
Monosomy 3 Is Linked to Resistance to MEK Inhibitors in Uveal Melanoma




Abstract
:1. Introduction
2. Results
2.1. Cell Lines with Monosomy 3 Show Higher Resistance to MEK Inhibition
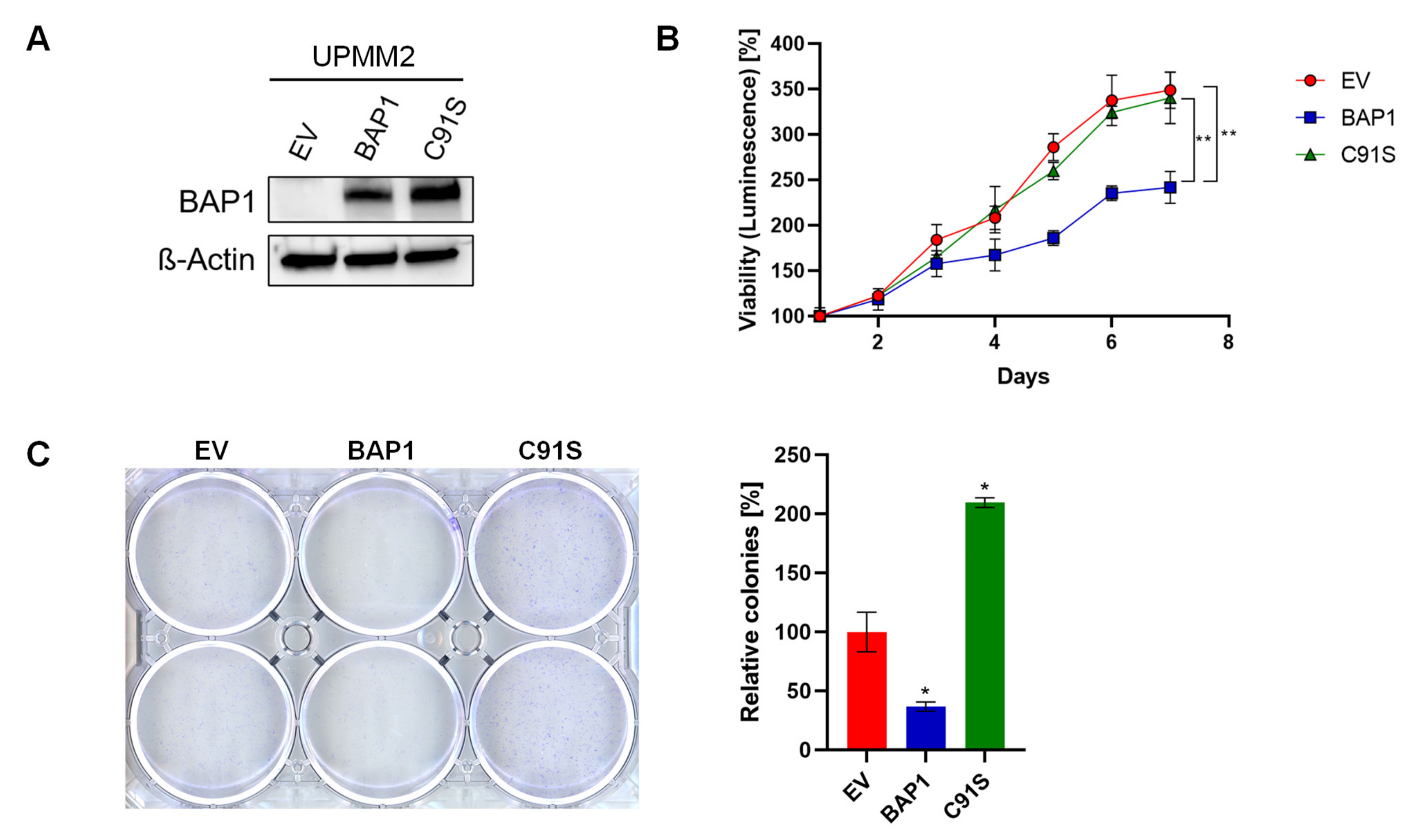
2.2. BAP1-Reconstitution in a M3 Cell Line Does Not Influence Sensitivity to MEKi
2.3. Monosomy 3 Does Not Seem to Be Associated with Higher Activation of the ERK/MAPK Pathway
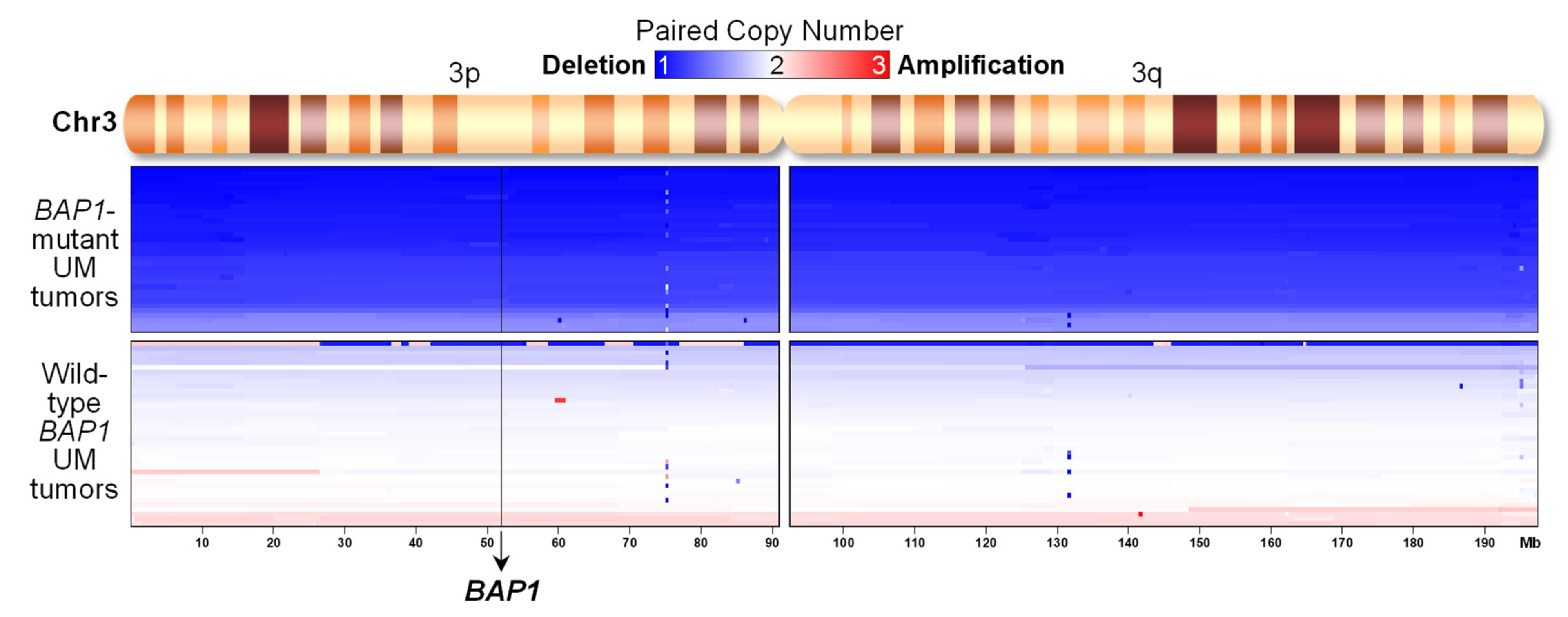
2.4. UM Tumors from TCGA with BAP1 Mutations Show Monosomy 3 whereas Those with Wild-Type BAP1 Show Neutral DNA Copy Numbers
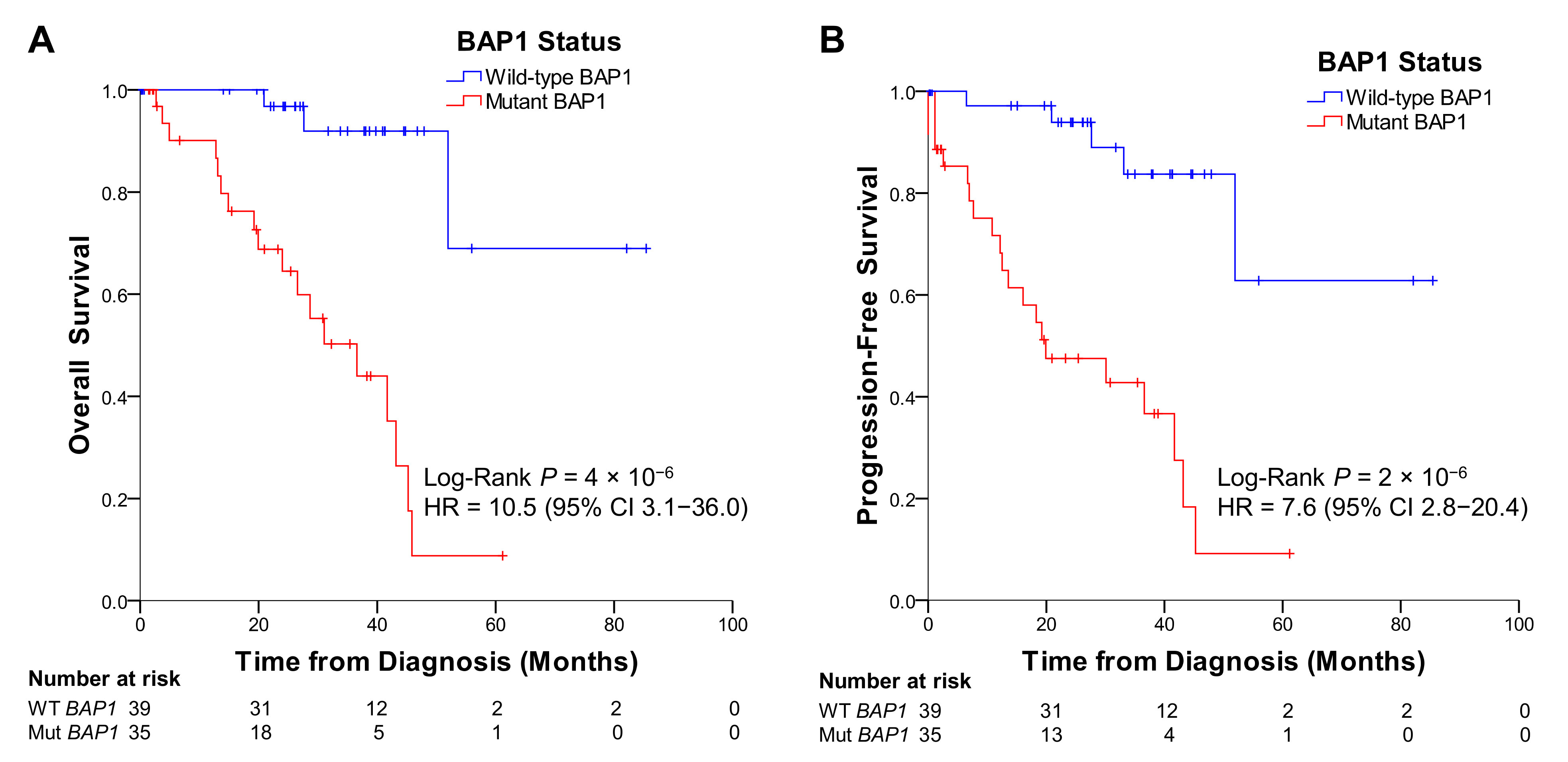
2.5. UM Patients from TCGA with BAP1 Mutations Show Poorer Overall and Progression-Free Survival than Patients without Mutations in BAP1
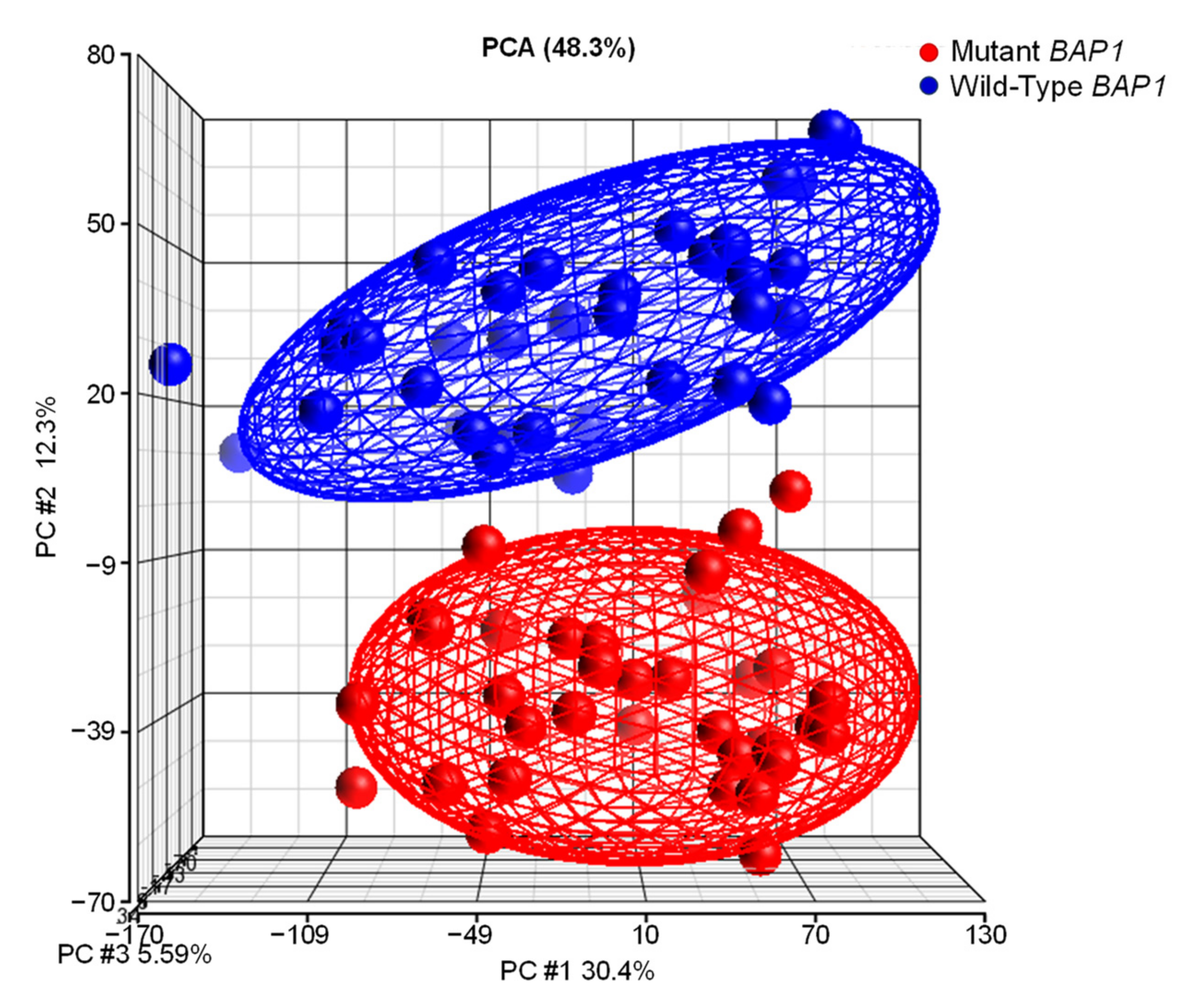
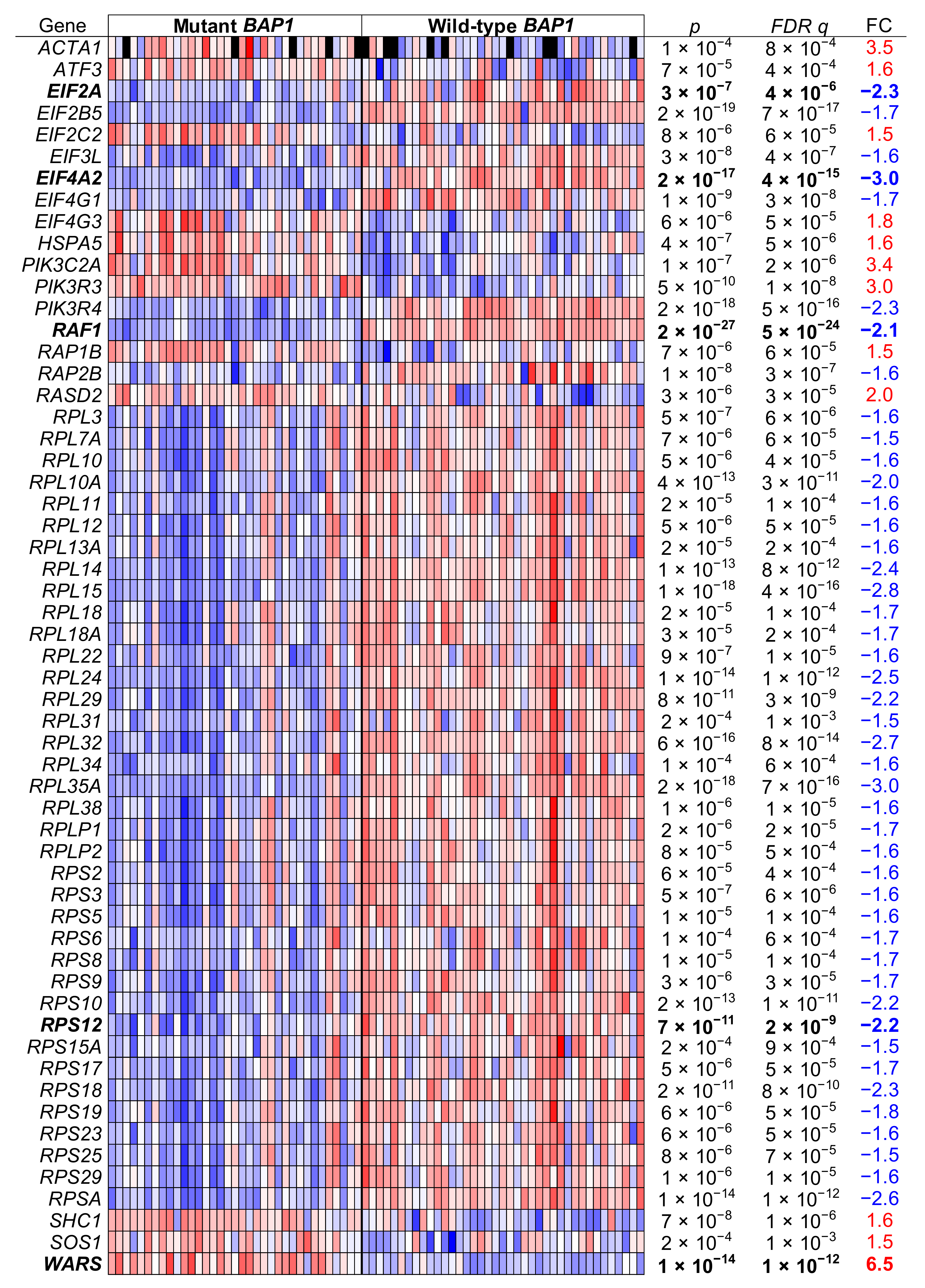
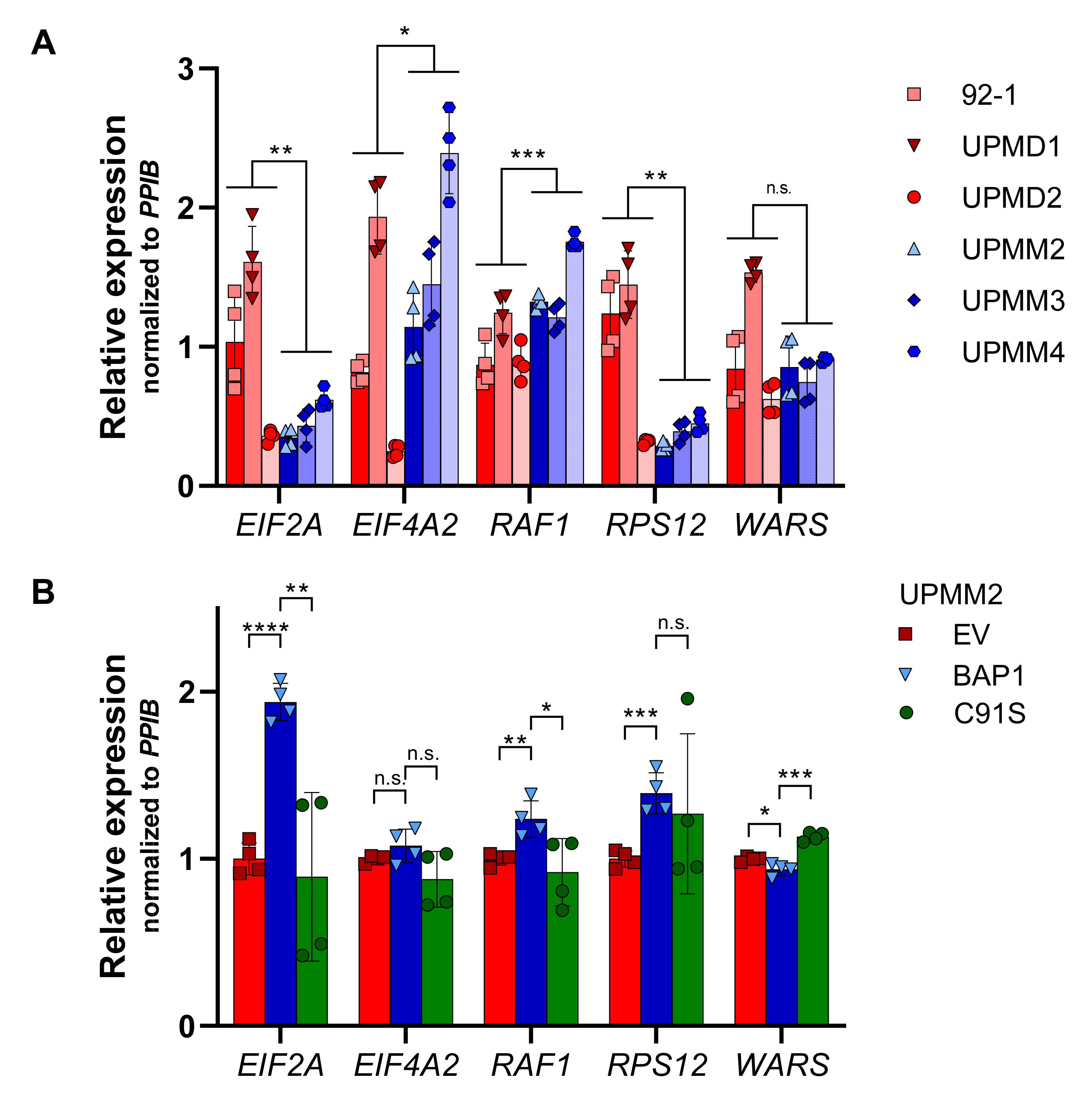
2.6. UM Patients with BAP1 Mutations Show a Completely Different Transcriptomics Program than Patients with Wild-Type BAP1
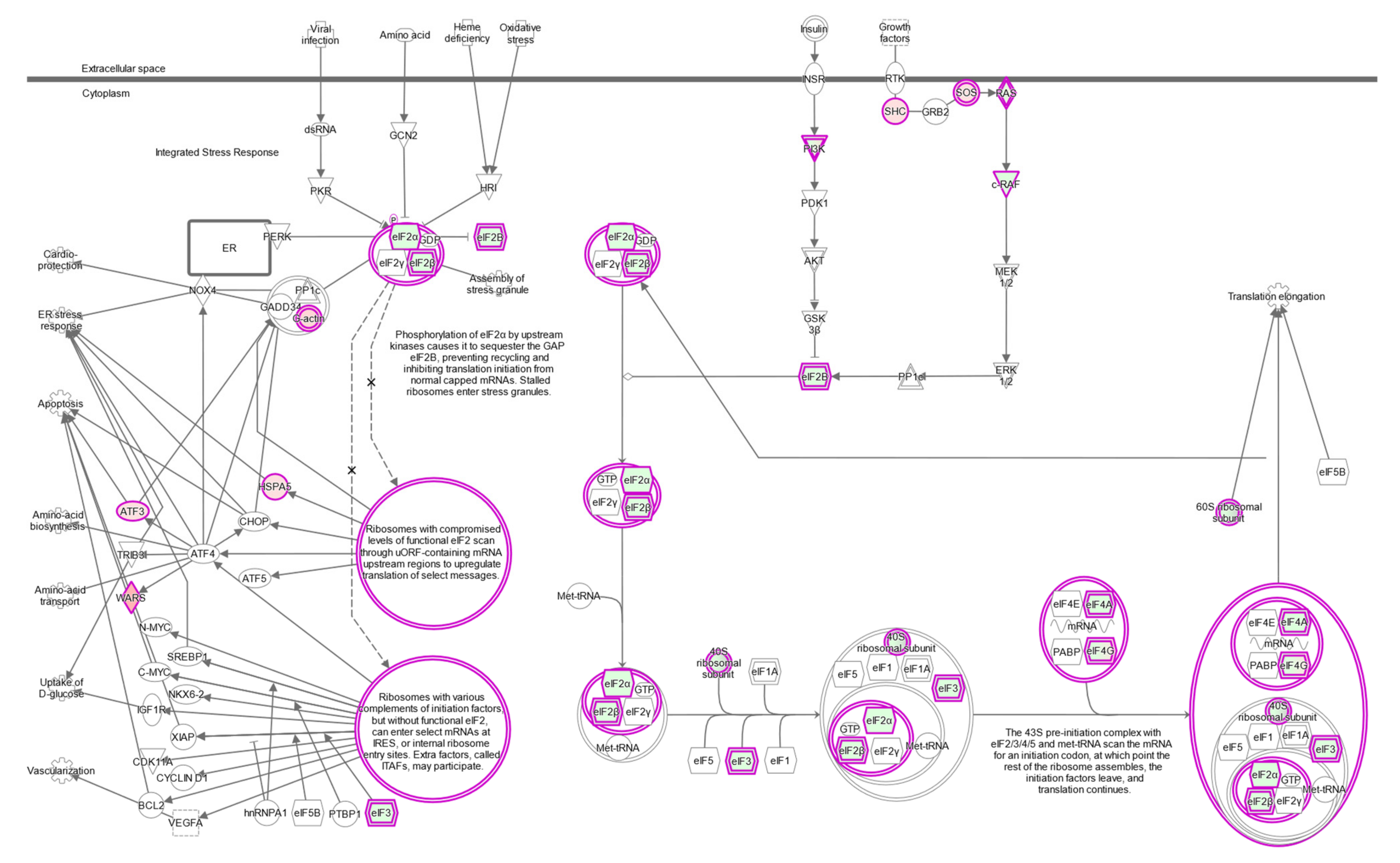
2.7. The eIF2 Signaling Is the Most Overrepresented Pathway in BAP1-Mutant UM Tumors
3. Discussion
4. Materials and Methods
4.1. Cell Lines
4.2. BAP1 Reconstitution
4.3. Drug Treatment
4.4. Cell Viability Assay
4.5. Proliferation Assay
4.6. Colony Formation Assay
4.7. Western Blot
4.8. The Cancer Genome Atlas (TCGA) Data Acquisition
4.9. UM Patient Survival Analysis
4.10. DNA Copy Number Analyses
4.11. Gene Expression Analyses
4.12. Quantitative Reverse-Transcribed PCR (qRT-PCR)
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| BAP1 | BRCA1-associated protein 1 |
| BRCA | Breast cancer gene |
| BSA | Bovine serum albumin |
| CI | Confidence intervals |
| D3 | Disomy 3 |
| EGFR | Endothelial growth factor receptor |
| eIF2α | Eukaryotic initiation factor 2α |
| ER | Endoplasmic reticulum |
| FBS | Fetal bovine serum |
| FC | Fold change |
| FDR | False discovery rate |
| GDC | Genomic Data Commons |
| ICC | Intrahepatic cholangiocarcinoma |
| IPA | Ingenuity pathway analysis |
| ISR | Integrated stress response |
| M3 | Monosomy 3 |
| MAPK | Mitogen-activated protein kinase |
| MEKi | MEK inhibitor(s) |
| PARP | Poly-ADP (adenosine diphosphate)-ribose polymerase |
| PCA | Principal component analysis |
| PERK | Double-stranded RNA-activated protein kinase (PKR)-like endoplasmic reticulum kinase |
| PPIB | Peptidylprolyl isomerase B |
| q(RT)-PCR | Quantitative (reverse-transcribed) PCR |
| RCC | Renal cell carcinoma |
| RPS12 | Ribosomal protein S12 |
| TCGA | The Cancer Genome Atlas |
| UM | Uveal melanoma |
| UPR | Unfolded protein response |
References
- Singh, A.D.; Bergman, L.; Seregard, S. Uveal melanoma: Epidemiologic aspects. Ophthalmol. Clin. N. Am. 2005, 18, 75–84. [Google Scholar] [CrossRef]
- Kujala, E.; Mäkitie, T.; Kivelä, T. Very long-term prognosis of patients with malignant uveal melanoma. Invest. Ophthalmol. Vis. Sci. 2003, 44, 4651–4659. [Google Scholar] [CrossRef] [PubMed]
- Gragoudas, E.S.; Egan, K.M.; Seddon, J.M.; Glynn, R.J.; Walsh, S.M.; Finn, S.M.; Munzenrider, J.E.; Spar, M.D. Survival of patents with metastases from uveal melanoma. Ophthalmology 1991, 98, 383–390. [Google Scholar] [CrossRef]
- Bedikian, A.Y.; Legha, S.S.; Mavligit, G.; Carrasco, C.H.; Khorana, S.; Plager, C.; Papadopoulos, N.; Benjamin, R.S. Treatment of uveal melanoma metastatic to the liver: A review of the M. D. Anderson cancer center experience and prognostic factors. Cancer 1995, 76, 1665–1670. [Google Scholar] [CrossRef]
- Diener-West, M.; Reynolds, S.M.; Agugliaro, D.J.; Caldwell, R.; Cumming, K.; Earle, J.D.; Hawkins, B.S.; Hayman, J.A.; Jaiyesimi, I.; Jampol, L.M.; et al. Development of metastatic disease after enrollment in the COMS trials for treatment of choroidal melanoma: Collaborative Ocular Melanoma Study Group Report No. 26. Arch. Ophthalmol. 2005, 123, 1639–1643. [Google Scholar] [CrossRef]
- Singh, A.D.; Turell, M.E.; Topham, A.K. Uveal melanoma: Trends in incidence, treatment, and survival. Ophthalmology 2011, 118, 1881–1885. [Google Scholar] [CrossRef]
- Rodriguez-Vidal, C.; Fernandez-Diaz, D.; Fernandez-Marta, B.; Lago-Baameiro, N.; Pardo, M.; Silva, P.; Paniagua, L.; Blanco-Teijeiro, M.J.; Piñeiro, A.; Bande, M. Treatment of metastatic uveal melanoma: Systematic review. Cancers 2020, 12, 2557. [Google Scholar] [CrossRef]
- Maemondo, M.; Inoue, A.; Kobayashi, K.; Sugawara, S.; Oizumi, S.; Isobe, H.; Gemma, A.; Harada, M.; Yoshizawa, H.; Kinoshita, I.; et al. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N. Engl. J. Med 2010, 362, 2380–2388. [Google Scholar] [CrossRef]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of poly (ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef]
- Chapman, P.B.; Hauschild, A.; Robert, C.; Haanen, J.B.; Ascierto, P.; Larkin, J.; Dummer, R.; Garbe, C.; Testori, A.; Maio, M.; et al. Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N. Engl. J. Med. 2011, 364, 2507–2516. [Google Scholar] [CrossRef]
- Planchard, D.; Smit, E.F.; Groen, H.J.M.; Mazieres, J.; Besse, B.; Helland, Å.; Giannone, V.; D’Amelio, A.M., Jr.; Zhang, P.; Mookerjee, B.; et al. Dabrafenib plus trametinib in patients with previously untreated BRAF(V600E)-mutant metastatic non-small-cell lung cancer: An open-label, phase 2 trial. Lancet Oncol. 2017, 18, 1307–1316. [Google Scholar] [CrossRef]
- Royer-Bertrand, B.; Torsello, M.; Rimoldi, D.; El Zaoui, I.; Cisarova, K.; Pescini-Gobert, R.; Raynaud, F.; Zografos, L.; Schalenbourg, A.; Speiser, D.; et al. Comprehensive Genetic Landscape of Uveal Melanoma by Whole-Genome Sequencing. Am. J. Hum. Genet. 2016, 99, 1190–1198. [Google Scholar] [CrossRef]
- Van Raamsdonk, C.D.; Bezrookove, V.; Green, G.; Bauer, J.; Gaugler, L.; O’Brien, J.M.; Simpson, E.M.; Barsh, G.S.; Bastian, B.C. Frequent somatic mutations of GNAQ in uveal melanoma and blue naevi. Nature 2009, 457, 599–602. [Google Scholar] [CrossRef]
- Van Raamsdonk, C.D.; Griewank, K.G.; Crosby, M.B.; Garrido, M.C.; Vemula, S.; Wiesner, T.; Obenauf, A.C.; Wackernagel, W.; Green, G.; Bouvier, N.; et al. Mutations in GNA11 in uveal melanoma. N. Engl. J. Med. 2010, 363, 2191–2199. [Google Scholar] [CrossRef]
- Robertson, A.G.; Shih, J.; Yau, C.; Gibb, E.A.; Oba, J.; Mungall, K.L.; Hess, J.M.; Uzunangelov, V.; Walter, V.; Danilova, L.; et al. Integrative analysis identifies four molecular and clinical subsets in uveal melanoma. Cancer Cell 2017, 32, 204–220.e215. [Google Scholar] [CrossRef] [PubMed]
- Dhillon, A.S.; Hagan, S.; Rath, O.; Kolch, W. MAP kinase signalling pathways in cancer. Oncogene 2007, 26, 3279–3290. [Google Scholar] [CrossRef]
- Schadendorf, D.; Long, G.V.; Stroiakovski, D.; Karaszewska, B.; Hauschild, A.; Levchenko, E.; Chiarion-Sileni, V.; Schachter, J.; Garbe, C.; Dutriaux, C.; et al. Three-year pooled analysis of factors associated with clinical outcomes across dabrafenib and trametinib combination therapy phase 3 randomised trials. Eur. J. Cancer 2017, 82, 45–55. [Google Scholar] [CrossRef]
- Ribas, A.; Daud, A.; Pavlick, A.C.; Gonzalez, R.; Lewis, K.D.; Hamid, O.; Gajewski, T.F.; Puzanov, I.; Wongchenko, M.; Rooney, I.; et al. Extended 5-Year Follow-up Results of a Phase Ib Study (BRIM7) of Vemurafenib and Cobimetinib in BRAF-Mutant Melanoma. Clin. Cancer Res. 2020, 26, 46–53. [Google Scholar] [CrossRef] [PubMed]
- Carvajal, R.D.; Piperno-Neumann, S.; Kapiteijn, E.; Chapman, P.B.; Frank, S.; Joshua, A.M.; Piulats, J.M.; Wolter, P.; Cocquyt, V.; Chmielowski, B.; et al. Selumetinib in combination with dacarbazine in patients with metastatic uveal melanoma: A phase III, multicenter, randomized trial (SUMIT). J. Clin. Oncol. 2018, 36, 1232–1239. [Google Scholar] [CrossRef] [PubMed]
- Shoushtari, A.N.; Kudchadkar, R.R.; Panageas, K.; Murthy, R.K.; Jung, M.; Shah, R.; O’Donnell, B.; Khawaja, T.T.; Shames, Y.; Prempeh-Keteku, N.A.; et al. A randomized phase 2 study of trametinib with or without GSK2141795 in patients with advanced uveal melanoma. J. Clin. Oncol. 2016, 34, 9511. [Google Scholar] [CrossRef]
- Prescher, G.; Bornfeld, N.; Hirche, H.; Horsthemke, B.; Jöckel, K.H.; Becher, R. Prognostic implications of monosomy 3 in uveal melanoma. Lancet 1996, 347, 1222–1225. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Pütter, C.; Weber, S.; Bornfeld, N.; Lohmann, D.R.; Zeschnigk, M. Prognostic significance of chromosome 3 alterations determined by microsatellite analysis in uveal melanoma: A long-term follow-up study. Br. J. Cancer 2012, 106, 1171–1176. [Google Scholar] [CrossRef]
- Onken, M.D.; Worley, L.A.; Ehlers, J.P.; Harbour, J.W. Gene expression profiling in uveal melanoma reveals two molecular classes and predicts metastatic death. Cancer Res. 2004, 64, 7205–7209. [Google Scholar] [CrossRef]
- Harbour, J.W.; Onken, M.D.; Roberson, E.D.; Duan, S.; Cao, L.; Worley, L.A.; Council, M.L.; Matatall, K.A.; Helms, C.; Bowcock, A.M. Frequent mutation of BAP1 in metastasizing uveal melanomas. Science 2010, 330, 1410–1413. [Google Scholar] [CrossRef]
- Misaghi, S.; Ottosen, S.; Izrael-Tomasevic, A.; Arnott, D.; Lamkanfi, M.; Lee, J.; Liu, J.; O’Rourke, K.; Dixit, V.M.; Wilson, A.C. Association of C-terminal ubiquitin hydrolase BRCA1-associated protein 1 with cell cycle regulator host cell factor 1. Mol. Cell. Biol. 2009, 29, 2181–2192. [Google Scholar] [CrossRef] [PubMed]
- Nishikawa, H.; Wu, W.; Koike, A.; Kojima, R.; Gomi, H.; Fukuda, M.; Ohta, T. BRCA1-associated protein 1 interferes with BRCA1/BARD1 RING heterodimer activity. Cancer Res. 2009, 69, 111–119. [Google Scholar] [CrossRef]
- Scheuermann, J.C.; de Ayala Alonso, A.G.; Oktaba, K.; Ly-Hartig, N.; McGinty, R.K.; Fraterman, S.; Wilm, M.; Muir, T.W.; Müller, J. Histone H2A deubiquitinase activity of the Polycomb repressive complex PR-DUB. Nature 2010, 465, 243–247. [Google Scholar] [CrossRef] [PubMed]
- Bott, M.; Brevet, M.; Taylor, B.S.; Shimizu, S.; Ito, T.; Wang, L.; Creaney, J.; Lake, R.A.; Zakowski, M.F.; Reva, B.; et al. The nuclear deubiquitinase BAP1 is commonly inactivated by somatic mutations and 3p21.1 losses in malignant pleural mesothelioma. Nat. Genet. 2011, 43, 668–672. [Google Scholar] [CrossRef] [PubMed]
- Jiao, Y.; Pawlik, T.M.; Anders, R.A.; Selaru, F.M.; Streppel, M.M.; Lucas, D.J.; Niknafs, N.; Guthrie, V.B.; Maitra, A.; Argani, P.; et al. Exome sequencing identifies frequent inactivating mutations in BAP1, ARID1A and PBRM1 in intrahepatic cholangiocarcinomas. Nat. Genet. 2013, 45, 1470–1473. [Google Scholar] [CrossRef]
- Peña-Llopis, S.; Vega-Rubín-de-Celis, S.; Liao, A.; Leng, N.; Pavía-Jiménez, A.; Wang, S.; Yamasaki, T.; Zhrebker, L.; Sivanand, S.; Spence, P.; et al. BAP1 loss defines a new class of renal cell carcinoma. Nat. Genet. 2012, 44, 751–759. [Google Scholar] [CrossRef]
- Matatall, K.A.; Agapova, O.A.; Onken, M.D.; Worley, L.A.; Bowcock, A.M.; Harbour, J.W. BAP1 deficiency causes loss of melanocytic cell identity in uveal melanoma. BMC Cancer 2013, 13, 371. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.A.; Bourne, T.D.; Murali, R. BAP1 protein loss by immunohistochemistry: A potentially useful tool for prognostic prediction in patients with uveal melanoma. Pathology 2013, 45, 651–656. [Google Scholar] [CrossRef]
- Lee, H.-J.; Pham, T.; Chang, M.T.; Barnes, D.; Cai, A.G.; Noubade, R.; Totpal, K.; Chen, X.; Tran, C.; Hagenbeek, T.; et al. The tumor suppressor BAP1 regulates the hippo pathway in pancreatic ductal adenocarcinoma. Cancer Res. 2020, 80, 1656. [Google Scholar] [CrossRef]
- Moore, A.R.; Ran, L.; Guan, Y.; Sher, J.J.; Hitchman, T.D.; Zhang, J.Q.; Hwang, C.; Walzak, E.G.; Shoushtari, A.N.; Monette, S.; et al. GNA11 Q209L Mouse model reveals RasGRP3 as an essential signaling node in uveal melanoma. Cell Rep. 2018, 22, 2455–2468. [Google Scholar] [CrossRef] [PubMed]
- Carbone, M.; Yang, H.; Pass, H.I.; Krausz, T.; Testa, J.R.; Gaudino, G. BAP1 and cancer. Nat. Rev. Cancer 2013, 13, 153–159. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.-X.; Yin, Y.; Cheng, J.-W.; Huang, A.; Hu, B.; Zhang, X.; Sun, Y.-F.; Wang, J.; Wang, Y.-P.; Ji, Y.; et al. BAP1 acts as a tumor suppressor in intrahepatic cholangiocarcinoma by modulating the ERK1/2 and JNK/c-Jun pathways. Cell Death Dis. 2018, 9, 1036. [Google Scholar] [CrossRef]
- Shahriyari, L.; Abdel-Rahman, M.; Cebulla, C. BAP1 expression is prognostic in breast and uveal melanoma but not colon cancer and is highly positively correlated with RBM15B and USP19. PLoS ONE 2019, 14, e0211507. [Google Scholar] [CrossRef]
- Koopmans, A.E.; Verdijk, R.M.; Brouwer, R.W.; van den Bosch, T.P.; van den Berg, M.M.; Vaarwater, J.; Kockx, C.E.; Paridaens, D.; Naus, N.C.; Nellist, M.; et al. Clinical significance of immunohistochemistry for detection of BAP1 mutations in uveal melanoma. Mod. Pathol. 2014, 27, 1321–1330. [Google Scholar] [CrossRef]
- Kalirai, H.; Dodson, A.; Faqir, S.; Damato, B.E.; Coupland, S.E. Lack of BAP1 protein expression in uveal melanoma is associated with increased metastatic risk and has utility in routine prognostic testing. Br. J. Cancer 2014, 111, 1373–1380. [Google Scholar] [CrossRef]
- Van de Nes, J.A.P.; Nelles, J.; Kreis, S.; Metz, C.H.D.; Hager, T.; Lohmann, D.R.; Zeschnigk, M. Comparing the Prognostic Value of BAP1 Mutation Pattern, Chromosome 3 Status, and BAP1 Immunohistochemistry in Uveal Melanoma. Am. J. Surg. Pathol. 2016, 40, 796–805. [Google Scholar] [CrossRef]
- Badhai, J.; Pandey, G.K.; Song, J.Y.; Krijgsman, O.; Bhaskaran, R.; Chandrasekaran, G.; Kwon, M.C.; Bombardelli, L.; Monkhorst, K.; Grasso, C.; et al. Combined deletion of Bap1, Nf2, and Cdkn2ab causes rapid onset of malignant mesothelioma in mice. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef]
- Nareyeck, G.; Zeschnigk, M.; Bornfeld, N.; Anastassiou, G. Novel cell lines derived by long-term culture of primary uveal melanomas. Ophthalmologica 2009, 223, 196–201. [Google Scholar] [CrossRef]
- Morgan, M.A.; Dolp, O.; Reuter, C.W.M. Cell-cycle–dependent activation of mitogen-activated protein kinase kinase (MEK-1/2) in myeloid leukemia cell lines and induction of growth inhibition and apoptosis by inhibitors of RAS signaling. Blood 2001, 97, 1823–1834. [Google Scholar] [CrossRef] [PubMed]
- Schröder, M.; Kaufman, R.J. THE mammalian unfolded protein response. Annu. Rev. Biochem. 2005, 74, 739–789. [Google Scholar] [CrossRef]
- Kadowaki, H.; Nishitoh, H. Signaling pathways from the endoplasmic reticulum and their roles in disease. Genes 2013, 4, 306–333. [Google Scholar] [CrossRef] [PubMed]
- Rozpedek, W.; Pytel, D.; Mucha, B.; Leszczynska, H.; Diehl, J.A.; Majsterek, I. The Role of the PERK/eIF2α/ATF4/CHOP Signaling pathway in tumor progression during endoplasmic reticulum stress. Curr. Mol. Med. 2016, 16, 533–544. [Google Scholar] [CrossRef] [PubMed]
- Corazao-Rozas, P.; Guerreschi, P.; André, F.; Gabert, P.E.; Lancel, S.; Dekiouk, S.; Fontaine, D.; Tardivel, M.; Savina, A.; Quesnel, B.; et al. Mitochondrial oxidative phosphorylation controls cancer cell’s life and death decisions upon exposure to MAPK inhibitors. Oncotarget 2016, 7, 39473–39485. [Google Scholar] [CrossRef] [PubMed]
- Beck, D.; Niessner, H.; Smalley, K.S.; Flaherty, K.; Paraiso, K.H.; Busch, C.; Sinnberg, T.; Vasseur, S.; Iovanna, J.L.; Drießen, S.; et al. Vemurafenib potently induces endoplasmic reticulum stress-mediated apoptosis in BRAFV600E melanoma cells. Sci. Signal. 2013, 6. [Google Scholar] [CrossRef] [PubMed]
- Leprivier, G.; Rotblat, B.; Khan, D.; Jan, E.; Sorensen, P.H. Stress-mediated translational control in cancer cells. Biochim. Biophys. Acta 2015, 1849, 845–860. [Google Scholar] [CrossRef]
- Stewart, S.A.; Dykxhoorn, D.M.; Palliser, D.; Mizuno, H.; Yu, E.Y.; An, D.S.; Sabatini, D.M.; Chen, I.S.; Hahn, W.C.; Sharp, P.A.; et al. Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 2003, 9, 493–501. [Google Scholar] [CrossRef]
- Morgenstern, J.P.; Land, H. Advanced mammalian gene transfer: High titre retroviral vectors with multiple drug selection markers and a complementary helper-free packaging cell line. Nucleic Acids Res. 1990, 18, 3587–3596. [Google Scholar] [CrossRef] [PubMed]
- Niersch, J.; Vega-Rubín-de-Celis, S.; Bazarna, A.; Mergener, S.; Jendrossek, V.; Siveke, J.T.; Peña-Llopis, S. A BAP1 synonymous mutation results in exon skipping, loss of function and worse patient prognosis. iScience 2021, 24, 102173. [Google Scholar] [CrossRef]
- Guzmán, C.; Bagga, M.; Kaur, A.; Westermarck, J.; Abankwa, D. ColonyArea: An ImageJ plugin to automatically quantify colony formation in clonogenic assays. PLoS ONE 2014, 9, e92444. [Google Scholar] [CrossRef]
- Peña-Llopis, S.; Brugarolas, J. Simultaneous isolation of high-quality DNA, RNA, miRNA and proteins from tissues for genomic applications. Nat. Protoc. 2013, 8, 2240–2255. [Google Scholar] [CrossRef] [PubMed]
- Peña-Llopis, S.; Wan, Y.; Martinez, E.D. Unique epigenetic gene profiles define human breast cancers with poor prognosis. Oncotarget 2016, 7, 85819–85831. [Google Scholar] [CrossRef]
- Peña-Llopis, S.; Vega-Rubin-de-Celis, S.; Schwartz, J.C.; Wolff, N.C.; Tran, T.A.; Zou, L.; Xie, X.J.; Corey, D.R.; Brugarolas, J. Regulation of TFEB and V-ATPases by mTORC1. EMBO J. 2011, 30, 3242–3258. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Canonical Pathways | FDR q < 10−3, FC > 1.5 | FDR q < 10−10, FC > 2 | ||
|---|---|---|---|---|
| p | Ratio | p | Ratio | |
| eIF2 Signaling | 4 × 10−10 | 0.251 | 5 × 10−6 | 0.053 |
| Axonal guidance signaling | 6 × 10−9 | 0.192 | 0.006 | 0.024 |
| Cardiac hypertrophy signaling (enhanced) | 10−7 | 0.183 | 3 × 10−4 | 0.030 |
| Regulation of eIF4 and p70S6K signaling | 4 × 10−7 | 0.241 | 0.009 | 0.035 |
| Synaptogenesis signaling pathway | 10−6 | 0.197 | 0.049 | 0.022 |
| Phospholipase C signaling | 3 × 10−6 | 0.201 | 0.008 | 0.029 |
| GP6 signaling pathway | 4 × 10−6 | 0.246 | 0.046 | 0.031 |
| Gap junction signaling | 9 × 10−6 | 0.209 | 0.006 | 0.033 |
| T cell receptor signaling | 10−5 | 0.252 | 0.028 | 0.036 |
| Thrombin signaling | 10−5 | 0.206 | 8 × 10−5 | 0.047 |
| Tec kinase signaling | 2 × 10−5 | 0.216 | 0.010 | 0.034 |
| CREB signaling in neurons | 2 × 10−5 | 0.161 | 0.004 | 0.023 |
| Role of NFAT in cardiac hypertrophy | 3 × 10−5 | 0.199 | 2 × 10−5 | 0.050 |
| Phagosome formation | 5 × 10−5 | 0.226 | 0.003 | 0.045 |
| Protein kinase A signaling | 5 × 10−5 | 0.170 | 0.011 | 0.024 |
| Cardiac hypertrophy signaling | 7 × 10−5 | 0.189 | 0.004 | 0.032 |
| Natural killer cell signaling | 8 × 10−5 | 0.198 | 0.005 | 0.035 |
| Glioblastoma multiforme signaling | 9 × 10−5 | 0.206 | 0.009 | 0.035 |
| HIF1α signaling | 2 × 10−4 | 0.190 | 0.023 | 0.028 |
| Hepatic fibrosis signaling pathway | 2 × 10−4 | 0.165 | 0.020 | 0.023 |
| Sphingosine-1-phosphate signaling | 4 × 10−4 | 0.215 | 0.037 | 0.033 |
| Adrenomedullin signaling pathway | 5 × 10−4 | 0.185 | 0.006 | 0.034 |
| Breast cancer regulation by stathmin1 | 5 × 10−4 | 0.150 | 0.001 | 0.025 |
| Glioma signaling | 6 × 10−4 | 0.212 | 0.008 | 0.042 |
| Molecular mechanisms of cancer | 6 × 10−4 | 0.159 | 0.026 | 0.022 |
| mTOR Signaling | 7 × 10−4 | 0.181 | 0.007 | 0.032 |
| Sperm motility | 7 × 10−4 | 0.176 | 0.012 | 0.029 |
| Endothelin-1 signaling | 7 × 10−4 | 0.182 | 3 × 10−4 | 0.044 |
| cAMP-mediated signaling | 8 × 10−4 | 0.176 | 0.035 | 0.026 |
| 14-3-3-mediated signaling | 10−3 | 0.202 | 0.002 | 0.047 |
| ERK/MAPK signaling | 0.001 | 0.180 | 0.020 | 0.029 |
| HGF signaling | 0.001 | 0.203 | 0.002 | 0.049 |
| Xenobiotic metabolism signaling | 0.001 | 0.166 | 9 × 10−4 | 0.035 |
| Production of nitric oxide and ROS in macrophages | 0.001 | 0.179 | 0.016 | 0.031 |
| CXCR4 signaling | 0.002 | 0.183 | 0.002 | 0.040 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mergener, S.; Siveke, J.T.; Peña-Llopis, S. Monosomy 3 Is Linked to Resistance to MEK Inhibitors in Uveal Melanoma. Int. J. Mol. Sci. 2021, 22, 6727. https://doi.org/10.3390/ijms22136727
Mergener S, Siveke JT, Peña-Llopis S. Monosomy 3 Is Linked to Resistance to MEK Inhibitors in Uveal Melanoma. International Journal of Molecular Sciences. 2021; 22(13):6727. https://doi.org/10.3390/ijms22136727
Chicago/Turabian StyleMergener, Svenja, Jens T. Siveke, and Samuel Peña-Llopis. 2021. "Monosomy 3 Is Linked to Resistance to MEK Inhibitors in Uveal Melanoma" International Journal of Molecular Sciences 22, no. 13: 6727. https://doi.org/10.3390/ijms22136727
APA StyleMergener, S., Siveke, J. T., & Peña-Llopis, S. (2021). Monosomy 3 Is Linked to Resistance to MEK Inhibitors in Uveal Melanoma. International Journal of Molecular Sciences, 22(13), 6727. https://doi.org/10.3390/ijms22136727