
eNAMPT Is Localised to Areas of Cartilage Damage in Patients with Hip Osteoarthritis and Promotes Cartilage Catabolism and Inflammation


{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
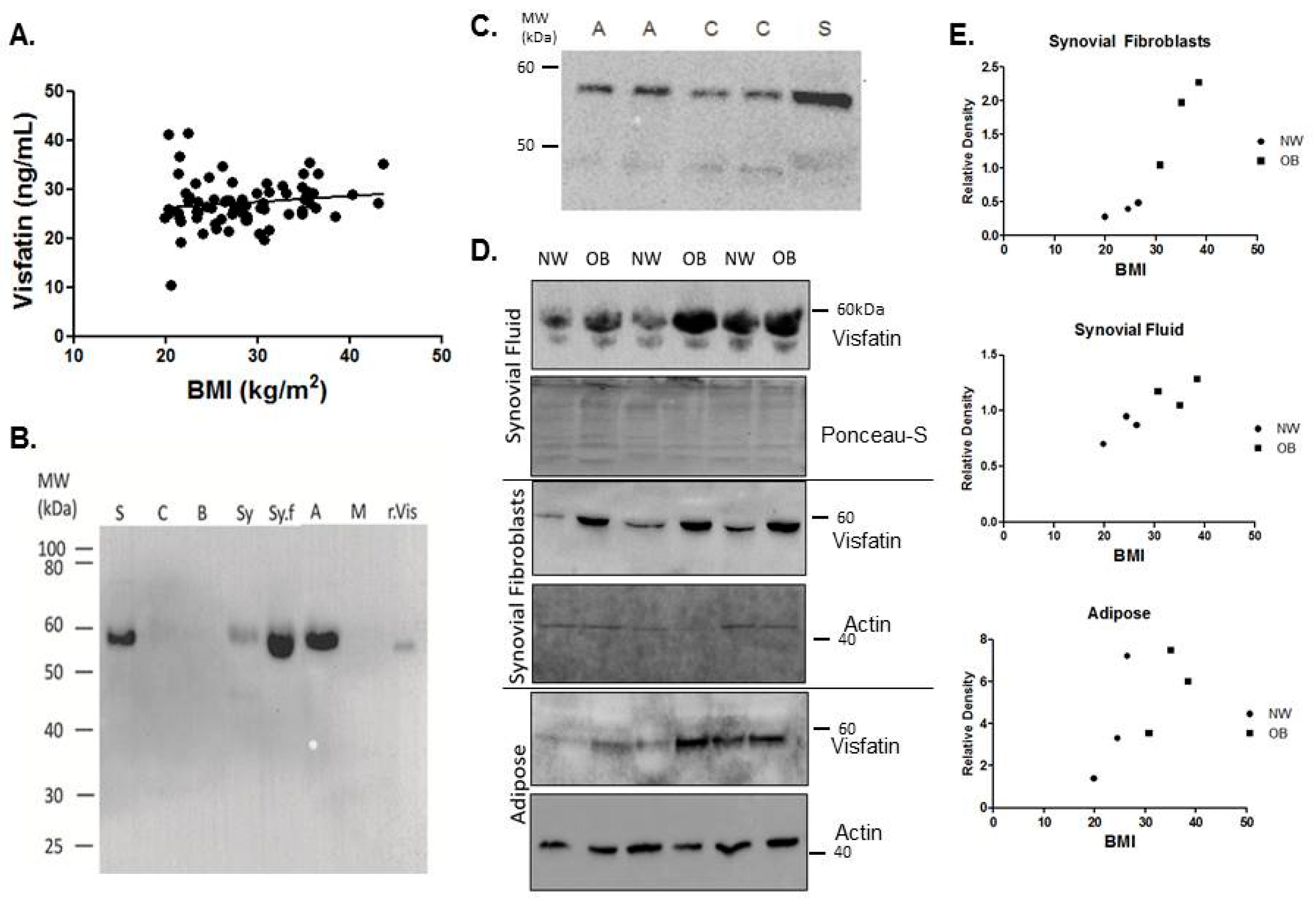
2.1. Visfatin Is Expressed Locally by the Tissues of the Hip OA Joint and Is Elevated in the Synovial Tissues of Obese Patients
2.2. Visfatin Induces the Production of Matrix Metalloproteases in Human Hip OA Cartilage and Is Co-Localised with MMP13 in Areas of Damage
2.3. Visfatin Induces the Secretion of Pro-Inflammatory Cytokines and Chemokines in Human Hip OA Cartilage
2.4. eNAMPT Does Not Increase Intracellular NAD+ Production in Primary Human Hip OA Chondrocytes
2.5. Visfatin Stimulates Loss of Proteoglycan and Exhibits Increased Expression and Co-Localisation with MMP13 in Areas of Cartilage Fibrillation
3. Discussion
4. Materials and Methods
4.1. Participant Recruitment and Sample Collection
4.2. Antibodies
4.3. Serum Visfatin Profiling by ELISA
4.4. Preparation of Cartilage Explants and Isolation of Primary Synovial Fibroblasts
4.5. Explant Stimulation
4.6. Determination of Matrix Metalloproteinase (MMP) and Proinflammatory Cytokine Production
4.7. Synthesis of the NAMPT Inhibitor N-(4-((4-(Phenylcarbamoyl)phenyl)sulfonyl)benzyl)imidazo[1,2-a]pyridine-6-carboxamide
4.8. Determination of Sulphated Glycoaminoglycan (sGAG)
4.9. Immunohistochemistry of the Femoral Head
4.10. NAD Activity Assay
4.11. Receptor Identification Screen
4.12. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Tonge, D.P.; Pearson, M.J.; Jones, S.W. The hallmarks of osteoarthritis and the potential to develop personalised disease-modifying pharmacological therapeutics. Osteoarthr. Cartil. 2014, 22, 609–621. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Philp, A.M.; Davis, E.T.; Jones, S.W. Developing anti-inflammatory therapeutics for patients with osteoarthritis. Rheumatology 2017, 56, 869–881. [Google Scholar] [CrossRef] [Green Version]
- Cicuttini, F.M.; Wluka, A.E. Osteoarthritis: Is OA a mechanical or systemic disease? Nat. Rev. Rheumatol. 2014, 10, 515–516. [Google Scholar] [CrossRef] [PubMed]
- Oliveria, S.A.; Felson, D.T.; Cirillo, P.A.; Reed, J.I.; Walker, A.M. Body weight, body mass index, and incident symptomatic osteoarthritis of the hand, hip, and knee. Epidemiology 1999, 10, 161–166. [Google Scholar] [CrossRef] [PubMed]
- Tsao, T.S.; Lodish, H.F.; Fruebis, J. ACRP30, a new hormone controlling fat and glucose metabolism. Eur. J. Pharmacol. 2002, 440, 213–221. [Google Scholar] [CrossRef]
- Dyck, D.J. Adipokines as regulators of muscle metabolism and insulin sensitivity. Appl. Physiol. Nutr. Metab. 2009, 34, 396–402. [Google Scholar] [CrossRef] [PubMed]
- Goralski, K.B.; McCarthy, T.C.; Hanniman, E.A.; Zabel, B.A.; Butcher, E.C.; Parlee, S.D.; Muruganandan, S.; Sinal, C.J. Chemerin, a novel adipokine that regulates adipogenesis and adipocyte metabolism. J. Biol. Chem. 2007, 282, 28175–28188. [Google Scholar] [CrossRef] [Green Version]
- Lu, J.Y.; Huang, K.C.; Chang, L.C.; Huang, Y.S.; Chi, Y.C.; Su, T.C.; Chen, C.L.; Yang, W.S. Adiponectin: A biomarker of obesity-induced insulin resistance in adipose tissue and beyond. J. Biomed. Sci. 2008, 15, 565–576. [Google Scholar] [CrossRef]
- Yang, W.S.; Chuang, L.M. Human genetics of adiponectin in the metabolic syndrome. J. Mol. Med. 2006, 84, 112–121. [Google Scholar] [CrossRef]
- Wilhelmsen, A.; Tsintzas, K.; Jones, S.W. Recent advances and future avenues in understanding the role of adipose tissue cross talk in mediating skeletal muscle mass and function with ageing. Geroscience 2021, 43, 85–110. [Google Scholar] [CrossRef]
- de Boer, T.N.; van Spil, W.E.; Huisman, A.M.; Polak, A.A.; Bijlsma, J.W.; Lafeber, F.P.; Mastbergen, S.C. Serum adipokines in osteoarthritis; comparison with controls and relationship with local parameters of synovial inflammation and cartilage damage. Osteoarthr. Cartil. 2012, 20, 846–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lago, R.; Gomez, R.; Otero, M.; Lago, F.; Gallego, R.; Dieguez, C.; Gomez-Reino, J.J.; Gualillo, O. A new player in cartilage homeostasis: Adiponectin induces nitric oxide synthase type II and pro-inflammatory cytokines in chondrocytes. Osteoarthr. Cartil. 2008, 16, 1101–1109. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, T.; Church, C.; Baker, D.J.; Jones, S.W. The role of adipokines in skeletal muscle inflammation and insulin sensitivity. J. Inflamm. 2018, 15, 9. [Google Scholar] [CrossRef] [Green Version]
- Nicholson, T.; Church, C.; Tsintzas, K.; Jones, R.; Breen, L.; Davis, E.T.; Baker, D.J.; Jones, S.W. Vaspin promotes insulin sensitivity of elderly muscle and is upregulated in obesity. J. Endocrinol. 2019, 241, 31–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- O’Leary, M.F.; Wallace, G.R.; Davis, E.T.; Murphy, D.P.; Nicholson, T.; Bennett, A.J.; Tsintzas, K.; Jones, S.W. Obese subcutaneous adipose tissue impairs human myogenesis, particularly in old skeletal muscle, via resistin-mediated activation of NFkappaB. Sci. Rep. 2018, 8, 15360. [Google Scholar] [CrossRef] [PubMed]
- Otero, M.; Gomez Reino, J.J.; Gualillo, O. Synergistic induction of nitric oxide synthase type II: In vitro effect of leptin and interferon-gamma in human chondrocytes and ATDC5 chondrogenic cells. Arthritis Rheum. 2003, 48, 404–409. [Google Scholar] [CrossRef] [PubMed]
- Philp, A.M.; Collier, R.L.; Grover, L.M.; Davis, E.T.; Jones, S.W. Resistin promotes the abnormal Type I collagen phenotype of subchondral bone in obese patients with end stage hip osteoarthritis. Sci. Rep. 2017, 7, 4042. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tong, K.M.; Chen, C.P.; Huang, K.C.; Shieh, D.C.; Cheng, H.C.; Tzeng, C.Y.; Chen, K.H.; Chiu, Y.C.; Tang, C.H. Adiponectin increases MMP-3 expression in human chondrocytes through AdipoR1 signaling pathway. J. Cell. Biochem. 2011, 112, 1431–1440. [Google Scholar] [CrossRef]
- Berry, P.A.; Jones, S.W.; Cicuttini, F.M.; Wluka, A.E.; Maciewicz, R.A. Temporal relationship between serum adipokines, biomarkers of bone and cartilage turnover, and cartilage volume loss in a population with clinical knee osteoarthritis. Arthritis Rheum. 2011, 63, 700–707. [Google Scholar] [CrossRef]
- Jia, S.H.; Li, Y.; Parodo, J.; Kapus, A.; Fan, L.; Rotstein, O.D.; Marshall, J.C. Pre-B cell colony-enhancing factor inhibits neutrophil apoptosis in experimental inflammation and clinical sepsis. J. Clin. Investig. 2004, 113, 1318–1327. [Google Scholar] [CrossRef]
- Laiguillon, M.C.; Houard, X.; Bougault, C.; Gosset, M.; Nourissat, G.; Sautet, A.; Jacques, C.; Berenbaum, F.; Sellam, J. Expression and function of visfatin (Nampt), an adipokine-enzyme involved in inflammatory pathways of osteoarthritis. Arthritis Res. Ther. 2014, 16, R38. [Google Scholar] [CrossRef] [Green Version]
- Revollo, J.R.; Grimm, A.A.; Imai, S. The regulation of nicotinamide adenine dinucleotide biosynthesis by Nampt/PBEF/visfatin in mammals. Curr. Opin. Gastroenterol. 2007, 23, 164–170. [Google Scholar] [CrossRef] [PubMed]
- Revollo, J.R.; Grimm, A.A.; Imai, S. The NAD biosynthesis pathway mediated by nicotinamide phosphoribosyltransferase regulates Sir2 activity in mammalian cells. J. Biol. Chem. 2004, 279, 50754–50763. [Google Scholar] [CrossRef] [Green Version]
- Fukuhara, A.; Matsuda, M.; Nishizawa, M.; Segawa, K.; Tanaka, M.; Kishimoto, K.; Matsuki, Y.; Murakami, M.; Ichisaka, T.; Murakami, H.; et al. Visfatin: A protein secreted by visceral fat that mimics the effects of insulin. Science 2005, 307, 426–430. [Google Scholar] [CrossRef] [PubMed]
- Miao, C.Y.; Li, Z.Y. The role of perivascular adipose tissue in vascular smooth muscle cell growth. Br. J. Pharmacol. 2012, 165, 643–658. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moschen, A.R.; Kaser, A.; Enrich, B.; Mosheimer, B.; Theurl, M.; Niederegger, H.; Tilg, H. Visfatin, an adipocytokine with proinflammatory and immunomodulating properties. J. Immunol. 2007, 178, 1748–1758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imai, S. “Clocks” in the NAD World: NAD as a metabolic oscillator for the regulation of metabolism and aging. Biochim. Biophys. Acta 2010, 1804, 1584–1590. [Google Scholar] [CrossRef] [Green Version]
- Esteghamati, A.; Alamdari, A.; Zandieh, A.; Elahi, S.; Khalilzadeh, O.; Nakhjavani, M.; Meysamie, A. Serum visfatin is associated with type 2 diabetes mellitus independent of insulin resistance and obesity. Diabetes Res. Clin. Pract. 2011, 91, 154–158. [Google Scholar] [CrossRef] [PubMed]
- Catalan, V.; Gomez-Ambrosi, J.; Rodriguez, A.; Ramirez, B.; Silva, C.; Rotellar, F.; Cienfuegos, J.A.; Salvador, J.; Fruhbeck, G. Association of increased visfatin/PBEF/NAMPT circulating concentrations and gene expression levels in peripheral blood cells with lipid metabolism and fatty liver in human morbid obesity. Nutr. Metab. Cardiovasc. Dis. 2011, 21, 245–253. [Google Scholar] [CrossRef]
- Chang, Y.H.; Chang, D.M.; Lin, K.C.; Shin, S.J.; Lee, Y.J. Visfatin in overweight/obesity, type 2 diabetes mellitus, insulin resistance, metabolic syndrome and cardiovascular diseases: A meta-analysis and systemic review. Diabetes Metab. Res. Rev. 2011, 27, 515–527. [Google Scholar] [CrossRef] [PubMed]
- Davutoglu, M.; Ozkaya, M.; Guler, E.; Garipardic, M.; Gursoy, H.; Karabiber, H.; Kilinc, M. Plasma visfatin concentrations in childhood obesity: Relationships with insulin resistance and anthropometric indices. Swiss Med. Wkly. 2009, 139, 22–27. [Google Scholar] [PubMed]
- Stastny, J.; Bienertova-Vasku, J.; Vasku, A. Visfatin and its role in obesity development. Diabetes Metab. Syndr. 2012, 6, 120–124. [Google Scholar] [CrossRef] [PubMed]
- Gosset, M.; Berenbaum, F.; Salvat, C.; Sautet, A.; Pigenet, A.; Tahiri, K.; Jacques, C. Crucial role of visfatin/pre-B cell colony-enhancing factor in matrix degradation and prostaglandin E2 synthesis in chondrocytes: Possible influence on osteoarthritis. Arthritis Rheum. 2008, 58, 1399–1409. [Google Scholar] [CrossRef] [PubMed]
- Otero, M.; Favero, M.; Dragomir, C.; Hachem, K.E.; Hashimoto, K.; Plumb, D.A.; Goldring, M.B. Human chondrocyte cultures as models of cartilage-specific gene regulation. Methods Mol. Biol. 2012, 806, 301–336. [Google Scholar]
- Shi, Y.; Ma, J.; Zhang, X.; Li, H.; Jiang, L.; Qin, J. Hypoxia combined with spheroid culture improves cartilage specific function in chondrocytes. Integr. Biol. 2015, 7, 289–297. [Google Scholar] [CrossRef]
- Tekari, A.; Luginbuehl, R.; Hofstetter, W.; Egli, R.J. Chondrocytes expressing intracellular collagen type II enter the cell cycle and co-express collagen type I in monolayer culture. J. Orthop. Res. 2014, 32, 1503–1511. [Google Scholar] [CrossRef]
- Duan, Y.; Hao, D.; Li, M.; Wu, Z.; Li, D.; Yang, X.; Qiu, G. Increased synovial fluid visfatin is positively linked to cartilage degradation biomarkers in osteoarthritis. Rheumatol. Int. 2012, 32, 985–990. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.P.; Bao, J.P.; Feng, J.; Hu, P.F.; Shi, Z.L.; Wu, L.D. Increased serum concentrations of visfatin and its production by different joint tissues in patients with osteoarthritis. Clin. Chem. Lab. Med. 2010, 48, 1141–1145. [Google Scholar] [CrossRef]
- Nanus, D.E.; Wijesinghe, S.N.; Pearson, M.J.; Hadjicharalambous, M.R.; Rosser, A.; Davis, E.T.; Lindsay, M.A.; Jones, S.W. Regulation of the Inflammatory Synovial Fibroblast Phenotype by Metastasis-Associated Lung Adenocarcinoma Transcript 1 Long Noncoding RNA in Obese Patients With Osteoarthritis. Arthritis Rheumatol. 2020, 72, 609–619. [Google Scholar] [CrossRef] [PubMed]
- Wijesinghe, S.N.; Nicholson, T.; Tsintzas, K.; Jones, S.W. Involvements of long noncoding RNAs in obesity-associated inflammatory diseases. Obes. Rev. 2021, 22, e13156. [Google Scholar] [CrossRef]
- Galasso, O.; Familiari, F.; De Gori, M.; Gasparini, G. Recent findings on the role of gelatinases (matrix metalloproteinase-2 and -9) in osteoarthritis. Adv. Orthop. 2012, 2012, 834208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, A.E.; Kunkel, S.L.; Shah, M.R.; Fu, R.; Mazarakis, D.D.; Haines, G.K.; Burdick, M.D.; Pope, R.M.; Strieter, R.M. Macrophage inflammatory protein-1 beta: A C-C chemokine in osteoarthritis. Clin. Immunol. Immunopathol. 1995, 77, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Pallu, S.; Francin, P.J.; Guillaume, C.; Gegout-Pottie, P.; Netter, P.; Mainard, D.; Terlain, B.; Presle, N. Obesity affects the chondrocyte responsiveness to leptin in patients with osteoarthritis. Arthritis Res. Ther. 2010, 12, R112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lohmander, L.S.; Neame, P.J.; Sandy, J.D. The structure of aggrecan fragments in human synovial fluid. Evidence that aggrecanase mediates cartilage degradation in inflammatory joint disease, joint injury, and osteoarthritis. Arthritis Rheum. 1993, 36, 1214–1222. [Google Scholar] [CrossRef]
- Bair, K.W.; Baumeister, T.; Buckmelter, A.J.; Clodfelter, K.H.; Dragovich, P.; Gosselin, F.; Han, B.; Lin, J.; Reynolds, D.J.; Roth, B. From PCT Int. Appl. (2012), WO2012031197. Available online: https://patentscope2.wipo.int/search/en/detail.jsf?docId=WO2012031197 (accessed on 17 June 2021).
- Farndale, R.W.; Sayers, C.A.; Barrett, A.J. A direct spectrophotometric microassay for sulfated glycosaminoglycans in cartilage cultures. Connect. Tissue Res. 1982, 9, 247–248. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Philp, A.M.; Butterworth, S.; Davis, E.T.; Jones, S.W. eNAMPT Is Localised to Areas of Cartilage Damage in Patients with Hip Osteoarthritis and Promotes Cartilage Catabolism and Inflammation. Int. J. Mol. Sci. 2021, 22, 6719. https://doi.org/10.3390/ijms22136719
Philp AM, Butterworth S, Davis ET, Jones SW. eNAMPT Is Localised to Areas of Cartilage Damage in Patients with Hip Osteoarthritis and Promotes Cartilage Catabolism and Inflammation. International Journal of Molecular Sciences. 2021; 22(13):6719. https://doi.org/10.3390/ijms22136719
Chicago/Turabian StylePhilp, Ashleigh M., Sam Butterworth, Edward T. Davis, and Simon W. Jones. 2021. "eNAMPT Is Localised to Areas of Cartilage Damage in Patients with Hip Osteoarthritis and Promotes Cartilage Catabolism and Inflammation" International Journal of Molecular Sciences 22, no. 13: 6719. https://doi.org/10.3390/ijms22136719
APA StylePhilp, A. M., Butterworth, S., Davis, E. T., & Jones, S. W. (2021). eNAMPT Is Localised to Areas of Cartilage Damage in Patients with Hip Osteoarthritis and Promotes Cartilage Catabolism and Inflammation. International Journal of Molecular Sciences, 22(13), 6719. https://doi.org/10.3390/ijms22136719