1. Introduction
Understanding biological responses to ionizing radiation is of crucial importance for cancer treatment using radiotherapy. Mechanistic Monte Carlo (MC) simulation of radiation’s effect on DNA in a water medium is a promising tool for relevant studies after decades of development [
1]. The central idea of such an approach is to obtain the initial DNA damage spectrum via mechanistic modeling of the radio-biological interactions at the atomic or molecular levels. This includes the development of track-structure codes [
2,
3,
4,
5,
6,
7,
8,
9,
10,
11,
12,
13,
14,
15] and the subsequent computation of DNA damages by incorporating DNA models [
5,
7,
16,
17,
18]. The track-structure simulation can be divided into the simulations of the physical stage and the chemical stage. The physical-stage simulation deals with the ionization, excitation, and elastic scattering processes between the ionizing radiation particles and the water media and records the 3D coordinates of energy deposition events. The chemical-stage simulation computes how the chemical radicals, produced after the physical-stage simulation, diffuse and react mutually with the recording of the residual radicals’ positions. The positions of these energy deposition events and radicals are then utilized to compute the initial DNA damage sites, followed by an analysis to characterize DNA strand break patterns.
Although many developments have been performed to generate state-of-the-art mechanistic MC simulation tools, it is still necessary to further improve the simulation methods to accommodate different scenarios [
8]. For instance, to make the code versatile for studies on the Oxygen Enhancement Ratio (OER) [
19] and the Fenton reaction effect [
20], it is desired to include more types of molecules other than free radicals generated by the initial radiation into the chemical-stage simulation. However, due to the computational complexity of the “many-body” problem and the long temporal duration of the chemical stage, a step-by-step simulation of these relevant processes on conventional CPU computational platforms can be extremely time consuming [
21]. Under the constraint of computational resources, studies typically suffer from a restricted simulation region or a shortened temporal duration [
22,
23,
24], limiting their broad applications.
To overcome these obstacles, Graphical Processing Unit (GPU)-based parallel computing can be a cost-effective option [
25,
26]. We developed an open-source, GPU-based microscopic MC simulation toolkit, gMicroMC [
9], with the first version available on GitHub (
https://github.com/utaresearch/gMicroMC (accessed on 17 June 2021)). We initially focused on boosting the chemical-stage simulation for radicals produced from water radiolysis, achieving a speedup of several hundred folds compared to CPU-based packages [
27]. Later, we supported the physical track simulation for energetic electrons and implemented a DNA model of a lymphocyte cell nucleus at the base-pair resolution for the computation of electron-induced DNA damages [
9]. Recently, we also included oxygen molecules in the chemical-stage simulation in a step-by-step manner, which enabled the study of the radiolytic depletion effect of dissolved oxygen molecules [
28]. With these efforts, we were able to quantitatively study multiple critical problems that are computationally demanding. For example, we performed comprehensive simulations with gMicroMC to answer how uncertainties from the simulation parameters affect the accuracy of the final DNA damage computations [
29]. We also studied the radiolytic depletion of oxygen under ultra-high dose rate radiation (FLASH) to investigate the fundamental mechanism behind FLASH radiotherapy with the developed oxygen module in gMicroMC [
28].
In this work, we reported our recent progress on two new and important features that we recently introduced to gMicroMC, namely: (1) enabling the physical-stage simulations of protons and heavy ions; and (2) considering the presence of the DNA structure and its chemical reactions with radicals in the chemical-stage simulation. It was expected that the first feature would contribute greatly to the mechanistic study of particle irradiation, such as particle radiotherapy [
30,
31]. The presence of the DNA structure in the chemical-stage simulation will allow us to realistically describe the indirect DNA damage process. With the GPU acceleration, we were able to afford computationally challenging simulations that included detailed physics modeling and chemical reactions that spanned over a large temporal duration, enabling more realistic simulations of the relevant processes.
4. Discussion
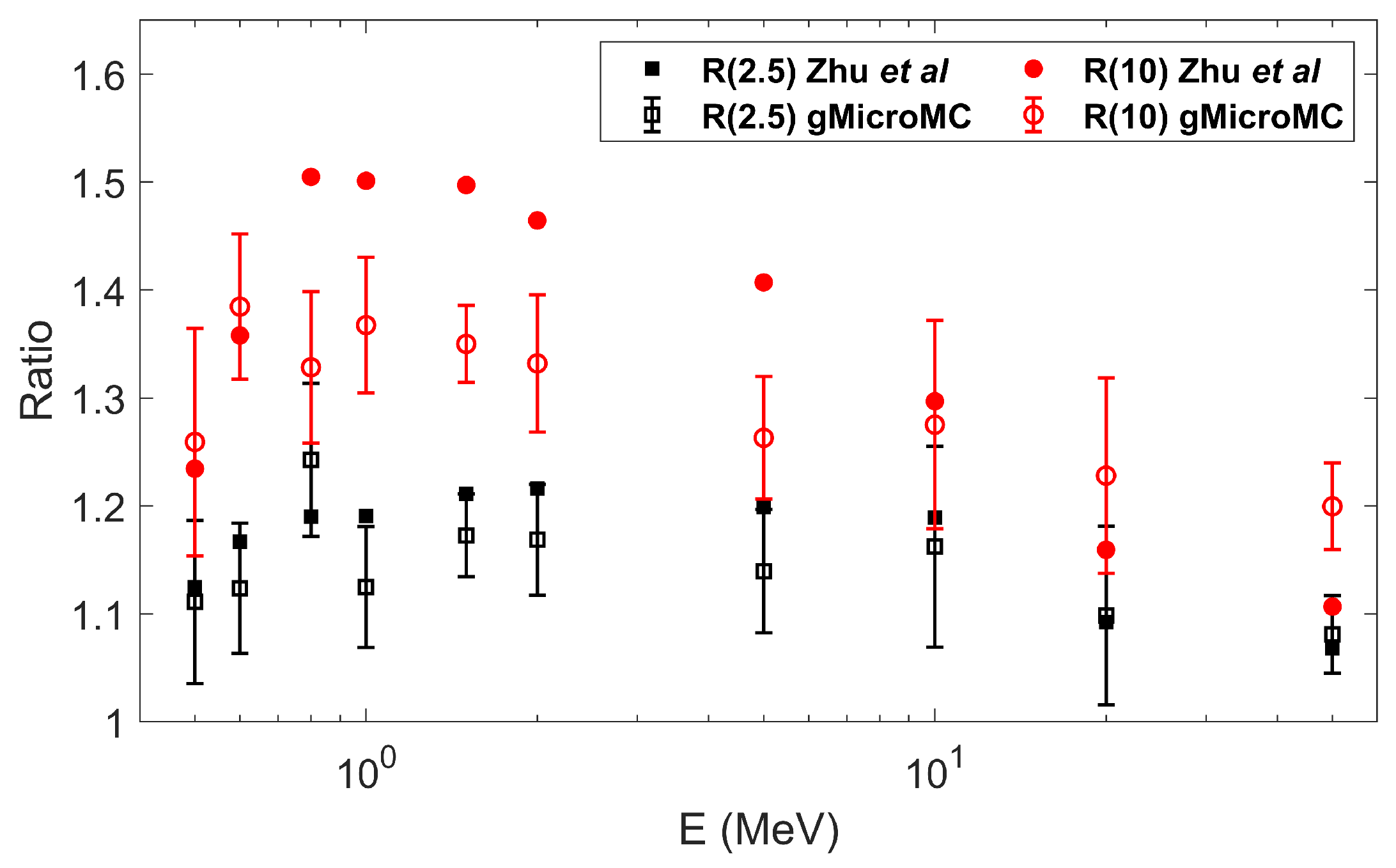
This is a continuous development work on gMicroMC. In this work, we successfully implemented the physical transport for proton and heavy ions and the concurrent transport of radicals and DNA in the chemical stage. For the former implementation, we considered the ionization, excitation, and charge effect during the transport and performed a series of case studies to validate the development. The obtained results matched well with the literature reports. We then computed the DNA DSBs for two proton energies, and the results agreed with those computed with the KURBUC model in Nikjoo et al.’s work [
45] within
. As for the latter, we considered the interaction of radicals with the DNA, histone protein, base, and sugar-phosphate groups during the chemical transport. To validate it, we first performed a self-consistency check for the evolution of chemical radicals and induced DNA DSBs under different checking frequencies for radical–DNA interactions. The result showed the high self-consistency of the developed module. We then performed a comprehensive study of the DSB yields as a function of the chemical stage duration and incident particle energies under the DNA concurrent transport frame. The results generally followed that from Zhu et al.’s work. The use of the GPU made the code have high computational efficiency. Running gMicroMC on a single GPU card, it took only 41 s to transport 100 protons with a kinetic energy of 10 MeV and around 8 min to transport
radicals up to 1 µs with the presence of a DNA model containing ∼
base pairs. The high computational performance of gMicroMC makes it suitable to simulate complex radiation scenarios such as proton FLASH radiotherapy, which is our next work. To benefit the community, we are also working on releasing the newly developed features of gMicroMC on GitHub (
https://github.com/utaresearch/gMicroMC (accessed on 17 June 2021)).
Despite the above success, there are also some limitations to our current development. In the physical transport of protons and heavy ions, we ignored the nuclear inelastic interaction and the nuclear and electromagnetic elastic scattering. Among them, the nuclear inelastic interaction can fragment the target and/or projectile nuclei, which is a main factor to remove primaries from the projectile beam. However, due to the complexity of the fragmentation process and its products, this process is typically not directly included in any mechanistic MC simulation tools at this moment [
46]. Since this work mainly focused on the novel GPU implementation of existing models, it will be our next work for a possible inclusion of the nuclear inelastic scattering process. As for the elastic scattering, it could change the direction of the primaries, hence affecting the track structure and radical dose distributions. However, elastic scattering mainly dominants interactions between the proton and water medium at a very low energy range, and the scattering angle is typically small [
46,
47]. Hence, we expect it will not affect the accuracy of the DNA damage computation greatly. Considering the powerful computational capacity of the GPU, it is promising to consider a more complete modeling of the physical interactions between ions and water molecules, making gMicroMC versatile for advanced applications.
It is also worth pointing out that we applied a low-energy five-pathway model and a high-energy three-pathway model to simulate the proton-induced excitation of a water molecule such that both very-low-energy and relativistic situations could be covered. Yet, this could cause a concern that some excitation pathways could be discontinuous at the model switching point. A previous study showed that the low-energy model could be extended up to 80 MeV with some proper parameter fitting [
34]. We hence made it an option in our package to only apply Equation (
19) to model the excitation process up to 80 MeV. In addition, for the two-model picture, although we currently set the model switching point at 500 keV to distinguish the slow and fast proton behavior following the same logic as stated in [
35], it deserves further study to investigate its impact on the subsequent radical yielding process.
Another important procedure that could affect the computational accuracy of the proton- and heavy ion-induced DNA damage is the modeling of the secondary electron transport, especially for the low-energy electrons (sub-keV range). Previous studies revealed their critical importance in determining the initial distribution of radicals and the consequent DNA damage patterns [
3]. However, due to the lack of sufficient experimental data, uncertainties about the simulation results could be introduced by the inaccurate modeling of this process. For instance, with different models adopted in gMicroMC and Geant4-DNA, the maximal discrepancies of the track length and penetration depth for electrons below 1 keV computed by the two packages were around 10 and 4 nm, respectively [
9]. In recent years, there has been much effort contributing to improving the accuracy of describing the low-energy electron transport process [
11,
12,
13,
14,
15,
48]. However, more efforts are required to fully elucidate this problem.
In addition, as discussed in our previous study [
29], the cross-section used to simulate the ionization and excitation processes from electrons could significantly impact the accuracy of the finally obtained DSB yields. In the case of protons and heavy ions, due to the lack of experimental data, the cross-sections and models could also be associated with large uncertainties, causing concerns about the robustness of the simulation results. To reduce these uncertainties, there have been multiple experiments and models [
13,
49,
50,
51,
52,
53,
54,
55,
56] developed in a more elaborate fashion. Yet, more studies are needed to more thoroughly understand the relevant processes.
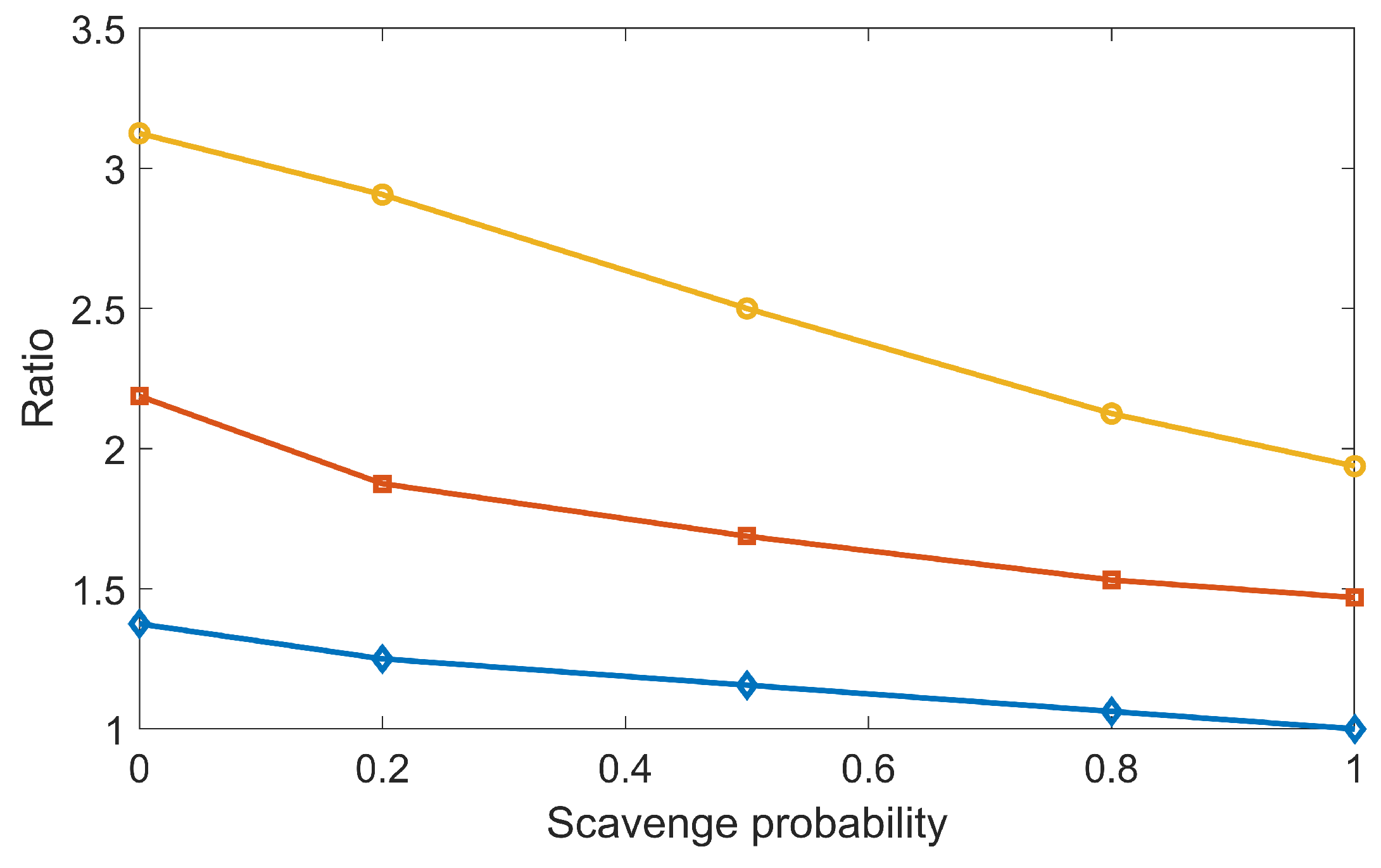
In our previous study [
29] with the overlay method, the DSB yields reduced when the chemical stage duration enlarged, which was against the trend obtained in this work with the concurrent DNA transport method (
Figure 7). This was due to the fact that in the overlay method, the radical–DNA interaction was only simulated after the chemical-stage simulation. The longer the chemical stage lasted, the more the
radicals were consumed during the mutual radical reactions, and the fewer the DSBs could be formed. Nonetheless, the controversial behavior between the concurrent and overlay frames reminds us to carefully check the parameters used in our simulation. One such parameter is the scavenging probability from histone proteins to chemical radicals, the value of which has not been well regulated by current experiments. We performed a new simulation to study its impact on DSB yield by gradually reducing the scavenging probability
from one to zero. Here,
means the total scavenging probability once radicals hit the histone proteins. Taking the DSB yields at
and
as a reference, we computed the relative DSB yields at other
s and
s. The results are shown in
Figure 8. Clearly, DSB yields increased when
decreased. However, even under the same
, when
differed,
could have different impacts on the relative DSB yield. For instance, the relative DSB yield was 1.4 for
, while it was 3.1 for
= 1 µs when
. This indicated that to make the simulation tool robust for various applications, we need to apply a proper cutoff to
and a detailed coordination of multiple parameters used in the chemical-stage simulation. This should be considered in future studies.
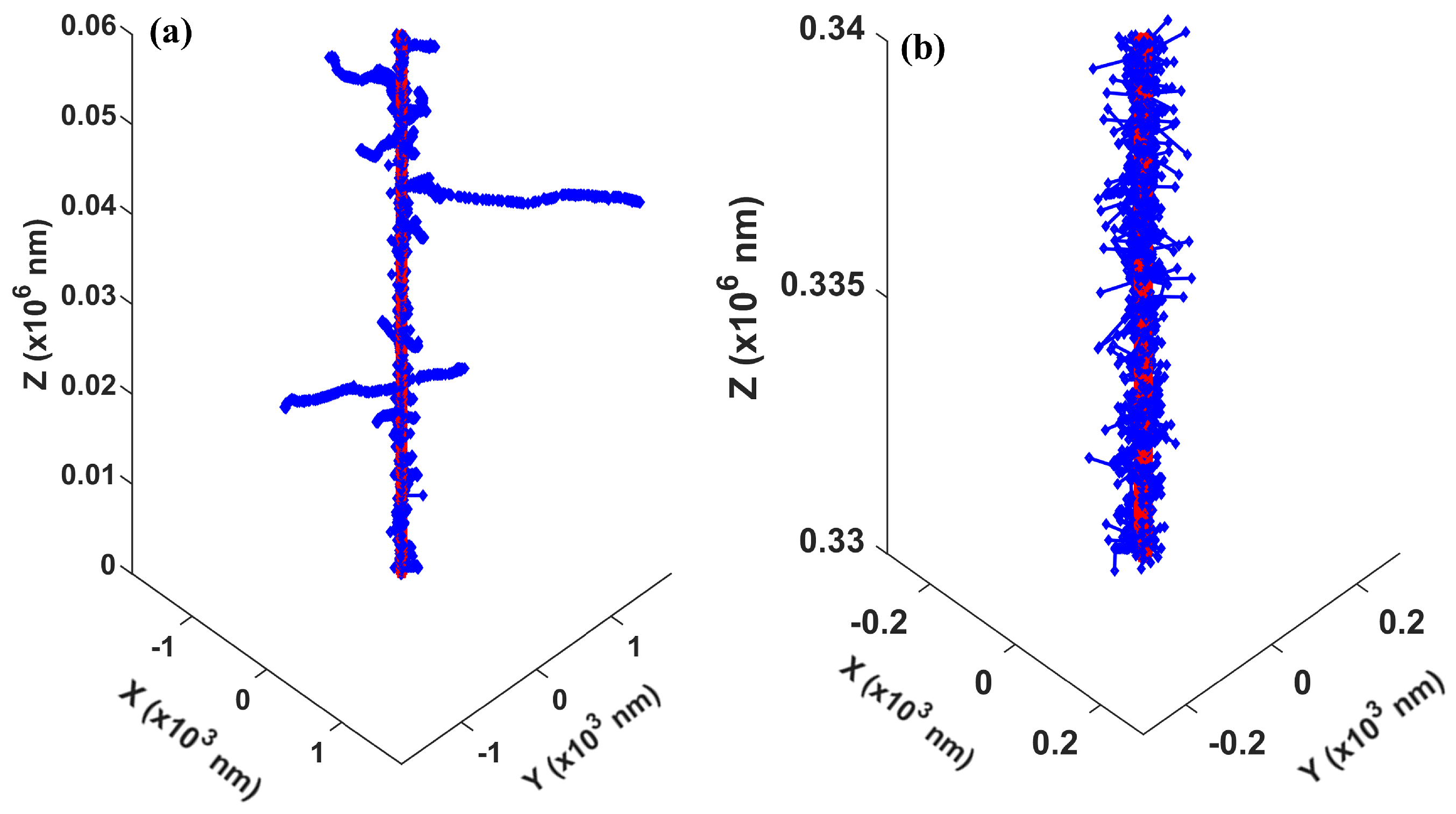
In our development of the concurrent transport module, we used a complete set of cellular DNA at the base-pair resolution to simulate the radical–DNA interactions. Previous studies have incorporated other DNA models, including the simple cylinder DNA [
3], other cellular DNAs [
7,
42,
57], and the DNA model at the atomic resolution [
58,
59]. Due to the different simulation setups and different DNA structures adopted, the absolute DSB yields from different studies were typically non-comparable [
60]. However, there were some common trends shared among different studies. For example, the DSB yields were found increasing and then decreasing when the LETs increased. The maximal yields of DSB occurred at the LET value around 60 keV/µm in [
57] and around 27.2 keV/µm (the LET value for 1 MeV protons [
22]) for gMicroMC and TOPAS-nBio, respectively. These consistencies motivated further studies with the concurrent simulation scheme while the expense was the lowered simulation efficiency. Our adoption of the GPU-based acceleration could shed light on this issue based on experiences from previous studies [
9,
27,
28,
61].
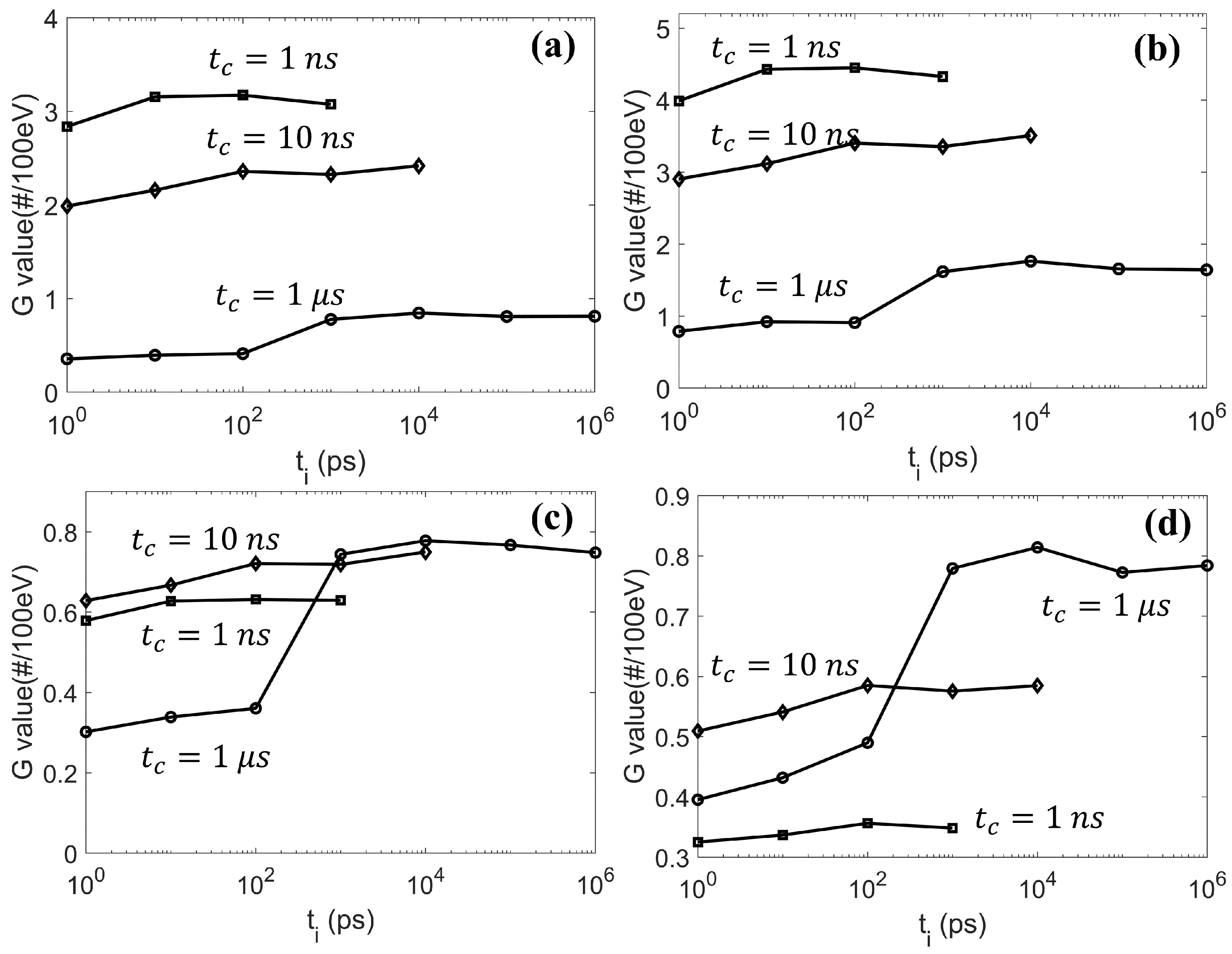
Finally, we performed a further study on the effect of
, which was introduced to balance the simulation efficiency and accuracy. In the
Results Section, we showed the obvious impact of
on the radical evolution. It would be interesting to see how it could consequently affect the final DNA damage. As the DSB is widely accepted as the most important factor in cell death, it is thus reasonable to use the DSB as a metric to evaluate the impact of
. We initiated a 4.5 keV electron with its position randomly sampled inside a sphere of radius 6.1 µm and its direction following the isotropic distribution. The sphere was concentric with the cell nucleus of our DNA model. We repeated the simulation until the accumulated dose inside the cell nucleus region reached 1 Gy, equivalent to simulating ∼ 2000 electrons. The generated radicals were then transported in the chemical stage along with considering the radical–DNA reactions. We calculated the resulting DNA DSBs as a function of
and show these in
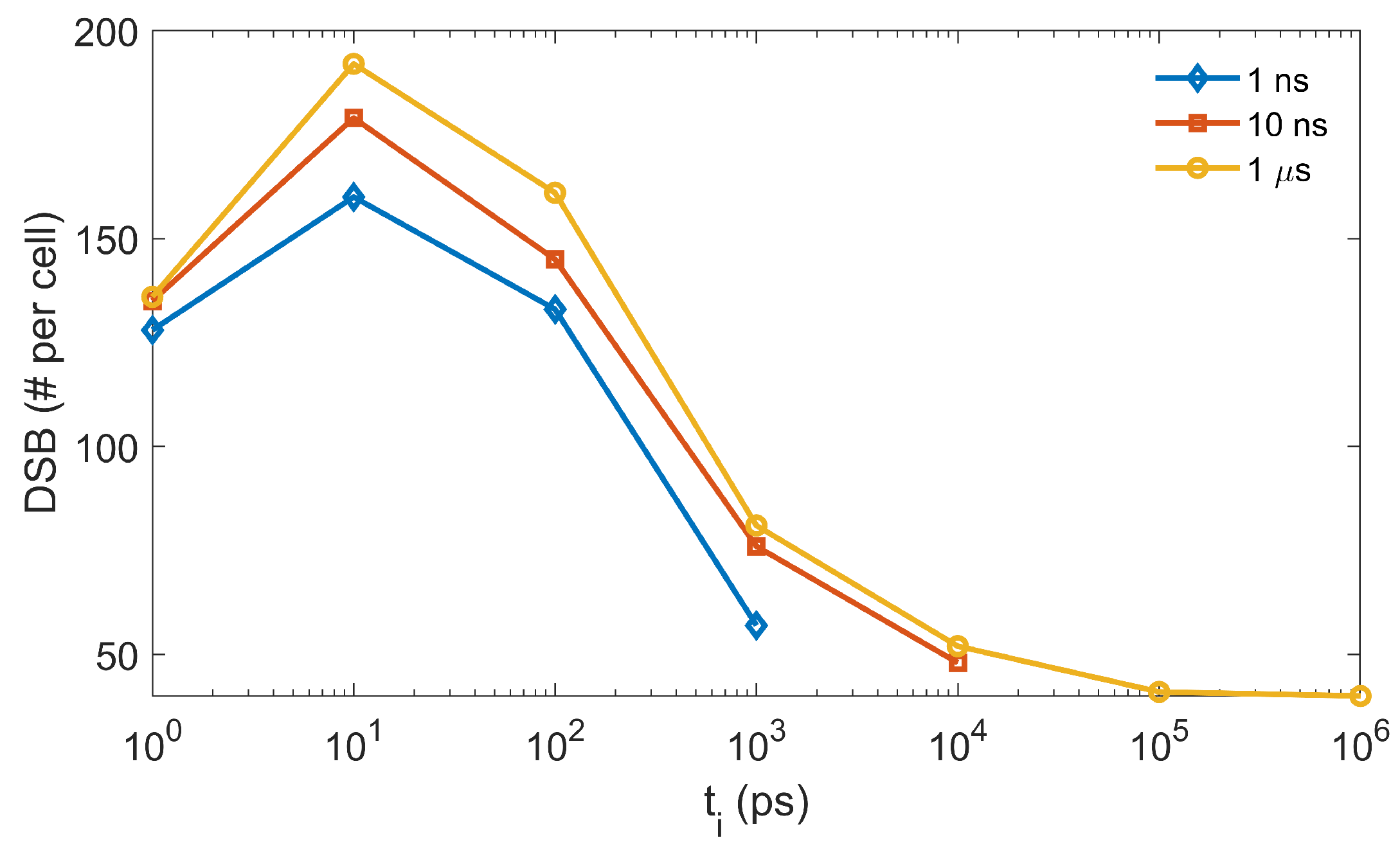
Figure 9. For the three
studied, the DSB lines showed a similar trend when
increased. The maximal DSB was obtained at
ps. The result could be interpreted as follows. At the beginning of the chemical stage, all generated radicals had a relatively dense distribution. When
slightly increased, the
radical could diffuse a longer distance away from its initial position before it reacted with the DNA, while its reaction probability with other radicals was not greatly affected. Hence, a sparser, but equivalent (or slightly reduced) number of DNA damage sites could be formed, which could lead to more generations of simple DSBs (composed of two damage sites) rather than the DSB+ (composed of multiple damage sites). In this way, the total DSB yields could increase. However, along with the further increase of
, the checking frequency for radical–radical reactions would be much higher than that for radical–DNA reactions. This could lead to a higher consumption of
radicals through radical–radical reactions than through DNA damaging reactions, which resulted in a reduced DSB yield.













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}