
Transcriptomic and Metabolic Profiling of High-Temperature Treated Storage Roots Reveals the Mechanism of Saccharification in Sweetpotato (Ipomoea batatas (L.) Lam.)

 , ,
, ,
Abstract
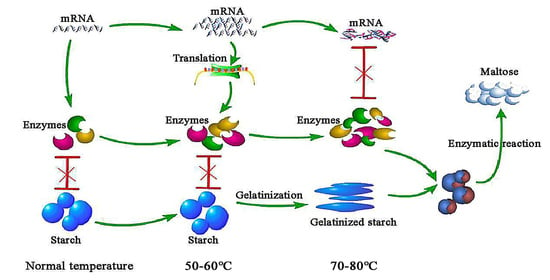
:
1. Introduction
2. Materials and Methods
2.1. Plant Materials and Treatments
2.2. Measurements of Glucose, Fructose, Sucrose, and Maltose
2.3. Measurements of Starch, Amylose, Total Amylase Activity, and Starch Gelatinization Temperature
2.4. Taste Evaluation of Sweetpotato
2.5. RNA Isolation cDNA Library Preparation and RNA-Seq
2.6. Analysis of Differentially Expressed Genes (DEGs)
2.7. Gene Ontology (GO) and Kyoto Encyclopedia of Genes and Genomes (KEGG) Enrichment Analysis
2.8. Real-Time Quantitative PCR (qRT-PCR) Validation
2.9. Metabolites Extraction and Identification
2.10. Qualitative and Quantitative Analysis of Metabolites
2.11. Statistical Analysis
3. Results
3.1. Changes of Sucrose, Fructose, Glucose, and Maltose
3.2. Differences of Starch Content, Amylose Content, and Starch Gelatinization Temperature
3.3. Changes of Total Amylase Activity
3.4. Taste Evaluation
3.5. Transcriptome Analysis
3.6. Identification of DEGs and Analysis
3.7. Enrichment of GO Terms and KEGG Pathway Analysis
3.8. Statistics Analysis of DAMs
3.9. KEGG Annotation and Enrichment Analysis of DAMs
3.10. Correlation Analysis of DEGs and DAMs
3.11. Enrichment Pathway Analysis of DEGs and DAMs
4. Discussion
4.1. Experimental Conception and Design
4.2. Changes of Genes and Metabolites Related to Sucrose Metabolism in Sweetpotato during High Temperature Saccharification
4.3. Transcription Factor Families (TF Families) and Inhibitors Related to Saccharification
4.4. Network Analysis of DEGs and DAMs
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Utomo:, J.S. Sweetpotato as a food crop. Bul. Palawija 2009, 17, 11–20. [Google Scholar]
- Nabubuya, A.; Namutebi, A.; Byaruhanga, Y.; Narvhus, J.; Wicklund, T. Influence of development, postharvest handling, and storage conditions on the carbohydrate components of sweetpotato (Ipomea batatas Lam.) roots. Food Sci. Nutr. 2017, 5, 1088–1097. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takahata, Y.; Noda, T.; Sato, T. Changes in carbohydrates and enzyme activities of sweetpotato lines during storage. J. Agric. Food Chem. 1995, 43, 1923–1928. [Google Scholar] [CrossRef]
- PICHA, D.H. HPLC determination of sugars in raw and baked sweet potatoes. J. Food Sci. 1985, 50, 1189–1190. [Google Scholar] [CrossRef]
- Morrison, T.A.; Pressey, R.; Kays, S.J. Changes in α- and β-amylase during storage of sweetpotato lines with varying starch hydrolysis potential. J. Am. Soc. Hortic. Sci. 1993, 118, 236–242. [Google Scholar] [CrossRef]
- Kitahara, K.; Nakamura, Y.; Otani, M.; Hamada, T.; Nakayachi, O.; Takahata, Y. Carbohydrate components in sweetpotato storage roots: Their diversities and genetic improvement. Breed. Sci. 2017, 67, 62–72. [Google Scholar] [CrossRef] [Green Version]
- Walter, W.M.; Purcell, A.E.; Nelson, A.M. Effects of amylolytic enzymes on “Moistness” and carbohydrate changes of baked sweetpotato cultivars. J. Food Sci. 1975, 40, 793–796. [Google Scholar] [CrossRef]
- Walter, W.M. Effect of curing on sensory properties and carbohydrate composition of baked sweetpotatoes. J. Food Sci. 1987, 52, 1026–1029. [Google Scholar] [CrossRef]
- Yoshiyuki, N.; Toshikazu, K.; Akiko, O.T.; Kenji, K. The effects of β-amylase activity and starch pasting temperature on maltose generation in steamed storage roots of sweetpotato. Jpn. Soc. FoodScience 2014, 61, 577–585. [Google Scholar]
- Jain, D.; Katyal, P. Optimization of gluco-amylase production from Aspergillus spp. For its use in saccharification of liquefied corn starch. 3 Biotech. 2018, 8, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Baba, T.; Nakama, H.; Tamaru, Y.; Kono, T. Changes in sugar and starch contents during storage of new type sweetpotato (low β-amylase activity in roots). Nippon Shokuhin Kogyo Gakkaishi 1987, 34, 249–253. [Google Scholar] [CrossRef]
- Kukimura, H.; Yoshida, T.; Komaki, K. New sweet potato cultivars, Benihayato and Satsumahikari, making a new turn for processing. JARQ 1988, 22, 7–13. [Google Scholar]
- Kumagai, T.; Umemura, Y.; Baba, T.; Iwanaga, M. The inheritance of β-amylase null in storage roots of sweetpotato, Ipomoea batatas (L.) Lam. Appl. Genet. 1990, 79, 369–376. [Google Scholar] [CrossRef]
- Ito, T.; Ando, T.; Ichikawa, K. Effects of cooking processes on the saccharification of sweetpotato (part 1): The relation between heating temperature and increase of sugar contents. J. Home Econ. Jpn. 1968, 19, 170–173. [Google Scholar]
- Nakamura, Y.; Ohara-Takada, A.; Kuranouchi, T.; Masuda, R.; Katayama, K. Mechanism for maltose generation by heating in the storage roots of sweet potato cultivar quick sweet containing starch with a low pasting temperature. Nippon Shokuhin Kagaku Kogaku Kaishi J. Jpn. Soc. Food Sci. Technol. 2014, 61, 62–69. [Google Scholar] [CrossRef] [Green Version]
- Noda, T. Relationships between chain length distribution of amylopectin and gelatinization properties within the same botanical origin for sweetpotato and buckwheat. Carbohyd. Polym. 1998, 37, 153–158. [Google Scholar] [CrossRef]
- Noda, T.; Kobayashi, T.; Suda, I. Effect of soil temperature on starch properties of sweetpotatoes. Carbohyd. Polym. 2001, 44, 239–246. [Google Scholar] [CrossRef]
- Nakayama, S.; Nakamura, Y. Purification and some properties of starch branching enzyme (Q-enzyme) from tuberous root of sweetpotato. Physiol. Plant. 1994, 91, 763–769. [Google Scholar] [CrossRef]
- Chan, C.; Chiang, C.; Lai, Y.; Huang, C.; Kao, S.; Liao, W.C. Changes in sugar composition during baking and their effects on sensory attributes of baked sweetpotatoes. J. Food Sci. Technol. 2014, 51, 4072–4077. [Google Scholar] [CrossRef] [Green Version]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 1–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. EdgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2009, 26, 139–140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Storey, J.D.; Tibshirani, R. Statistical significance for genomewide studies. Proc. Natl. Acad. Sci. USA 2003, 100, 9440–9445. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ashburner, M.; Ball, C.A.; Blake, J.A.; Botstein, D.; Butler, H.; Cherry, J.M.; Davis, A.P.; Dolinski, K.; Dwight, S.S.; Eppig, J.T. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gerlich, M.; Neumann, S. KEGG: Kyoto encyclopedia of genes and genomes. Nuclc. Acids Res. 2000, 1, 27–30. [Google Scholar]
- Park, S.; Kim, Y.; Ji, C.Y.; Park, S.; Jeong, J.C.; Lee, H.; Kwak, S. Stable internal reference genes for the normalization of Real-Time PCR in different sweetpotato cultivars subjected to abiotic stress conditions. PLoS ONE 2012, 7, e51502. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using Real-Time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Smith, C.A.; Want, E.J.; O’Maille, G.; Abagyan, R.; Siuzdak, G. XCMS: Processing mass spectrometry data for metabolite profiling using nonlinear peak alignment, matching, and identification. Anal. Chem 2006, 78, 779–787. [Google Scholar] [CrossRef]
- Roberts, L.D.; Souza, A.L.; Gerszten, R.E.; Clish, C.B. Targeted metabolomics. Curr. Protoc. Mol. Biol. 2012, 98, 30.2.1–30.2.24. [Google Scholar] [CrossRef]
- Xia, J.; Psychogios, N.; Young, N.; Wishart, D.S. MetaboAnalyst: A web server for metabolomic data analysis and interpretation. Nucleic Acids Res. 2009, 37, W652–W660. [Google Scholar] [CrossRef] [Green Version]
- Bylesjö, M.; Eriksson, D.; Kusano, M.; Moritz, T.; Trygg, J. Data integration in plant biology: The O2PLS method for combined modeling of transcript and metabolite data. Plant J. 2007, 52, 1181–1191. [Google Scholar] [CrossRef]
- Shen, F.S.; Xiang, C.; Wu, L.H.; Li, B.; Luo, Z.G. Analysis on the characteristics of soluble sugar components in sweetpotato storage root and its relationship with taste. Sci. Agric. Sin. 2021, 54, 34–45. [Google Scholar]
- Takahata, Y.; Noda, T.; Nagata, T. Effect of β-amylase stability and starch gelatinization during heating on varietal differences in maltose content in sweetpotatoes. J. Agric. Food Chem 1994, 42, 2564–2569. [Google Scholar] [CrossRef]
- Dubos, C.; Stracke, R.; Grotewold, E.; Weisshaar, B.; Martin, C.; Lepiniec, L. MYB transcription factors in Arabidopsis. Trends Plant. Sci. 2010, 15, 573–581. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Ho, T.D.; Ho, S.; Yu, S. Three novel MYB proteins with one DNA binding repeat mediate sugar and hormone regulation of α-amylase gene expression. Plant Cell 2002, 14, 1963–1980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bakshi, M.; Oelmüller, R. WRKY transcription factors. Plant. Signal. Behav. 2014, 9, e27700. [Google Scholar] [CrossRef] [Green Version]
- Song, Y.; Ai, C.; Jing, S.; Yu, D. Research progress on functional analysis of rice WRKY genes. Rice Sci. 2010, 17, 60–72. [Google Scholar] [CrossRef]
- Hammargren, J.; Rosenquist, S.; Jansson, C.; Knorpp, C. A novel connection between nucleotide and carbohydrate metabolism in mitochondria: Sugar regulation of the Arabidopsis nucleoside diphosphate kinase 3a gene. Plant Cell Rep. 2008, 27, 529–534. [Google Scholar] [CrossRef]
- Ishiguro, S.; Nakamura, K. Characterization of a cDNA encoding a novel DNA-binding protein, SPF1, that recognizes SP8 sequences in the 5’ upstream regions of genes coding for sporamin and beta-amylase from sweetpotato. Mol. Gen. Genet. 1994, 244, 563–571. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Zhang, Z.; Hanzlik, S.; Cook, E.; Shen, Q.J. Salicylic acid inhibits gibberellin-induced alpha-amylase expression and seed germination via a pathway involving an abscisic-acid-inducible WRKY gene. Plant. Mol. Biol. 2007, 64, 293–303. [Google Scholar] [CrossRef] [PubMed]
- Kumari, B.; Sharma, P.; Nath, A.K. A-amylase inhibitor in local Himalyan collections of Colocasia: Isolation, purification, characterization and selectivity towards α-amylases from various sources. Pestic. Biochem. Phys. 2012, 103, 49–55. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, J.; Hou, J.; Yao, Y.; Lin, Y.; Ou, Y.; Song, B.; Xie, C. The potato amylase inhibitor gene SbAI regulates cold-induced sweetening in potato tubers by modulating amylase activity. Plant Biotechnol. J. 2014, 12, 984–993. [Google Scholar] [CrossRef] [PubMed]
- Barbosa, A.E.; Albuquerque, É.V.; Silva, M.C.; Souza, D.S.; Oliveira-Neto, O.B.; Valencia, A.; Rocha, T.L.; Grossi-de-Sa, M.F. A-amylase inhibitor-1 gene from Phaseolus vulgaris expressed in Coffea arabica plants inhibits α-amylases from the coffee berry borer pest. BMC Biotechnol. 2010, 10, 44. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, P.M.; Reiner, D.; Moore, A.E.; Lee, R.; Epstein, M.M.; Higgins, T.J.V. Comparison of the α-Amylase inhibitor-1 from common bean (Phaseolus vulgaris) varieties and transgenic expression in other legumes—Post-Translational modifications and immunogenicity. J. Agric. Food Chem. 2011, 59, 6047–6054. [Google Scholar] [CrossRef] [PubMed]
- Brohée, S.; Faust, K.; Lima-Mendez, G.; Vanderstocken, G.; van Helden, J. Network Analysis Tools: From biological networks to clusters and pathways. Nat. Protoc. 2008, 3, 1616–1629. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Terms | S1 | |||||
|---|---|---|---|---|---|---|
| Number of DAMs | q Value | |||||
| Y25 | Z13 | X27 | Y25 | Z13 | X27 | |
| ABC transporters | 10 | 7 | 9 | 0.0060 | 0.1358 | 0.0266 |
| beita-Alanine metabolism | 2 | 2 | 2 | 0.8594 | 0.9314 | 0.7337 |
| Aminoacyl-tRNA biosynthesis | 4 | 2 | 4 | 0.4053 | 0.9314 | 0.3486 |
| Starch and sucrose metabolism | 1 | 1 | 2 | 0.8594 | 0.9314 | 0.5925 |
| 2-Oxocarboxylic acid metabolism | 4 | 3 | 5 | 0.8594 | 0.9314 | 0.6510 |
| Porphyrin and chlorophyll metabolism | 3 | 1 | 3 | 0.4053 | 0.9314 | 0.3486 |
| Histidine metabolism | 3 | 2 | 3 | 0.8594 | 0.9314 | 0.7337 |
| Terms | S2 | |||||
| ABC transporters | 4 | 5 | 8 | 0.7417 | 0.6227 | 0.2009 |
| Starch and sucrose metabolism | 2 | 1 | 2 | 0.3624 | 0.8954 | 0.7974 |
| Cyanoamino acid metabolism | 1 | 2 | 2 | 0.8803 | 0.6227 | 0.7974 |
| Arginine and proline metabolism | 5 | 3 | 4 | 0.2321 | 0.8884 | 0.7974 |
| S1 | S2 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Types | ID | Log2 (fc) | Types | ID | Log2 (fc) | ||||
| Y25 | Z13 | X27 | Y25 | Z13 | X27 | ||||
| MYB | G12956 | −4.63 | −1.21 | −0.68 | MYB | G31499 | 7.46 | 3.53 | 2.09 |
| G31499 | −4.51 | 1.91 | 0.78 | G3991 | −4.32 | −3.93 | −2.68 | ||
| G33975 | −4.21 | −5.26 | −1.38 | G18652 | −3.19 | 2.15 | −0.92 | ||
| G26043 | −3.45 | −2.17 | −1.70 | G18587 | −2.98 | −3.10 | −1.55 | ||
| G16218 | −2.99 | −3.19 | −1.62 | G36436 | −2.92 | 2.00 | −1.32 | ||
| MSTRG.35719 | −2.42 | −2.50 | −2.47 | G18679 | −2.35 | −2.28 | −1.74 | ||
| G4815 | −1.95 | 3.89 | −3.59 | G34548 | −2.23 | −1.52 | −1.00 | ||
| G34342 | 1.37 | 1.99 | 0.95 | ||||||
| MSTRG.9428 | 1.50 | 1.34 | 0.89 | HSF | G21417 | −3.52 | −2.80 | −0.50 | |
| G30675 | 2.19 | 1.98 | 0.80 | ||||||
| G36436 | 2.67 | 1.32 | −0.42 | ERF | G27649 | −5.31 | −8.96 | −0.43 | |
| MSTRG.50858 | −3.09 | −1.58 | −0.87 | ||||||
| HSF | G38291 | −10.37 | −2.31 | −4.74 | G42381 | 1.52 | 2.35 | 0.77 | |
| G21545 | −3.19 | −1.19 | −0.22 | ||||||
| G31518 | −2.56 | −1.81 | −1.65 | bHLH | G21240 | −6.45 | −6.16 | −1.85 | |
| G23088 | −2.21 | −2.02 | −2.21 | G12959 | −5.03 | −2.54 | −2.78 | ||
| G3345 | −1.84 | −0.41 | −2.67 | G46039 | −4.92 | −4.56 | −2.77 | ||
| G25273 | 1.83 | 1.38 | 0.68 | G2655 | −4.50 | −10.36 | 9.15 | ||
| G18195 | 1.90 | 1.60 | −0.43 | G27729 | −4.36 | −3.89 | −0.51 | ||
| G29441 | −3.52 | −3.09 | −1.60 | ||||||
| ERF | G23031 | −11.73 | −4.86 | −3.35 | G1375 | −3.46 | −1.46 | −1.55 | |
| G28150 | −4.72 | −4.38 | −8.23 | G15876 | −2.65 | −1.20 | −1.35 | ||
| G32004 | −4.37 | −3.65 | −3.21 | G29812 | −2.58 | −3.09 | −2.03 | ||
| MSTRG.47017 | −2.38 | −2.92 | −1.65 | ||||||
| bHLH | G6860 | −9.73 | −9.61 | −1.51 | G12549 | −2.13 | −2.03 | −0.85 | |
| G16325 | −5.74 | −3.65 | −2.34 | G42307 | −2.07 | −1.97 | −1.40 | ||
| G17961 | −4.53 | −2.97 | −1.30 | G19671 | −1.98 | −3.58 | −1.97 | ||
| G25250 | −2.99 | −3.28 | −2.20 | G16702 | −1.87 | −1.26 | −1.44 | ||
| G19671 | −2.18 | −1.93 | −1.58 | G16207 | −1.76 | −1.87 | −1.49 | ||
| G31307 | 3.32 | 2.23 | 0.63 | ||||||
| WRKY | G11050 | −10.12 | −9.37 | −8.13 | |||||
| WRKY | MSTRG.69878 | −3.38 | −2.42 | −2.38 | G15453 | −3.26 | −3.66 | −1.01 | |
| G7445 | 2.95 | 2.35 | 0.91 | G41003 | −2.88 | −2.17 | −1.24 | ||
| G45220 | 4.68 | 3.27 | −0.73 | G7665 | −2.67 | −3.23 | −2.09 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, C.; Kou, M.; Arisha, M.H.; Tang, W.; Ma, M.; Yan, H.; Wang, X.; Wang, X.; Zhang, Y.; Liu, Y.; et al. Transcriptomic and Metabolic Profiling of High-Temperature Treated Storage Roots Reveals the Mechanism of Saccharification in Sweetpotato (Ipomoea batatas (L.) Lam.). Int. J. Mol. Sci. 2021, 22, 6641. https://doi.org/10.3390/ijms22136641
Li C, Kou M, Arisha MH, Tang W, Ma M, Yan H, Wang X, Wang X, Zhang Y, Liu Y, et al. Transcriptomic and Metabolic Profiling of High-Temperature Treated Storage Roots Reveals the Mechanism of Saccharification in Sweetpotato (Ipomoea batatas (L.) Lam.). International Journal of Molecular Sciences. 2021; 22(13):6641. https://doi.org/10.3390/ijms22136641
Chicago/Turabian StyleLi, Chen, Meng Kou, Mohamed Hamed Arisha, Wei Tang, Meng Ma, Hui Yan, Xin Wang, Xiaoxiao Wang, Yungang Zhang, Yaju Liu, and et al. 2021. "Transcriptomic and Metabolic Profiling of High-Temperature Treated Storage Roots Reveals the Mechanism of Saccharification in Sweetpotato (Ipomoea batatas (L.) Lam.)" International Journal of Molecular Sciences 22, no. 13: 6641. https://doi.org/10.3390/ijms22136641