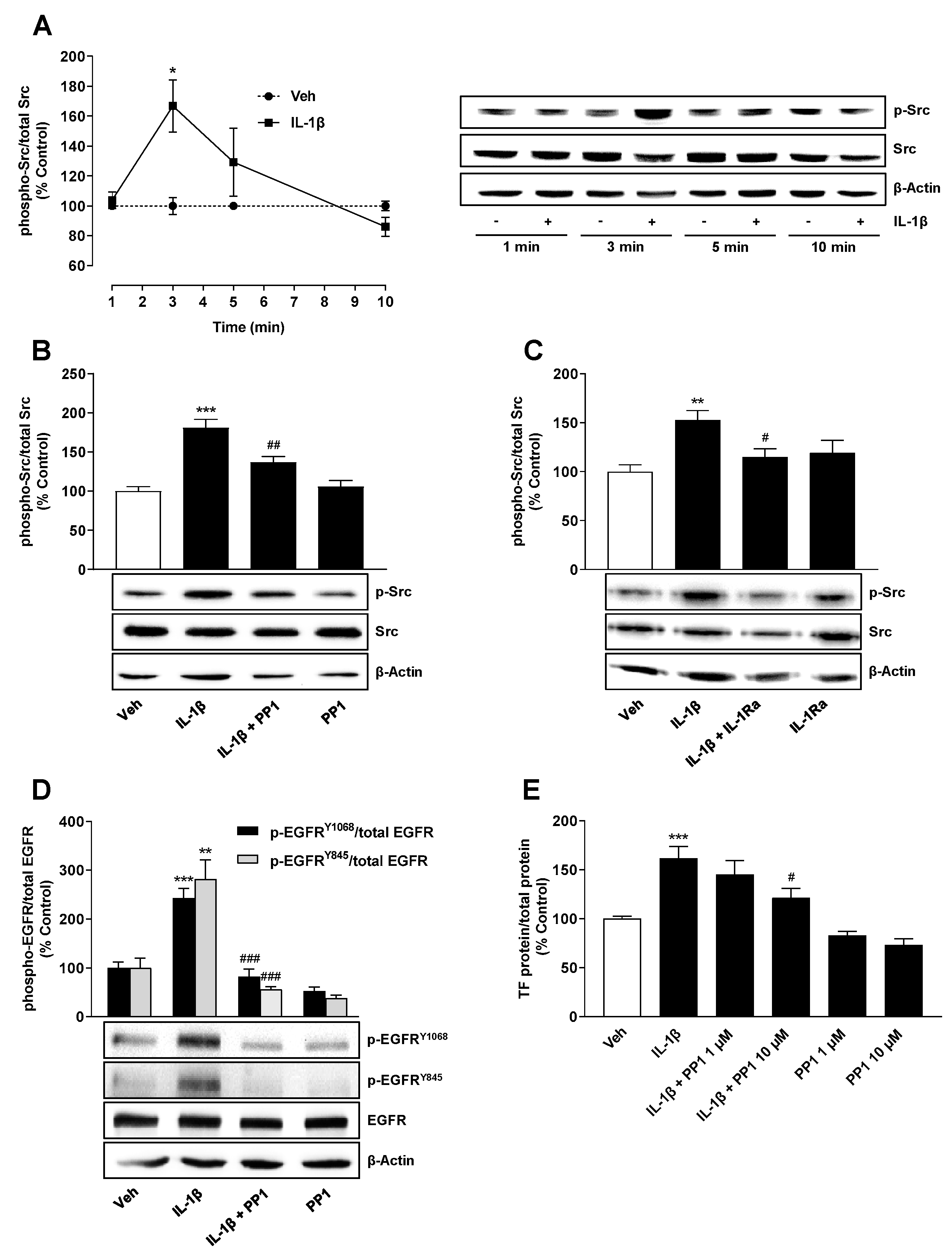
2.4. Src Is Involved in IL-1β-Mediated Transactivation of EGFR Leading to TF Upregulation
2.5. IL-1β Induces TF Expression via Both a Src/EGFR/p42/44 MAPK-Dependent and a Src/EGFR-Independent Pathway Mediated by Activation of p38 MAPK and JNK
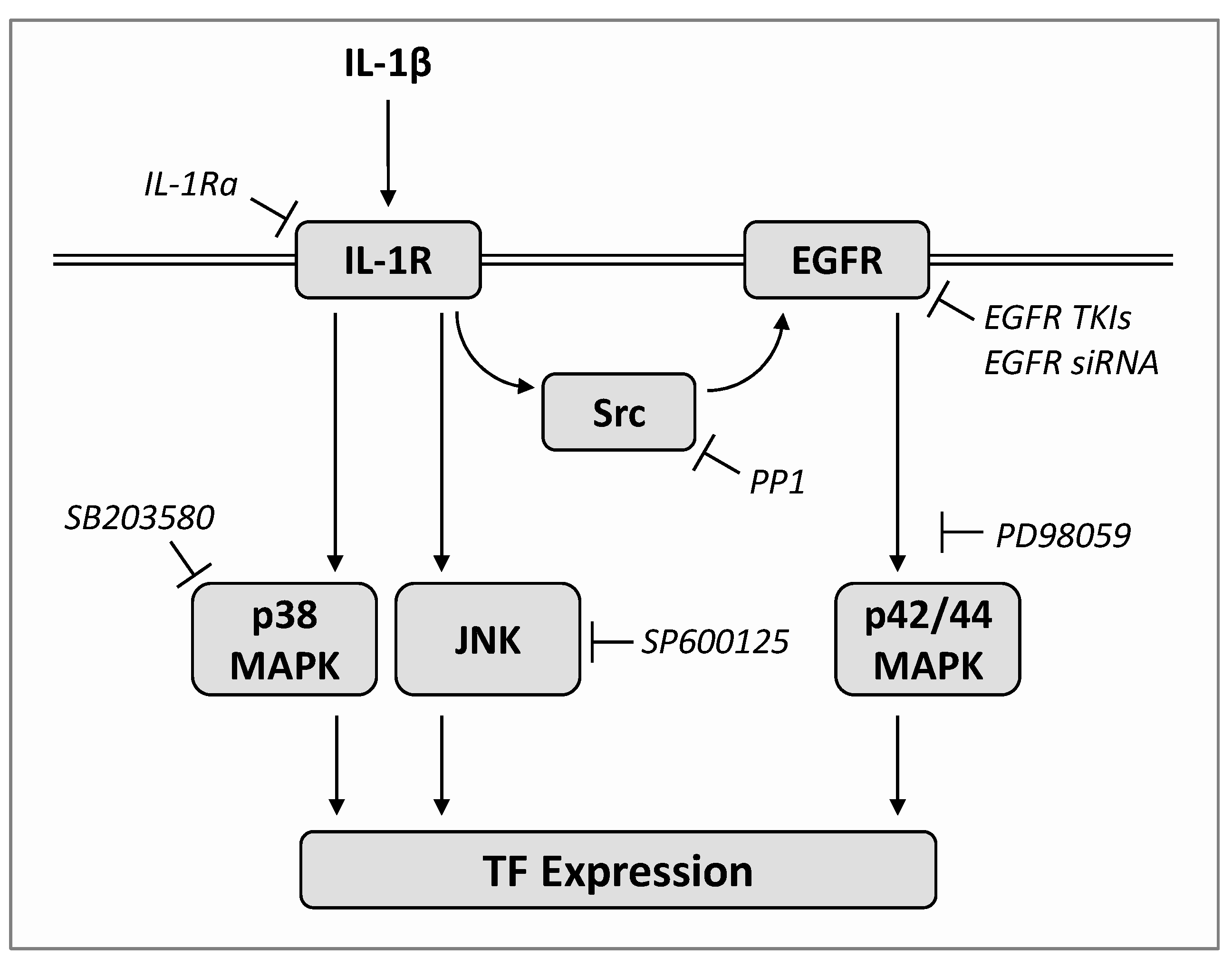
3. Discussion
4. Materials and Methods
4.1. Materials
4.2. Cell Culture
4.3. Determination of TF Protein
4.4. Western Blot Analysis
4.5. Quantitative Reverse Transcriptase Polymerase Chain Reaction (RT-PCR)
4.6. SiRNA Transfections
4.7. Cellular Viability Assay
4.8. Statistics
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Nemerson, Y.; Pitlick, F.A. Purification and characterization of the protein component of tissue factor. Biochemistry 1970, 9, 5100–5105. [Google Scholar] [CrossRef]
- Bjorklid, E.; Storm, E. Purification and some properties of the protein component of tissue thromboplastin from human brain. Biochem. J. 1977, 165, 89–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harlos, K.; Martin, D.M.; O’Brien, D.P.; Jones, E.Y.; Stuart, D.I.; Polikarpov, I.; Miller, A.; Tuddenham, E.G.; Boys, C.W. Crystal structure of the extracellular region of human tissue factor. Nature 1994, 370, 662–666. [Google Scholar] [CrossRef]
- Rak, J.; Milsom, C.; Yu, J. Tissue factor in cancer. Curr. Opin. Hematol. 2008, 15, 522–528. [Google Scholar] [CrossRef]
- Leppert, U.; Eisenreich, A. The role of tissue factor isoforms in cancer biology. Int. J. Cancer 2015, 137, 497–503. [Google Scholar] [CrossRef] [Green Version]
- Van Dreden, P.; Εlalamy, Ι.; Gerotziafas, G.T. The Role of Tissue Factor in Cancer-Related Hypercoagulability, Tumor Growth, Angiogenesis and Metastasis and Future Therapeutic Strategies. Crit. Rev. Oncog. 2017, 22, 219–248. [Google Scholar] [CrossRef]
- Hisada, Y.; Mackman, N. Tissue Factor and Cancer: Regulation, Tumor Growth, and Metastasis. Semin. Thromb. Hemost. 2019, 45, 385–395. [Google Scholar] [CrossRef]
- Jiang, X.; Bailly, M.A.; Panetti, T.S.; Cappello, M.; Konigsberg, W.H.; Bromberg, M.E. Formation of tissue factor-factor VIIa-factor Xa complex promotes cellular signaling and migration of human breast cancer cells. J. Thromb. Haemost. 2004, 2, 93–101. [Google Scholar] [CrossRef] [Green Version]
- Hu, L.; Xia, L.; Zhou, H.; Wu, B.; Mu, Y.; Wu, Y.; Yan, J. TF/FVIIa/PAR2 promotes cell proliferation and migration via PKCα and ERK-dependent c-Jun/AP-1 pathway in colon cancer cell line SW620. Tumour. Biol. 2013, 34, 2573–2581. [Google Scholar] [CrossRef]
- Koomägi, R.; Volm, M. Tissue-factor expression in human non-small-cell lung carcinoma measured by immunohistochemistry: Correlation between tissue factor and angiogenesis. Int. J. Cancer 1998, 79, 19–22. [Google Scholar] [CrossRef]
- Goldin-Lang, P.; Tran, Q.V.; Fichtner, I.; Eisenreich, A.; Antoniak, S.; Schulze, K.; Coupland, S.E.; Poller, W.; Schultheiss, H.P.; Rauch, U. Tissue factor expression pattern in human non-small cell lung cancer tissues indicate increased blood thrombogenicity and tumor metastasis. Oncol. Rep. 2008, 20, 123–128. [Google Scholar] [CrossRef] [Green Version]
- Sawada, M.; Miyake, S.; Ohdama, S.; Matsubara, O.; Masuda, S.; Yakumaru, K.; Yoshizawa, Y. Expression of tissue factor in non-small-cell lung cancers and its relationship to metastasis. Br. J. Cancer 1999, 79, 472–477. [Google Scholar] [CrossRef]
- Regina, S.; Valentin, J.B.; Lachot, S.; Lemarié, E.; Rollin, J.; Gruel, Y. Increased tissue factor expression is associated with reduced survival in non-small cell lung cancer and with mutations of TP53 and PTEN. Clin. Chem. 2009, 55, 1834–1842. [Google Scholar] [CrossRef] [Green Version]
- Milsom, C.C.; Yu, J.L.; Mackman, N.; Micallef, J.; Anderson, G.M.; Guha, A.; Rak, J.W. Tissue factor regulation by epidermal growth factor receptor and epithelial-to-mesenchymal transitions: Effect on tumor initiation and angiogenesis. Cancer Res. 2008, 68, 10068–10076. [Google Scholar] [CrossRef] [Green Version]
- Rong, Y.; Belozerov, V.E.; Tucker-Burden, C.; Chen, G.; Durden, D.L.; Olson, J.J.; Van Meir, E.G.; Mackman, N.; Brat, D.J. Epidermal growth factor receptor and PTEN modulate tissue factor expression in glioblastoma through JunD/activator protein-1 transcriptional activity. Cancer Res. 2009, 69, 2540–2549. [Google Scholar] [CrossRef] [Green Version]
- Magnus, N.; Garnier, D.; Rak, J. Oncogenic epidermal growth factor receptor up-regulates multiple elements of the tissue factor signaling pathway in human glioma cells. Blood 2010, 116, 815–818. [Google Scholar] [CrossRef] [Green Version]
- Yu, J.L.; Xing, R.; Milsom, C.; Rak, J. Modulation of the oncogene-dependent tissue factor expression by kinase suppressor of ras 1. Thromb. Res. 2010, 126, e6–e10. [Google Scholar] [CrossRef] [Green Version]
- Garnier, D.; Magnus, N.; Lee, T.H.; Bentley, V.; Meehan, B.; Milsom, C.; Montermini, L.; Kislinger, T.; Rak, J. Cancer cells induced to express mesenchymal phenotype release exosome-like extracellular vesicles carrying tissue factor. J. Biol. Chem. 2012, 287, 43565–43572. [Google Scholar] [CrossRef] [Green Version]
- Hu, C.; Huang, L.; Gest, C.; Xi, X.; Janin, A.; Soria, C.; Li, H.; Lu, H. Opposite regulation by PI3K/Akt and MAPK/ERK pathways of tissue factor expression, cell-associated procoagulant activity and invasiveness in MDA-MB-231 cells. J. Hematol. Oncol. 2012, 5, 16. [Google Scholar] [CrossRef] [Green Version]
- Kato, S.; Pinto, M.; Carvajal, A.; Espinoza, N.; Monsó, C.; Bravo, L.; Villalon, M.; Cuello, M.; Quest, A.F.; Suenaga, A.; et al. Tissue factor is regulated by epidermal growth factor in normal and malignant human endometrial epithelial cells. Thromb. Haemost. 2005, 94, 444–453. [Google Scholar] [CrossRef] [Green Version]
- Wan, Y.; Belt, A.; Wang, Z.; Voorhees, J.; Fisher, G. Transmodulation of epidermal growth factor receptor mediates IL-1β-induced MMP-1 expression in cultured human keratinocytes. Int. J. Mol. Med. 2001, 7, 329–334. [Google Scholar] [CrossRef]
- Tanida, S.; Joh, T.; Itoh, K.; Kataoka, H.; Sasaki, M.; Ohara, H.; Nakazawa, T.; Nomura, T.; Kinugasa, Y.; Ohmoto, H.; et al. The mechanism of cleavage of EGFR ligands induced by inflammatory cytokines in gastric cancer cells. Gastroenterology 2004, 127, 559–569. [Google Scholar] [CrossRef]
- Cheng, C.Y.; Kuo, C.T.; Lin, C.C.; Hsieh, H.L.; Yang, C.M. IL-1β induces expression of matrix metalloproteinase-9 and cell migration via a c-Src-dependent, growth factor receptor transactivation in A549 cells. Br. J. Pharmacol. 2010, 160, 1595–1610. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Guerrero, E.; Chen, E.; Kockx, M.; An, S.W.; Chong, B.H.; Khachigian, L.M. IL-1beta signals through the EGF receptor and activates Egr-1 through MMP-ADAM. PLoS ONE 2012, 7, e39811. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.H.; Syu, S.H.; Liu, K.J.; Chu, P.Y.; Yang, W.C.; Lin, P.; Shieh, W.Y. Interleukin-1 beta transactivates epidermal growth factor receptor via the CXCL1-CXCR2 axis in oral cancer. Oncotarget 2015, 6, 38866–38880. [Google Scholar] [CrossRef] [Green Version]
- Broekman, W.; Amatngalim, G.D.; de Mooij-Eijk, Y.; Oostendorp, J.; Roelofs, H.; Taube, C.; Stolk, J.; Hiemstra, P.S. TNF-α and IL-1β-activated human mesenchymal stromal cells increase airway epithelial wound healing in vitro via activation of the epidermal growth factor receptor. Respir. Res. 2016, 17, 3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Massip-Copiz, M.; Clauzure, M.; Valdivieso, Á.G.; Santa-Coloma, T.A. Epiregulin (EREG) is upregulated through an IL-1β autocrine loop in Caco-2 epithelial cells with reduced CFTR function. J. Cell. Biochem. 2018, 119, 2911–2922. [Google Scholar] [CrossRef]
- Bevilacqua, M.P.; Pober, J.S.; Majeau, G.R.; Cotran, R.S.; Gimbrone, M.A., Jr. Interleukin 1 (IL-1) induces biosynthesis and cell surface expression of procoagulant activity in human vascular endothelial cells. J. Exp. Med. 1984, 160, 618–623. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Archipoff, G.; Beretz, A.; Freyssinet, J.M.; Klein-Soyer, C.; Brisson, C.; Cazenave, J.P. Heterogeneous regulation of constitutive thrombomodulin or inducible tissue-factor activities on the surface of human saphenous-vein endothelial cells in culture following stimulation by interleukin-1, tumour necrosis factor, thrombin or phorbol ester. Biochem. J. 1991, 273, 679–684. [Google Scholar] [CrossRef] [PubMed]
- Puhlmann, M.; Weinreich, D.M.; Farma, J.M.; Carroll, N.M.; Turner, E.M.; Alexander, H.R., Jr. Interleukin-1β induced vascular permeability is dependent on induction of endothelial tissue factor (TF) activity. J. Transl. Med. 2005, 3, 37. [Google Scholar] [CrossRef] [Green Version]
- Scholl, A.; Ivanov, I.; Hinz, B. Inhibition of interleukin-1β-induced endothelial tissue factor expression by the synthetic cannabinoid WIN 55;212-2. Oncotarget 2016, 7, 61438–61757. [Google Scholar] [CrossRef] [Green Version]
- Osnes, L.T.; Westvik, A.B.; Joø, G.B.; Okkenhaug, C.; Kierulf, P. Inhibition of IL-1 induced tissue factor (TF) synthesis and procoagulant activity (PCA) in purified human monocytes by IL-4, IL-10 and IL-13. Cytokine 1996, 8, 822–827. [Google Scholar] [CrossRef] [PubMed]
- Balkwill, F.; Mantovani, A. Inflammation and cancer: Back to Virchow? Lancet 2001, 357, 539–545. [Google Scholar] [CrossRef]
- De Vita, F.; Orditura, M.; Auriemma, A.; Infusino, S.; Catalano, G. Serum concentrations of proinflammatory cytokines in advanced non small cell lung cancer patients. J. Exp. Clin. Cancer Res. 1998, 17, 413–417. [Google Scholar] [PubMed]
- Lin, C.C.; Kuo, C.T.; Cheng, C.Y.; Wu, C.Y.; Lee, C.W.; Hsieh, H.L.; Lee, I.T.; Yang, C.M. IL-1β promotes A549 cell migration via MAPKs/AP-1- and NF-κB-dependent matrix metalloproteinase-9 expression. Cell Signal. 2009, 21, 1652–1662. [Google Scholar] [CrossRef] [PubMed]
- Eda, H.; Burnette, B.L.; Shimada, H.; Hope, H.R.; Monahan, J.B. Interleukin-1β-induced interleukin-6 production in A549 cells is mediated by both phosphatidylinositol 3-kinase and interleukin-1 receptor-associated kinase-4. Cell Biol. Int. 2011, 35, 355–358. [Google Scholar] [CrossRef]
- Shah, S.; Mostafa, M.M.; McWhae, A.; Traves, S.L.; Newton, R. Negative Feed-forward Control of Tumor Necrosis Factor (TNF) by Tristetraprolin (ZFP36) Is Limited by the Mitogen-activated Protein Kinase Phosphatase, Dual-specificity Phosphatase 1 (DUSP1): IMPLICATIONS FOR REGULATION BY GLUCOCORTICOIDS. J. Biol. Chem. 2016, 291, 110–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aarreberg, L.D.; Esser-Nobis, K.; Driscoll, C.; Shuvarikov, A.; Roby, J.A.; Gale, M., Jr. Interleukin-1β Induces mtDNA Release to Activate Innate Immune Signaling via cGAS-STING. Mol. Cell. 2019, 74, 801.e6–815.e6. [Google Scholar] [CrossRef]
- Kopetz, S. Targeting SRC and epidermal growth factor receptor in colorectal cancer: Rationale and progress into the clinic. Gastrointest. Cancer Res. 2007, 1, S37–S41. [Google Scholar]
- Chen, Z.; Oh, D.; Dubey, A.K.; Yao, M.; Yang, B.; Groves, J.T.; Sheetz, M. EGFR family and Src family kinase interactions: Mechanics matters? Curr. Opin. Cell. Biol. 2018, 51, 97–102. [Google Scholar] [CrossRef]
- Daub, H.; Wallasch, C.; Lankenau, A.; Herrlich, A.; Ullrich, A. Signal characteristics of G protein-transactivated EGF receptor. EMBO J. 1997, 16, 7032–7044. [Google Scholar] [CrossRef] [PubMed]
- Buteau, J.; Foisy, S.; Joly, E.; Prentki, M. Glucagon-like peptide 1 induces pancreatic β-cell proliferation via transactivation of the epidermal growth factor receptor. Diabetes 2003, 52, 124–132. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhuang, S.; Schnellmann, R.G. H2O2-induced transactivation of EGF receptor requires Src and mediates ERK1/2, but not Akt, activation in renal cells. Am. J. Physiol. Renal Physiol. 2004, 286, F858–F865. [Google Scholar]
- Biscardi, J.S.; Maa, M.C.; Tice, D.A.; Cox, M.E.; Leu, T.H.; Parsons, S.J. c-Src-mediated phosphorylation of the epidermal growth factor receptor on Tyr845 and Tyr1101 is associated with modulation of receptor function. J. Biol. Chem. 1999, 274, 8335–8343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amos, S.; Martin, P.M.; Polar, G.A.; Parsons, S.J.; Hussaini, I.M. Phorbol 12-myristate 13-acetate induces epidermal growth factor receptor transactivation via protein kinase Cdelta/c-Src pathways in glioblastoma cells. J. Biol. Chem. 2005, 280, 7729–7738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Kalyankrishna, S.; Wislez, M.; Thilaganathan, N.; Saigal, B.; Wei, W.; Ma, L.; Wistuba, I.I.; Johnson, F.M.; Kurie, J.M. SRC-family kinases are activated in non-small cell lung cancer and promote the survival of epidermal growth factor receptor-dependent cell lines. Am. J. Pathol. 2007, 170, 366–376. [Google Scholar] [CrossRef] [Green Version]
- Gschwind, A.; Zwick, E.; Prenzel, N.; Leserer, M.; Ullrich, A. Cell communication networks: Epidermal growth factor receptor transactivation as the paradigm for interreceptor signal transmission. Oncogene 2001, 20, 1594–1600. [Google Scholar] [CrossRef] [Green Version]
- Steffel, J.; Akhmedov, A.; Greutert, H.; Lüscher, T.F.; Tanner, F.C. Histamine induces tissue factor expression: Implications for acute coronary syndromes. Circulation 2005, 112, 341–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gebhard, C.; Breitenstein, A.; Akhmedov, A.; Gebhard, C.E.; Camici, G.G.; Lüscher, T.F.; Tanner, F.C. Amphetamines induce tissue factor and impair tissue factor pathway inhibitor: Role of dopamine receptor type 4. Eur. Heart J. 2010, 31, 1780–1791. [Google Scholar] [CrossRef]
- Ding, L.; Ma, W.; Littmann, T.; Camp, R.; Shen, J. The P2Y2 nucleotide receptor mediates tissue factor expression in human coronary artery endothelial cells. J. Biol. Chem. 2011, 286, 27027–27038. [Google Scholar] [CrossRef] [Green Version]
- Stähli, B.E.; Camici, G.G.; Steffel, J.; Akhmedov, A.; Shojaati, K.; Graber, M.; Lüscher, T.F.; Tanner, F.C. Paclitaxel enhances thrombin-induced endothelial tissue factor expression via c-Jun terminal NH2 kinase activation. Circ. Res. 2006, 99, 149–155. [Google Scholar] [CrossRef] [Green Version]
- Dong, J.; Ramachandiran, S.; Tikoo, K.; Jia, Z.; Lau, S.S.; Monks, T.J. EGFR-independent activation of p38 MAPK and EGFR-dependent activation of ERK1/2 are required for ROS-induced renal cell death. Am. J. Physiol. Renal Physiol. 2004, 287, F1049–F1058. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Han, T.; Zhuo, M.; Wu, L.L.; Yuan, C.; Wu, L.; Lei, W.; Jiao, F.; Wang, L.W. Elevated COX-2 Expression Promotes Angiogenesis Through EGFR/p38-MAPK/Sp1-Dependent Signalling in Pancreatic Cancer. Sci. Rep. 2017, 7, 470. [Google Scholar] [CrossRef] [Green Version]
- Hashimoto, A.; Kurosaki, M.; Gotoh, N.; Shibuya, M.; Kurosaki, T. Shc regulates epidermal growth factor-induced activation of the JNK signaling pathway. J. Biol. Chem. 1999, 274, 20139–20143. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schettino, C.; Bareschino, M.A.; Ricci, V.; Ciardiello, F. Erlotinib: An EGF receptor tyrosine kinase inhibitor in non-small-cell lung cancer treatment. Expert Rev. Respir. Med. 2008, 2, 167–178. [Google Scholar] [CrossRef]
- Ko, J.C.; Ciou, S.C.; Jhan, J.Y.; Cheng, C.M.; Su, Y.J.; Chuang, S.M.; Lin, S.T.; Chang, C.C.; Lin, Y.W. Roles of MKK1/2-ERK1/2 and phosphoinositide 3-kinase-AKT signaling pathways in erlotinib-induced Rad51 suppression and cytotoxicity in human non-small cell lung cancer cells. Mol. Cancer Res. 2009, 7, 1378–1389. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.; Hackshaw, A.; Feng, Q.; Fu, X.; Zhang, Y.; Mao, C.; Tang, J. Comparison of gefitinib, erlotinib and afatinib in non-small cell lung cancer: A meta-analysis. Int. J. Cancer 2017, 140, 2805–2819. [Google Scholar] [CrossRef] [PubMed]
- Mackman, N. Regulation of the tissue factor gene. Thromb. Haemost. 1997, 78, 747–754. [Google Scholar] [CrossRef] [PubMed]
- Ohsawa, M.; Koyama, T.; Nara, N.; Hirosawa, S. Induction of tissue factor expression in human monocytic cells by protease inhibitors through activating activator protein-1 (AP-1) with phosphorylation of Jun-N-terminal kinase and p38. Thromb. Res. 2003, 112, 313–320. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mechelke, T.; Wittig, F.; Ramer, R.; Hinz, B. Interleukin-1β Induces Tissue Factor Expression in A549 Cells via EGFR-Dependent and -Independent Mechanisms. Int. J. Mol. Sci. 2021, 22, 6606. https://doi.org/10.3390/ijms22126606
Mechelke T, Wittig F, Ramer R, Hinz B. Interleukin-1β Induces Tissue Factor Expression in A549 Cells via EGFR-Dependent and -Independent Mechanisms. International Journal of Molecular Sciences. 2021; 22(12):6606. https://doi.org/10.3390/ijms22126606
Chicago/Turabian StyleMechelke, Tobias, Felix Wittig, Robert Ramer, and Burkhard Hinz. 2021. "Interleukin-1β Induces Tissue Factor Expression in A549 Cells via EGFR-Dependent and -Independent Mechanisms" International Journal of Molecular Sciences 22, no. 12: 6606. https://doi.org/10.3390/ijms22126606