
Nuclear Export Inhibitor KPT-8602 Synergizes with PARP Inhibitors in Escalating Apoptosis in Castration Resistant Cancer Cells

 , ,
, ,  and
and
Abstract
:1. Introduction
2. Results
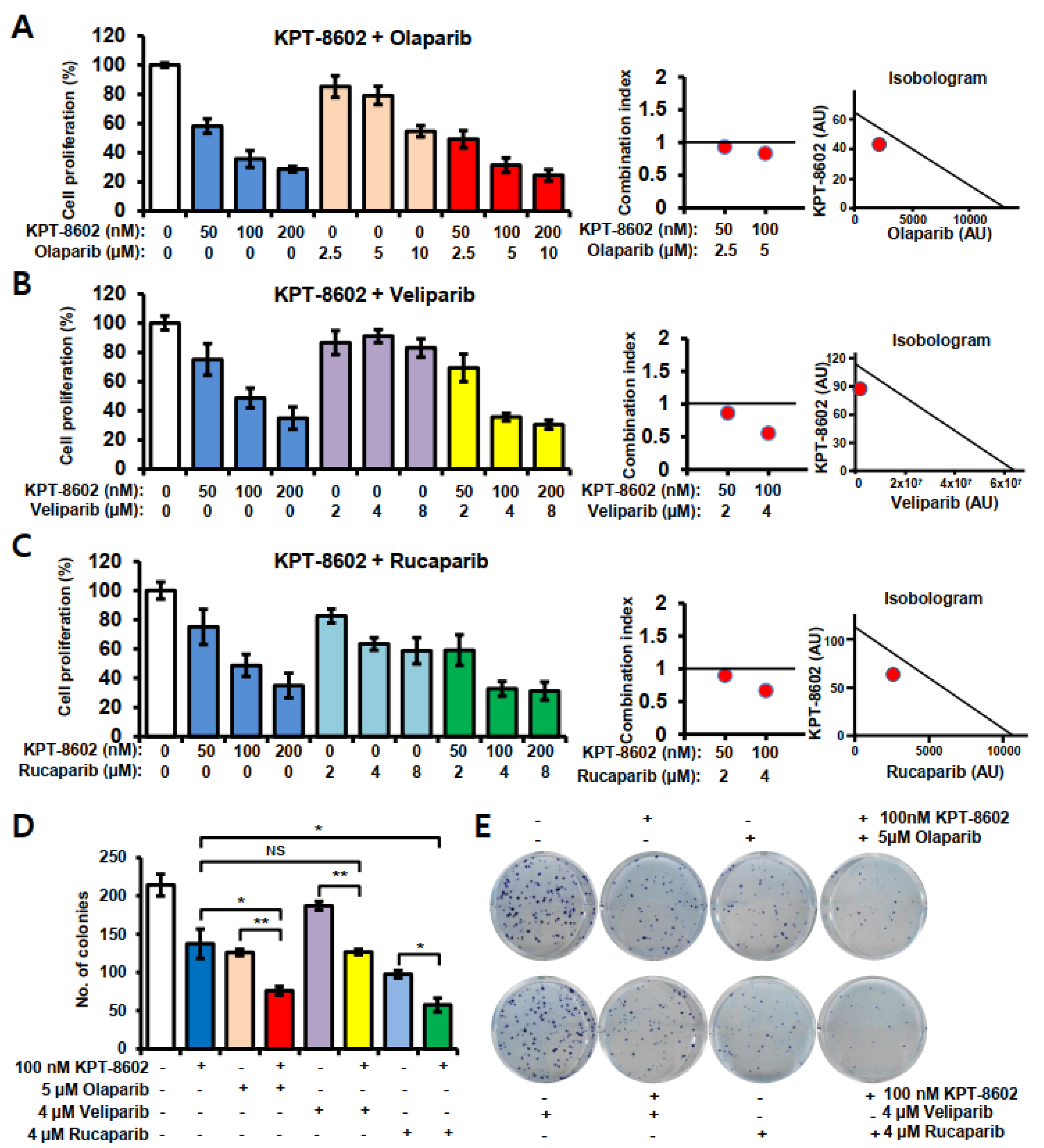
2.1. KPT-8602 with PARP Inhibitors Synergistically Inhibits the Growth of Prostate Cancer Cells
2.2. KPT-8602 Treatment with PARPi Enhanced Apoptotic Cell Death in 22rv1 Cells
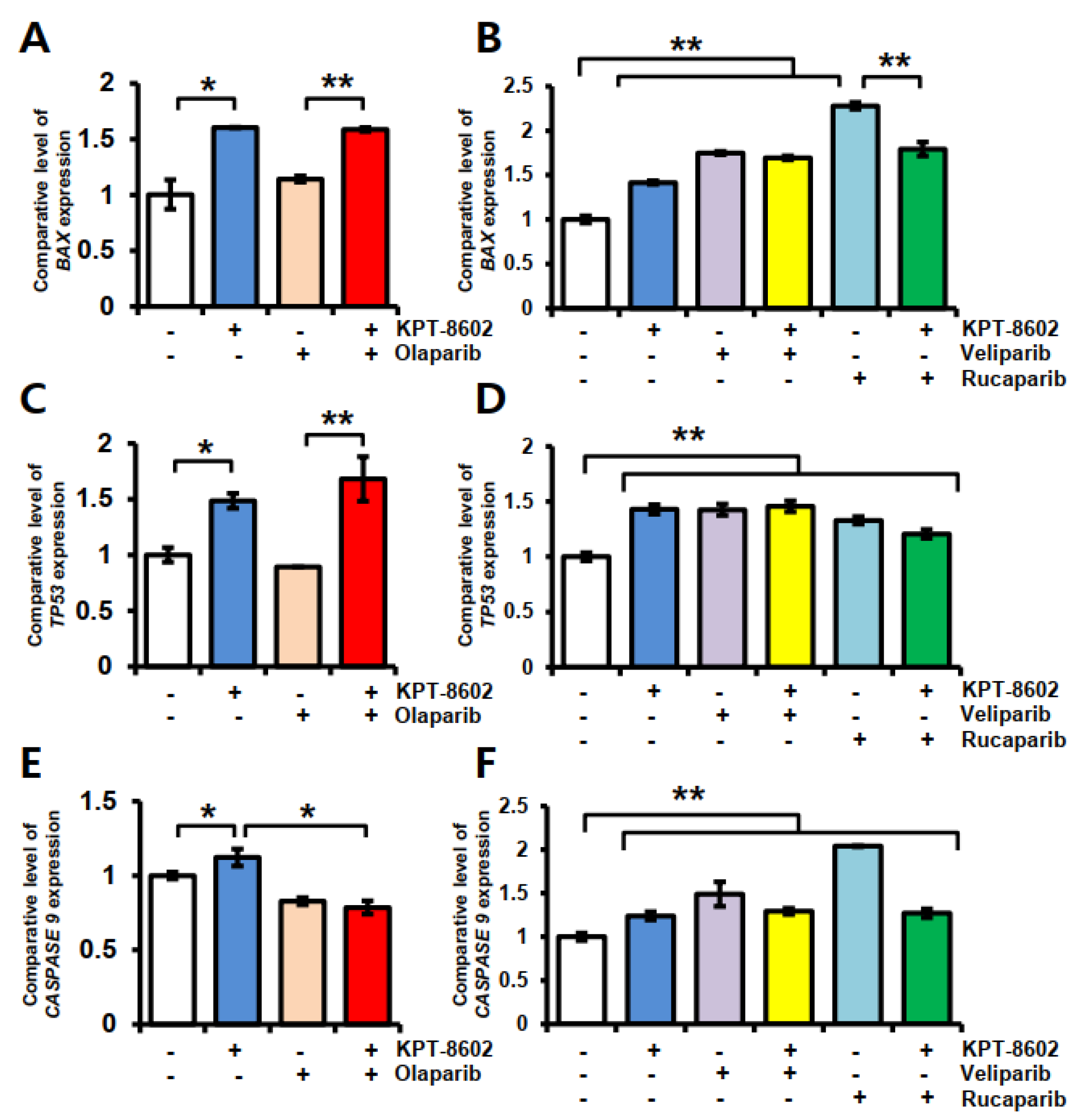
2.3. KPT-8602 and PARP Inhibitors Treatment Causes Upregulation of Apoptosis Related Genes in 22rv1 Cells
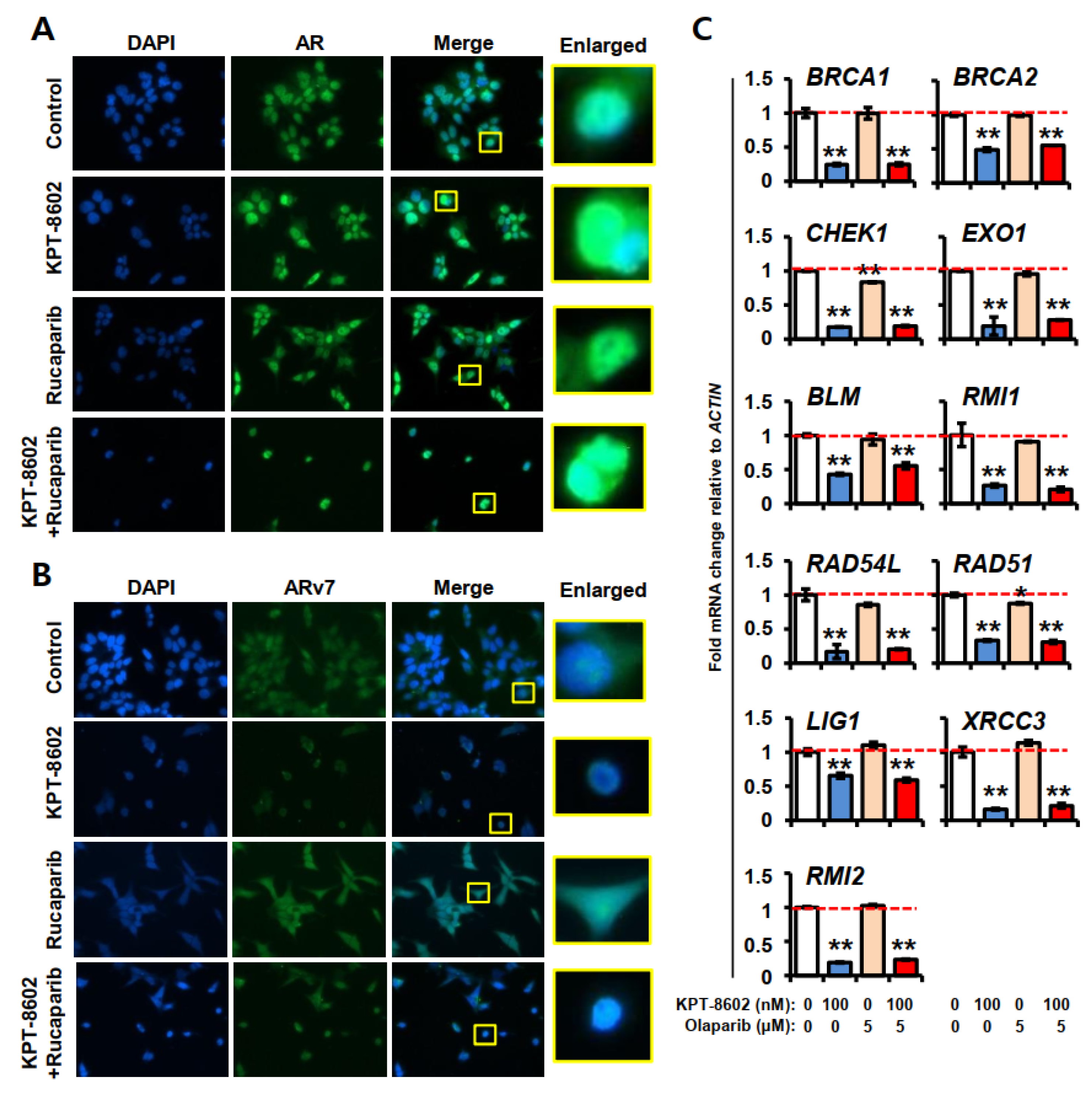
2.4. KPT-8602 Treatment with or without PARPi Down-Regulates AR and Its Target Genes
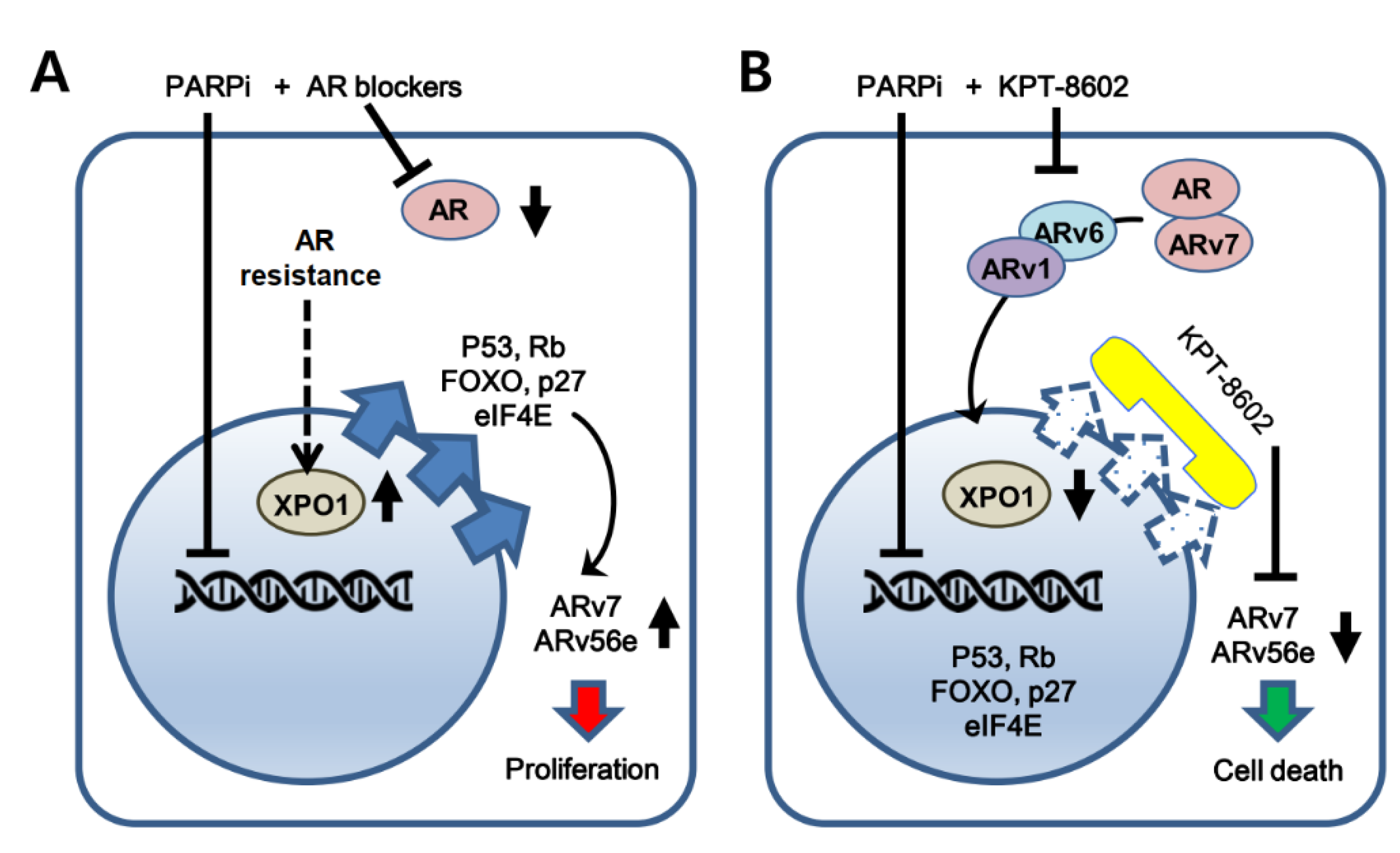
3. Discussion
4. Materials and Methods
4.1. Cell Lines, Reagents and Antibodies
4.2. Growth Inhibition Assay
4.3. Colony Formation Assay
4.4. Apoptosis Assay
4.5. Real-Time RT-qPCR
4.6. Overexpression of XPO1
4.7. Immunofluorescence Analysis
4.8. Western Blot Analysis
4.9. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Jemal, A.; Culp, M.B.; Ma, J.; Islami, F.; Fedewa, S.A. Prostate Cancer Incidence 5 Years After US Preventive Services Task Force Recommendations Against Screening. J. Natl. Cancer Inst. 2020, 113, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Jiang, X.; Liang, X.; Jiang, G. Molecular and cellular mechanisms of castration resistant prostate cancer. Oncol. Lett. 2018, 15, 6063–6076. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karantanos, T.; Corn, P.G.; Thompson, T.C. Prostate cancer progression after androgen deprivation therapy: Mechanisms of castrate resistance and novel therapeutic approaches. Oncogene 2013, 32, 5501–5511. [Google Scholar] [CrossRef] [PubMed]
- Donovan, M.J.; Osman, I.; Khan, F.M.; Vengrenyuk, Y.; Capodieci, P.; Koscuiszka, M.; Anand, A.; Cordon-Cardo, C.; Costa, J.; Scher, H.I. Androgen receptor expression is associated with prostate cancer-specific survival in castrate patients with metastatic disease. BJU Int. 2010, 105, 462–467. [Google Scholar] [CrossRef]
- Yang, C.C.; Fazli, L.; Loguercio, S.; Zharkikh, I.; Aza-Blanc, P.; Gleave, M.E.; Wolf, D.A. Downregulation of c-SRC kinase CSK promotes castration resistant prostate cancer and pinpoints a novel disease subclass. Oncotarget 2015, 6, 22060–22071. [Google Scholar] [CrossRef] [Green Version]
- Sircar, K.; Yoshimoto, M.; Monzon, F.A.; Koumakpayi, I.H.; Katz, R.L.; Khanna, A.; Alvarez, K.; Chen, G.; Darnel, A.D.; Aprikian, A.G.; et al. PTEN genomic deletion is associated with p-Akt and AR signalling in poorer outcome, hormone refractory prostate cancer. J. Pathol. 2009, 218, 505–513. [Google Scholar] [CrossRef]
- de la Taille, A.; Rubin, M.A.; Chen, M.W.; Vacherot, F.; de Medina, S.G.; Burchardt, M.; Buttyan, R.; Chopin, D. Beta-catenin-related anomalies in apoptosis-resistant and hormone-refractory prostate cancer cells. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2003, 9, 1801–1807. [Google Scholar]
- Azoulay, S.; Terry, S.; Chimingqi, M.; Sirab, N.; Faucon, H.; Diez de Medina, S.G.; Moutereau, S.; Maille, P.; Soyeux, P.; Abbou, C.; et al. Comparative expression of Hedgehog ligands at different stages of prostate carcinoma progression. J. Pathol. 2008, 216, 460–470. [Google Scholar] [CrossRef]
- Isali, I.; Al-Sadawi, M.A.A.; Qureshi, A.; Khalifa, A.O.; Agrawal, M.K.; Shukla, S. Growth factors involve in cellular proliferation, differentiation and migration during prostate cancer metastasis. Int. J. Cell Biol. Physiol. 2019, 2, 1–13. [Google Scholar]
- Wozney, J.L.; Antonarakis, E.S. Growth factor and signaling pathways and their relevance to prostate cancer therapeutics. Cancer Metastasis Rev. 2014, 33, 581–594. [Google Scholar] [CrossRef] [Green Version]
- Crawford, E.D.; Schellhammer, P.F.; McLeod, D.G.; Moul, J.W.; Higano, C.S.; Shore, N.; Denis, L.; Iversen, P.; Eisenberger, M.A.; Labrie, F. Androgen Receptor Targeted Treatments of Prostate Cancer: 35 Years of Progress with Antiandrogens. J. Urol. 2018, 200, 956–966. [Google Scholar] [CrossRef]
- Chung, J.H.; Dewal, N.; Sokol, E.; Mathew, P.; Whitehead, R.; Millis, S.Z.; Frampton, G.M.; Bratslavsky, G.; Pal, S.K.; Lee, R.J.; et al. Prospective Comprehensive Genomic Profiling of Primary and Metastatic Prostate Tumors. JCO Precis. Oncol. 2019, 3. [Google Scholar] [CrossRef]
- Robinson, D.; Van Allen, E.M.; Wu, Y.M.; Schultz, N.; Lonigro, R.J.; Mosquera, J.M.; Montgomery, B.; Taplin, M.E.; Pritchard, C.C.; Attard, G.; et al. Integrative clinical genomics of advanced prostate cancer. Cell 2015, 161, 1215–1228. [Google Scholar] [CrossRef] [Green Version]
- Pritchard, C.C.; Mateo, J.; Walsh, M.F.; De Sarkar, N.; Abida, W.; Beltran, H.; Garofalo, A.; Gulati, R.; Carreira, S.; Eeles, R.; et al. Inherited DNA-Repair Gene Mutations in Men with Metastatic Prostate Cancer. N. Engl. J. Med. 2016, 375, 443–453. [Google Scholar] [CrossRef]
- Fisher, A.E.; Hochegger, H.; Takeda, S.; Caldecott, K.W. Poly(ADP-ribose) polymerase 1 accelerates single-strand break repair in concert with poly(ADP-ribose) glycohydrolase. Mol. Cell. Biol. 2007, 27, 5597–5605. [Google Scholar] [CrossRef] [Green Version]
- Helleday, T. The underlying mechanism for the PARP and BRCA synthetic lethality: Clearing up the misunderstandings. Mol. Oncol. 2011, 5, 387–393. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.H.; Han, M.E.; Oh, S.O. The molecular mechanism for nuclear transport and its application. Anat. Cell Biol. 2017, 50, 77–85. [Google Scholar] [CrossRef]
- Vousden, K.H.; Vande Woude, G.F. The ins and outs of p53. Nat. Cell Biol. 2000, 2, E178–E180. [Google Scholar] [CrossRef]
- Mutka, S.C.; Yang, W.Q.; Dong, S.D.; Ward, S.L.; Craig, D.A.; Timmermans, P.B.; Murli, S. Identification of nuclear export inhibitors with potent anticancer activity in vivo. Cancer Res. 2009, 69, 510–517. [Google Scholar] [CrossRef] [Green Version]
- Fung, H.Y.; Chook, Y.M. Atomic basis of CRM1-cargo recognition, release and inhibition. Semin. Cancer Biol. 2014, 27, 52–61. [Google Scholar] [CrossRef] [Green Version]
- Cagatay, T.; Chook, Y.M. Karyopherins in cancer. Curr. Opin. Cell Biol. 2018, 52, 30–42. [Google Scholar] [CrossRef]
- Azmi, A.S.; Uddin, M.H.; Mohammad, R.M. The nuclear export protein XPO1—From biology to targeted therapy. Nat. Rev. Clin. Oncol. 2021, 18, 152–169. [Google Scholar] [CrossRef]
- Aboukameel, A.; Muqbil, I.; Baloglu, E.; Senapedis, W.; Landesman, Y.; Argueta, C.; Kauffman, M.; Chang, H.; Kashyap, T.; Shacham, S.; et al. Down-regulation of AR splice variants through XPO1 suppression contributes to the inhibition of prostate cancer progression. Oncotarget 2018, 9, 35327–35342. [Google Scholar] [CrossRef]
- Dhillon, P.K.; Barry, M.; Stampfer, M.J.; Perner, S.; Fiorentino, M.; Fornari, A.; Ma, J.; Fleet, J.; Kurth, T.; Rubin, M.A.; et al. Aberrant cytoplasmic expression of p63 and prostate cancer mortality. Cancer Epidemiol. Biomark. Prev. 2009, 18, 595–600. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Z.C.; Liu, J.W.; Yang, C.; Zhao, M.; Xiong, Z.Q. XPO1 inhibitor KPT-330 synergizes with Bcl-xL inhibitor to induce cancer cell apoptosis by perturbing rRNA processing and Mcl-1 protein synthesis. Cell Death Dis. 2019, 10, 395. [Google Scholar] [CrossRef]
- Gravina, G.L.; Mancini, A.; Sanita, P.; Vitale, F.; Marampon, F.; Ventura, L.; Landesman, Y.; McCauley, D.; Kauffman, M.; Shacham, S.; et al. KPT-330, a potent and selective exportin-1 (XPO-1) inhibitor, shows antitumor effects modulating the expression of cyclin D1 and survivin [corrected] in prostate cancer models. BMC Cancer 2015, 15, 941. [Google Scholar] [CrossRef] [Green Version]
- Silberstein, J.L.; Taylor, M.N.; Antonarakis, E.S. Novel Insights into Molecular Indicators of Response and Resistance to Modern Androgen-Axis Therapies in Prostate Cancer. Curr. Urol. Rep. 2016, 17, 29. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antonarakis, E.S.; Lu, C.; Wang, H.; Luber, B.; Nakazawa, M.; Roeser, J.C.; Chen, Y.; Mohammad, T.A.; Chen, Y.; Fedor, H.L.; et al. AR-V7 and resistance to enzalutamide and abiraterone in prostate cancer. N. Engl. J. Med. 2014, 371, 1028–1038. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cheng, C.; Sharp, P.A. Regulation of CD44 alternative splicing by SRm160 and its potential role in tumor cell invasion. Mol. Cell. Biol. 2006, 26, 362–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matter, N.; Herrlich, P.; Konig, H. Signal-dependent regulation of splicing via phosphorylation of Sam68. Nature 2002, 420, 691–695. [Google Scholar] [CrossRef] [PubMed]
- Qin, T.; Huang, G.; Chi, L.; Sui, S.; Song, C.; Li, N.; Sun, S.; Li, N.; Zhang, M.; Zhao, Z.; et al. Exceptionally high UBE2C expression is a unique phenomenon in basal-like type breast cancer and is regulated by BRCA1. Biomed. Pharmacother. 2017, 95, 649–655. [Google Scholar] [CrossRef]
- Mateo, J.; Porta, N.; Bianchini, D.; McGovern, U.; Elliott, T.; Jones, R.; Syndikus, I.; Ralph, C.; Jain, S.; Varughese, M.; et al. Olaparib in patients with metastatic castration-resistant prostate cancer with DNA repair gene aberrations (TOPARP-B): A multicentre, open-label, randomised, phase 2 trial. Lancet. Oncol. 2020, 21, 162–174. [Google Scholar] [CrossRef]
- Zhang, J.; Chism, D.D.; Tagawa, S.T.; Monk, P.; Alter, R.S.; Reichman, W.; Senapedis, W.; Baloglu, E.; Shacham, S.; Kauffman, M.G. Eltanexor (KPT-8602), a second-generation selective inhibitor of nuclear export (SINE) compound, in patients with metastatic castration-resistant prostate cancer (mCRPC). J. Clin. Oncol. 2019, 37, 197. [Google Scholar] [CrossRef]
- Hing, Z.A.; Fung, H.Y.; Ranganathan, P.; Mitchell, S.; El-Gamal, D.; Woyach, J.A.; Williams, K.; Goettl, V.M.; Smith, J.; Yu, X.; et al. Next-generation XPO1 inhibitor shows improved efficacy and in vivo tolerability in hematological malignancies. Leukemia 2016, 30, 2364–2372. [Google Scholar] [CrossRef] [Green Version]
- Etchin, J.; Berezovskaya, A.; Conway, A.S.; Galinsky, I.A.; Stone, R.M.; Baloglu, E.; Senapedis, W.; Landesman, Y.; Kauffman, M.; Shacham, S.; et al. KPT-8602, a second-generation inhibitor of XPO1-mediated nuclear export, is well tolerated and highly active against AML blasts and leukemia-initiating cells. Leukemia 2017, 31, 143–150. [Google Scholar] [CrossRef]
- Sramkoski, R.M.; Pretlow, T.G., 2nd; Giaconia, J.M.; Pretlow, T.P.; Schwartz, S.; Sy, M.S.; Marengo, S.R.; Rhim, J.S.; Zhang, D.; Jacobberger, J.W. A new human prostate carcinoma cell line, 22Rv1. Vitr. Cell. Dev. Biol. Anim. 1999, 35, 403–409. [Google Scholar] [CrossRef]
- Turner, J.G.; Dawson, J.; Sullivan, D.M. Nuclear export of proteins and drug resistance in cancer. Biochem. Pharmacol. 2012, 83, 1021–1032. [Google Scholar] [CrossRef] [Green Version]
- Ishizawa, J.; Kojima, K.; Hail, N., Jr.; Tabe, Y.; Andreeff, M. Expression, function, and targeting of the nuclear exporter chromosome region maintenance 1 (CRM1) protein. Pharmacol. Ther. 2015, 153, 25–35. [Google Scholar] [CrossRef] [Green Version]
- Kaufman, B.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmana, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; Hubert, A.; et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2015, 33, 244–250. [Google Scholar] [CrossRef]
- Mateo, J.; Carreira, S.; Sandhu, S.; Miranda, S.; Mossop, H.; Perez-Lopez, R.; Nava Rodrigues, D.; Robinson, D.; Omlin, A.; Tunariu, N.; et al. DNA-Repair Defects and Olaparib in Metastatic Prostate Cancer. N. Engl. J. Med. 2015, 373, 1697–1708. [Google Scholar] [CrossRef]
- Weston, V.J.; Oldreive, C.E.; Skowronska, A.; Oscier, D.G.; Pratt, G.; Dyer, M.J.; Smith, G.; Powell, J.E.; Rudzki, Z.; Kearns, P.; et al. The PARP inhibitor olaparib induces significant killing of ATM-deficient lymphoid tumor cells in vitro and in vivo. Blood 2010, 116, 4578–4587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhan, Y.; Zhang, G.; Wang, X.; Qi, Y.; Bai, S.; Li, D.; Ma, T.; Sartor, O.; Flemington, E.K.; Zhang, H.; et al. Interplay between Cytoplasmic and Nuclear Androgen Receptor Splice Variants Mediates Castration Resistance. Mol. Cancer Res. MCR 2017, 15, 59–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bergerat, J.P.; Ceraline, J. Pleiotropic functional properties of androgen receptor mutants in prostate cancer. Hum. Mutat. 2009, 30, 145–157. [Google Scholar] [CrossRef] [PubMed]
- Feng, Q.; He, B. Androgen Receptor Signaling in the Development of Castration-Resistant Prostate Cancer. Front. Oncol. 2019, 9, 858. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryan, C.J.; Smith, A.; Lal, P.; Satagopan, J.; Reuter, V.; Scardino, P.; Gerald, W.; Scher, H.I. Persistent prostate-specific antigen expression after neoadjuvant androgen depletion: An early predictor of relapse or incomplete androgen suppression. Urology 2006, 68, 834–839. [Google Scholar] [CrossRef] [PubMed]
- Visakorpi, T.; Hyytinen, E.; Koivisto, P.; Tanner, M.; Keinanen, R.; Palmberg, C.; Palotie, A.; Tammela, T.; Isola, J.; Kallioniemi, O.P. In vivo amplification of the androgen receptor gene and progression of human prostate cancer. Nat. Genet. 1995, 9, 401–406. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Li, W.; Zhang, Y.; Yuan, X.; Xu, K.; Yu, J.; Chen, Z.; Beroukhim, R.; Wang, H.; Lupien, M.; et al. Androgen receptor regulates a distinct transcription program in androgen-independent prostate cancer. Cell 2009, 138, 245–256. [Google Scholar] [CrossRef] [Green Version]
- Stockley, J.; Markert, E.; Zhou, Y.; Robson, C.N.; Elliott, D.J.; Lindberg, J.; Leung, H.Y.; Rajan, P. The RNA-binding protein Sam68 regulates expression and transcription function of the androgen receptor splice variant AR-V7. Sci. Rep. 2015, 5, 13426. [Google Scholar] [CrossRef]
- Jones, D.; Wade, M.; Nakjang, S.; Chaytor, L.; Grey, J.; Robson, C.N.; Gaughan, L. FOXA1 regulates androgen receptor variant activity in models of castrate-resistant prostate cancer. Oncotarget 2015, 6, 29782–29794. [Google Scholar] [CrossRef] [Green Version]
- Li, L.; Karanika, S.; Yang, G.; Wang, J.; Park, S.; Broom, B.M.; Manyam, G.C.; Wu, W.; Luo, Y.; Basourakos, S.; et al. Androgen receptor inhibitor-induced "BRCAness" and PARP inhibition are synthetically lethal for castration-resistant prostate cancer. Sci. Signal. 2017, 10, 7479. [Google Scholar] [CrossRef] [Green Version]
- Polkinghorn, W.R.; Parker, J.S.; Lee, M.X.; Kass, E.M.; Spratt, D.E.; Iaquinta, P.J.; Arora, V.K.; Yen, W.F.; Cai, L.; Zheng, D.; et al. Androgen receptor signaling regulates DNA repair in prostate cancers. Cancer Discov. 2013, 3, 1245–1253. [Google Scholar] [CrossRef] [Green Version]
- Virtanen, V.; Paunu, K.; Ahlskog, J.K.; Varnai, R.; Sipeky, C.; Sundvall, M. PARP Inhibitors in Prostate Cancer-The Preclinical Rationale and Current Clinical Development. Genes 2019, 10, 565. [Google Scholar] [CrossRef] [Green Version]
- Kashyap, T.; Argueta, C.; Unger, T.; Klebanov, B.; Debler, S.; Senapedis, W.; Crochiere, M.L.; Lee, M.S.; Kauffman, M.; Shacham, S.; et al. Selinexor reduces the expression of DNA damage repair proteins and sensitizes cancer cells to DNA damaging agents. Oncotarget 2018, 9, 30773–30786. [Google Scholar] [CrossRef] [Green Version]
- Gravina, G.L.; Mancini, A.; Colapietro, A.; Marampon, F.; Sferra, R.; Pompili, S.; Biordi, L.A.; Iorio, R.; Flati, V.; Argueta, C.; et al. Pharmacological treatment with inhibitors of nuclear export enhances the antitumor activity of docetaxel in human prostate cancer. Oncotarget 2017, 8, 111225–111245. [Google Scholar] [CrossRef] [Green Version]
- Mendonca, J.; Sharma, A.; Kim, H.S.; Hammers, H.; Meeker, A.; De Marzo, A.; Carducci, M.; Kauffman, M.; Shacham, S.; Kachhap, S. Selective inhibitors of nuclear export (SINE) as novel therapeutics for prostate cancer. Oncotarget 2014, 5, 6102–6112. [Google Scholar] [CrossRef] [Green Version]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name of the Primers | Direction | Sequences (5′-3′) | References |
|---|---|---|---|
| AR | Forward | GACTTCACCGCACCTGATG | Aboukameel et al. 2018 [23] |
| Reverse | AATGGGCAAAACATGGTCCC | ||
| ARv7 | Forward | TGTCCATCTTGTCGTCTTCGG | |
| Reverse | TGCAATTGCCAACCCGGAAT | ||
| PSA | Forward | GTCCCGGTTGTCTTCCTCAC | |
| Reverse | CTCCCACAATCCGAGACAGG | ||
| FOXA1 | Forward | ACCAGCGACTGGAACAGCTA | |
| Reverse | GTCATGTTGCCGCTCGTAGT | ||
| UBE2C | Forward | TCCTGTCTCTCTGCCAACGC | |
| Reverse | TTGTCTGATTCAGGGAAGGCA | ||
| ACTIN | Forward | GCACAGAGCCTCGCCTT | |
| Reverse | TCATCATCCATGGTGAGCTG | ||
| 18 S | Forward | GCAATTATTCCCCATGAACG | |
| Reverse | GGCCTCACTAAACCATCCAA | ||
| BRCA1 | Forward | GGAACCTGTCTCCACAAAGTGT | This study |
| Reverse | ACCTGTGTCAAGCTGAAAAGC | This study | |
| BRCA2 | Forward | AGTTCCCTCTGCGTGTTCTC | This study |
| Reverse | GGGTATGAGCCATCCACCAT | This study | |
| BLM | Forward | GAGTCTGCGTGCGAGGATTA | This study |
| Reverse | CAGGTGTTTTTGCTACTGACACA | This study | |
| XRCC3 | Forward | GAAGAGGAGTGCGGAACCC | This study |
| Reverse | CTGTGCACATCCTGCTGAGA | This study | |
| EXO1 | Forward | TCCATTGTGAAAAGACCAAGAAGTG | This study |
| Reverse | CCATTTACCAGGTCAGGCAC | This study | |
| RMI1 | Forward | GCGGCGGTTCCTGTCCTTA | This study |
| Reverse | TTGAAACCTCCACTGCTCAGAA | This study | |
| RMI2 | Forward | CAGGGTAGTGATGGCGGACC | This study |
| Reverse | CCACTCCCATCACCATCACAT | This study | |
| RAD54L | Forward | TGGTCCTACACTCTTAGCCG | This study |
| Reverse | TCTCACTGCTGGATTTCCGT | This study | |
| CHEK1 | Forward | CAGTGGTGGGCAAAGGACAGT | This study |
| Reverse | GTCTACGGCACGCTTCATATCT | This study | |
| LIG1 | Forward | CGAAGAAAAGTGCTGGACAGG | This study |
| Reverse | TTTACCCTCTTTCTTGGGGTGG | This study | |
| RAD51 | Forward | GCTGGGAACTGCAACTCATCT | This study |
| Reverse | GCTGCATCTGCATTGCCATTA | This study | |
| GAPDH | Forward | CCACATCGCTCAGACACCAT | This study |
| Reverse | ACCAGAGTTAAAAGCAGCCCT | This study | |
| XPO1 | Forward | ACGAGGAAGGAAGGAGCAGT | This study |
| Reverse | CGAGCTGCATGGTCTGCTAA | This study | |
| CASPASE 9 | Forward | TGTTCAGGCCCCATATGATCG | This study |
| Reverse | CAACTTTGCTGCTTGCCTGT | This study | |
| BAX | Forward | AGGTCTTTTTCCGAGTGGCA | This study |
| Reverse | CCCGGAGGAAGTCCAATGTC | This study | |
| BCL2 | Forward | CCTGGCTGTCTCTGAAGACTC | This study |
| Reverse | GGGGCAGGCATGTTGACTTC | This study | |
| TP53 | Forward | TGACACGCTTCCCTGGATTG | This study |
| Reverse | TTTTCAGGAAGTAGTTTCCATAGGT | This study |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Uddin, M.H.; Li, Y.; Khan, H.Y.; Muqbil, I.; Aboukameel, A.; Sexton, R.E.; Reddy, S.; Landesman, Y.; Kashyap, T.; Azmi, A.S.; et al. Nuclear Export Inhibitor KPT-8602 Synergizes with PARP Inhibitors in Escalating Apoptosis in Castration Resistant Cancer Cells. Int. J. Mol. Sci. 2021, 22, 6676. https://doi.org/10.3390/ijms22136676
Uddin MH, Li Y, Khan HY, Muqbil I, Aboukameel A, Sexton RE, Reddy S, Landesman Y, Kashyap T, Azmi AS, et al. Nuclear Export Inhibitor KPT-8602 Synergizes with PARP Inhibitors in Escalating Apoptosis in Castration Resistant Cancer Cells. International Journal of Molecular Sciences. 2021; 22(13):6676. https://doi.org/10.3390/ijms22136676
Chicago/Turabian StyleUddin, Md. Hafiz, Yiwei Li, Husain Yar Khan, Irfana Muqbil, Amro Aboukameel, Rachel E. Sexton, Shriya Reddy, Yosef Landesman, Trinayan Kashyap, Asfar S. Azmi, and et al. 2021. "Nuclear Export Inhibitor KPT-8602 Synergizes with PARP Inhibitors in Escalating Apoptosis in Castration Resistant Cancer Cells" International Journal of Molecular Sciences 22, no. 13: 6676. https://doi.org/10.3390/ijms22136676
APA StyleUddin, M. H., Li, Y., Khan, H. Y., Muqbil, I., Aboukameel, A., Sexton, R. E., Reddy, S., Landesman, Y., Kashyap, T., Azmi, A. S., & Heath, E. I. (2021). Nuclear Export Inhibitor KPT-8602 Synergizes with PARP Inhibitors in Escalating Apoptosis in Castration Resistant Cancer Cells. International Journal of Molecular Sciences, 22(13), 6676. https://doi.org/10.3390/ijms22136676