
Hydrogen Bonds, Topologies, Energy Frameworks and Solubilities of Five Sorafenib Salts


Abstract
:1. Introduction
2. Results
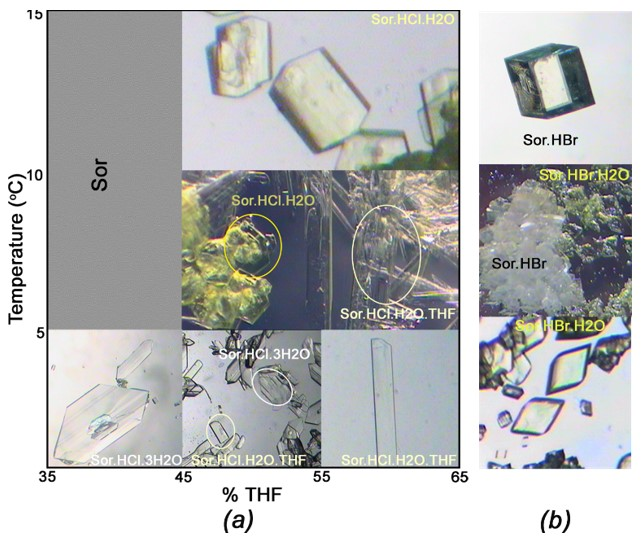
2.1. Concomitantly Crystallization
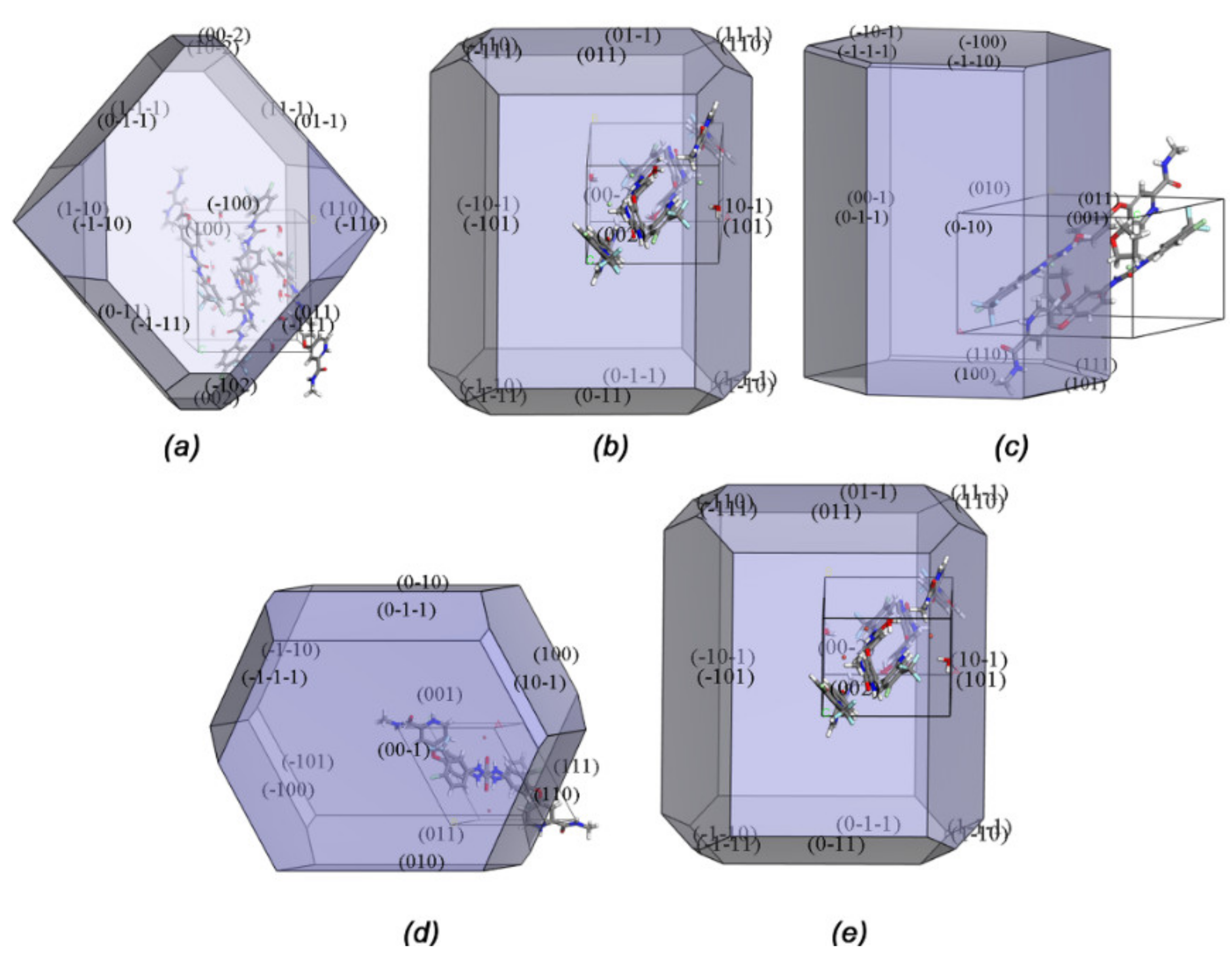
2.2. Morphology
2.3. Crystallization
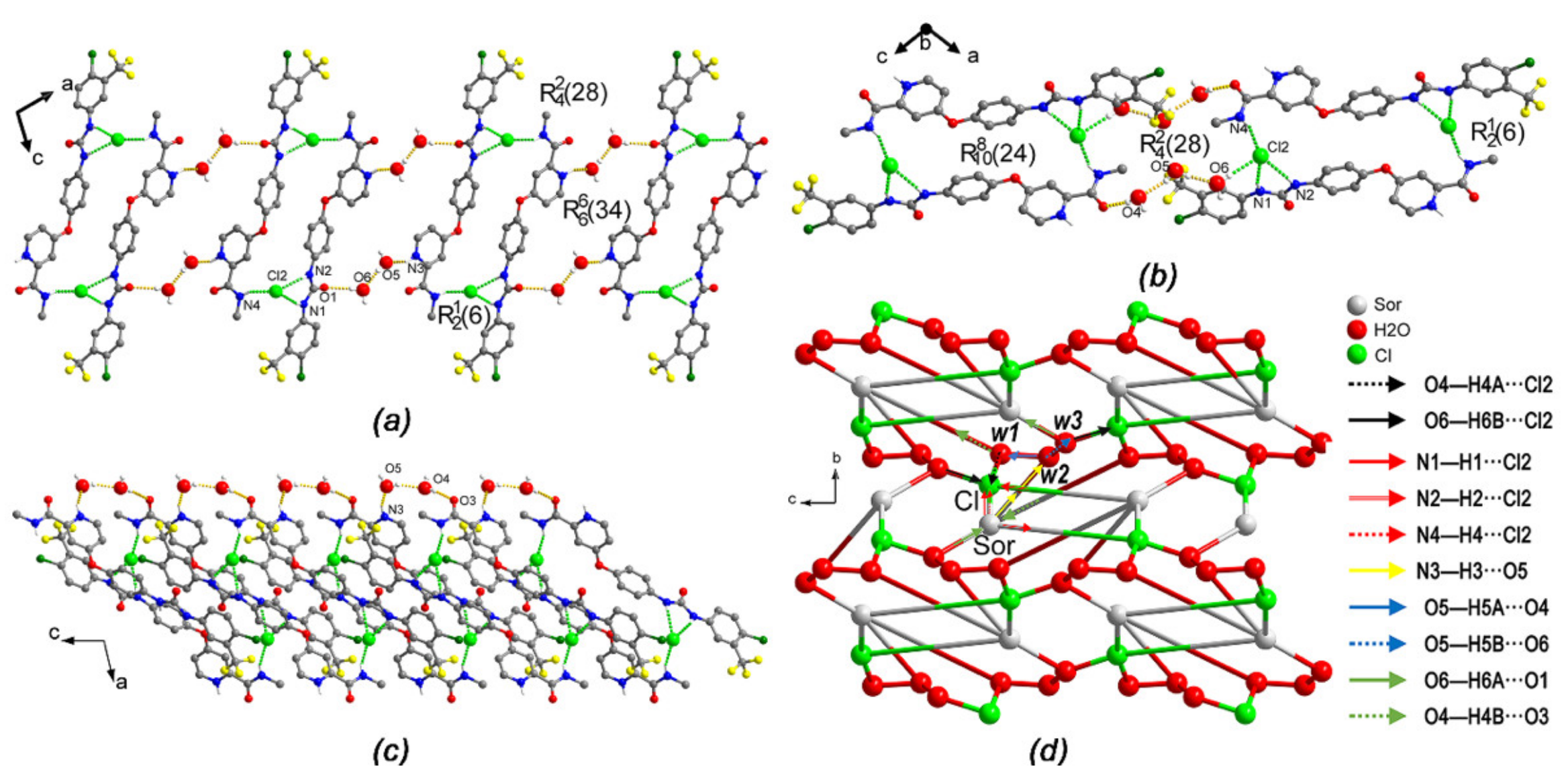
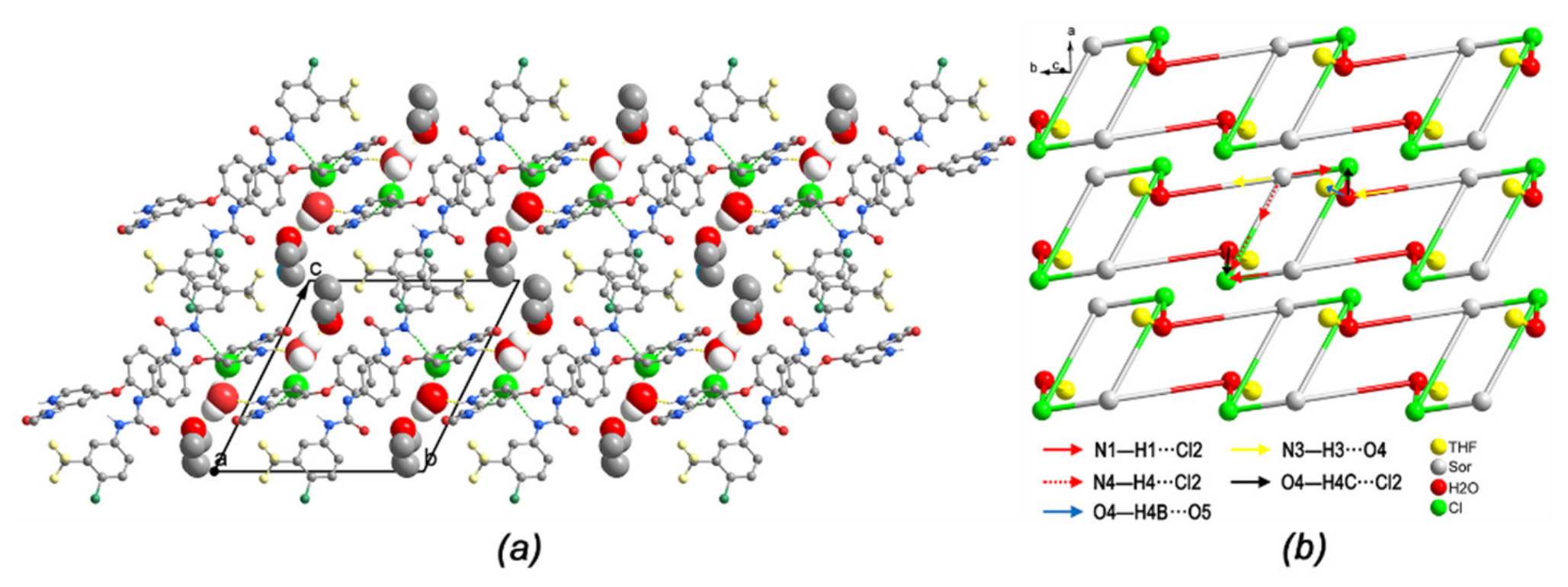
2.4. Hydrogen Bond
2.5. Energy Framework
2.6. Solubilities and Dissolution Rates of Sor and Its Salts
3. Materials and Methods
3.1. Synthesis and Crystallization
3.2. Powder X-ray Diffraction (PXRD)
3.3. Single-Crystal X-ray Diffraction (SC-XRD)
3.4. Solubility and Dissolution Measurement
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Gadaleta-Caldarola, G.; Infusino, S.; Divella, R.; Mazzocca, A.; Rose, F.D.; Filippelli, G.; Brandi, M. Sorafenib: 10 years after the first pivotal trial. Future Oncol. 2015, 11, 1863–1880. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, S.; Carter, C.; Lynch, M.; Lowinger, T.; Dumas, J.; Smith, R.A.; Schwartz, B.; Simantov, R.; Kelley, S. Discovery and development of sorafenib: A multikinase inhibitor for treating cancer. Nat. Rev. Drug Discov. 2006, 5, 835–844. [Google Scholar] [CrossRef] [PubMed]
- Wilhelm, S.M.; Carter, C.; Tang, L.Y.; Wilkie, D.; McNabola, A.; Rong, H.; Chen, C.; Zhang, X.M.; Vincent, P.; McHugh, M.; et al. BAY 43-9006 exhibits broad spectrum oral antitumor activity and targets the RAF/MEK/ERK pathway and receptor tyrosine kinases involved in tumor progression and angiogenesis. Cancer Res. 2004, 64, 7099–7109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bondì, M.L.; Scala, A.; Sortino, G.; Amore, E.; Botto, C.; Azzolina, A.; Balasus, D.; Cervello, M.; Mazzaglia, A. Nanoassemblies Based on Supramolecular Complexes of Nonionic Amphiphilic Cyclodextrin and Sorafenib as Effective Weapons to Kill Human HCC Cells. Biomacromolecules 2015, 16, 3784–3791. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Chen, Z.; Chen, Y.; Lu, J.; Li, Y.; Wang, S.; Wu, G.; Qian, F. Improving Oral Bioavailability of Sorafenib by Optimizing the “Spring” and “Parachute” Based on Molecular Interaction Mechanisms. Mol. Pharm. 2016, 13, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Kansara, H.; Panola, R.; Mishra, A. Techniques used to Enhance Bioavailability of BCS Class II Drugs: A Review. Int. J. Drug Dev. Res. 2015, 7, 82–93. [Google Scholar]
- Serajuddin, A.T.M. Salt formation to improve drug solubility. Adv. Drug Deliv. Rev. 2007, 59, 603–616. [Google Scholar] [CrossRef] [PubMed]
- Rajput, L. Stable Crystalline Salts of Haloperidol: A Highly Water-Soluble Mesylate Salt. Cryst. Growth Des. 2014, 14, 5196–5205. [Google Scholar] [CrossRef]
- Gidwani, R.M.; Wakchaure, V.S.; Striegel, H.G.; Albrecht, W. Polymorphorphs of 4-[4-[[4-Chloro-3-(trifluoromethyl)phenyl]carbamoylamino]phenoxy]-n-methyl-pyridine-2-carboxamide. WO Patent. 2010/142678A2, 16 December 2010. [Google Scholar]
- Jaryal, J.S.; Sathyanarayana, S.; Thaper, R.K.; Prasad, M. Polymorphs of Sorafenib Acid Addition Salts. U.S. Patent. 2012/0264789A1, 18 October 2012. [Google Scholar]
- Turner, M.J.; Thomas, S.P.; Shi, M.W.; Jayatilaka, D.; Spackman, M.A. Energy frameworks: Insights into interaction anisotropy and the mechanical properties of molecular crystals. Chem. Commun. 2015, 51, 3735–3738. [Google Scholar] [CrossRef] [PubMed]
- Munshi, P.; Venugopala, K.N.; Jayashree, B.S.; Guru, T.N. Concomitant Polymorphism in 3-Acetylcoumarin: Role of Weak C-H⋯O and C-H⋯π Interactions. Cryst. Growth Des. 2004, 4, 1105–1107. [Google Scholar] [CrossRef]
- Leităo, M.L.P.; Canotilho, J.; Cruz, M.S.C.; Pereira, J.C.; Sousa, A.T.; Redinha, J.S. Study of Polymorphism from DSC Melting Curves; Polymorphs of Terfenadine. J. Therm. Anal. Calorim. 2002, 68, 397–412. [Google Scholar]
- Phan, C.U.; Shen, J.; Liu, J.; Mao, J.; Hu, X.; Tang, G. Isomorphous Crystals Formed by the Similar Supramolecular Motifs in Sorafenib Hydrochloride and Regorafenib Hydrochloride Salts. Crystals 2019, 9, 649. [Google Scholar] [CrossRef] [Green Version]
- Etter, M.C. Encoding and decoding hydrogen-bond patterns of organic compounds. Acc. Chem. Res. 1990, 23, 120–126. [Google Scholar] [CrossRef]
- Etter, M.C.; MacDonald, J.C.; Bernstein, J. Graph-set analysis of hydrogen-bond patterns in organic crystals. Acta Crystallogr. 1990, B46, 256–262. [Google Scholar] [CrossRef] [PubMed]
- Bernstein, J.; Davis, R.E.; Shimoni, L.; Chang, N.L. Patterns in Hydrogen Bonding: Functionality and Graph Set Analysis in Crystals. Angew. Chem. Int. Ed. Engl. 1995, 34, 1555–1573. [Google Scholar] [CrossRef]
- APEX3, SADABS and SAINT 2016.
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.; Puschmann, H. OLEX2: A complete structure solution, refinement and analysis program. J. Appl. Crystallogr. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Sheldrick, G.M. SHELXT—Integrated space-group and crystal-structure determination. Acta Crystallogr. A Found. Adv. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. C Struct. Chem. 2015, 71, 3–8. [Google Scholar] [CrossRef] [PubMed]
- Brandenburg, K.; Putz, H. DIAMOND; Crystal Impact GbR: Bonn, Germany, 2005. [Google Scholar]
- Blatov, V.A. Multipurpose crystallochemical analysis with the program package TOPOS. IUCr Comput. Comm. Newsl. 2006, 7, 4–38. [Google Scholar]
- Baburin, I.A.; Blatov, V.A. Three-dimensional hydrogen-bonded frameworks in organic crystals: A topological study. Acta Crystallogr. Sect. B 2007, 63, 791–802. [Google Scholar] [CrossRef] [PubMed]
- Accelrys Inc. Materials Studio; Accelrys Inc.: San Diego, CA, USA, 2003. [Google Scholar]
- Turner, M.J.; McKinnon, J.J.; Wolff, S.K.; Grimwood, D.J.; Spackman, P.R.; Jayatilaka, D.; Spackman, M.A. CrystalExplorer 17. University of Western Australia. 2017. Available online: http://hirshfeldsurface.net (accessed on 10 December 2019).






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (I) | (II) | (III) | (IV) | (V) | |
| Chemical formula | Cl·C21H17ClF3N4O3·3(H2O) | (Cl)·(C21H17ClF3N4O3)·(H2O) | Cl·C21H17ClF3N4O3·H2O·C4H8O | Br·C21H17ClF3N4O3 | Br·C21H17ClF3N4O3·H2O |
| Mr | 555.33 | 2077.20 | 591.40 | 545.74 | 563.76 |
| Crystal system, space group | Monoclinic, P21/c | Monoclinic, P21/n | Triclinic, P | Triclinic, | Monoclinic, P21/n |
| a, b, c (Å) | 13.9736 (5), 12.7166 (5), 14.6215 (5) | 11.3049 (3), 8.5008 (2), 23.1013 (6) | 9.1309 (3), 13.0397 (4), 13.2554 (5) | 9.545 (3), 10.840 (3), 12.353 (4) | 11.3349 (15), 8.7524 (10), 23.048 (3) |
| α, β, γ (°) | 90, 103.294 (1), 90 | 90, 93.826 (1), 90 | 63.472 (1), 89.255 (1), 88.985 (1) | 77.633 (13), 87.153 (16), 64.131 (12) | 90, 93.167 (5), 90 |
| V (Å3) | 2528.57 (16) | 2215.10 (10) | 1411.84 (8) | 1121.9 (6) | 2283.1 (5) |
| Z | 4 | 1 | 2 | 2 | 4 |
| D—H···A | D—H | H⋯A | D⋯A | D—H⋯A |
| N1—H1···Cl2 | 0.86 | 2.42 | 3.2473 (14) | 161 |
| N2—H2···Cl2 | 0.86 | 2.44 | 3.2527 (14) | 158 |
| N4—H4···Cl2 i | 0.86 | 2.41 | 3.2122 (16) | 156 |
| O4—H4A···Cl2 ii | 0.85 | 2.38 | 3.2049 (17) | 163 |
| O4—H4B···O3 iii | 0.85 | 1.96 | 2.796 (2) | 169 |
| O5—H5A···O4 | 0.85 | 1.96 | 2.764 (3) | 158 |
| O5—H5B···O6 | 0.85 | 1.89 | 2.737 (3) | 172 |
| O6—H6A···O1 | 0.85 | 2.02 | 2.869 (2) | 175 |
| N3—H3···O5 iv | 0.92 (2) | 1.77 (3) | 2.631 (2) | 154 (2) |
| O6—H6B···Cl2 v | 0.89 (4) | 2.34 (4) | 3.225 (2) | 172 (3) |
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
| N1—H1⋯Cl2 | 0.88 | 2.35 | 3.1934 (16) | 161 |
| N2—H2⋯Cl2 | 0.88 | 2.47 | 3.2872 (15) | 155 |
| N4—H4⋯Cl2 i | 0.88 | 2.45 | 3.2569 (16) | 153 |
| O4—H4B⋯O3 | 0.97 (4) | 2.12 (7) | 2.774 (3) | 124 (6) |
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
| N4—H4⋯Br1 i | 0.88 | 2.59 | 3.3853 (18) | 151 |
| N1—H1⋯Br1 | 0.88 | 2.49 | 3.331 (2) | 161 |
| O4—H4B⋯O3 ii | 0.94 (2) | 2.23 (5) | 2.868 (3) | 125 (4) |
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
| O4—H4B⋯O5 | 0.85 | 1.83 | 2.676 (4) | 171 |
| O4—H4C⋯Cl2 | 0.85 | 2.32 | 3.118 (2) | 156 |
| N4—H4⋯Cl2 ii | 0.86 | 2.42 | 3.206 (2) | 152 |
| N1—H1⋯Cl2 i | 0.86 | 2.38 | 3.224 (2) | 168 |
| N3—H3⋯O4 | 0.89 (3) | 1.79 (3) | 2.641 (3) | 159 (2) |
| D—H⋯A | D—H | H⋯A | D⋯A | D—H⋯A |
| N1—H1⋯Br1 i | 0.88 | 2.48 | 3.324 (2) | 160 |
| N3—H3⋯O1 ii | 0.88 | 2.01 | 2.860 (2) | 162 |
| N4—H4A⋯Br1 | 0.88 | 2.48 | 3.325 (3) | 161 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Phan, C.; Shen, J.; Yu, K.; Liu, J.; Tang, G. Hydrogen Bonds, Topologies, Energy Frameworks and Solubilities of Five Sorafenib Salts. Int. J. Mol. Sci. 2021, 22, 6682. https://doi.org/10.3390/ijms22136682
Phan C, Shen J, Yu K, Liu J, Tang G. Hydrogen Bonds, Topologies, Energy Frameworks and Solubilities of Five Sorafenib Salts. International Journal of Molecular Sciences. 2021; 22(13):6682. https://doi.org/10.3390/ijms22136682
Chicago/Turabian StylePhan, Chiuyen, Jie Shen, Kaxi Yu, Jiyong Liu, and Guping Tang. 2021. "Hydrogen Bonds, Topologies, Energy Frameworks and Solubilities of Five Sorafenib Salts" International Journal of Molecular Sciences 22, no. 13: 6682. https://doi.org/10.3390/ijms22136682
APA StylePhan, C., Shen, J., Yu, K., Liu, J., & Tang, G. (2021). Hydrogen Bonds, Topologies, Energy Frameworks and Solubilities of Five Sorafenib Salts. International Journal of Molecular Sciences, 22(13), 6682. https://doi.org/10.3390/ijms22136682