
Expression and Function of C1orf132 Long-Noncoding RNA in Breast Cancer Cell Lines and Tissues

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
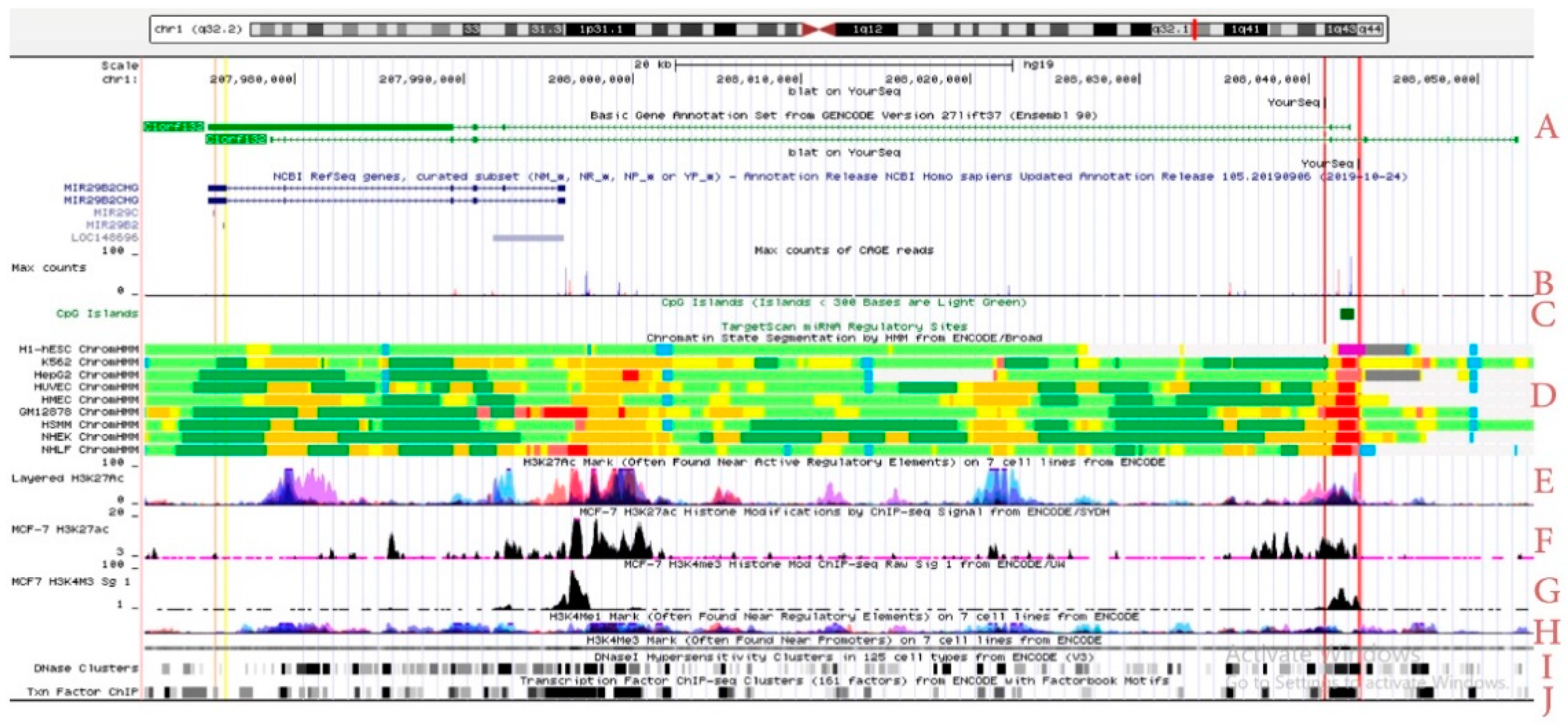
2.1. C1orf132 Locus and Its Transcripts
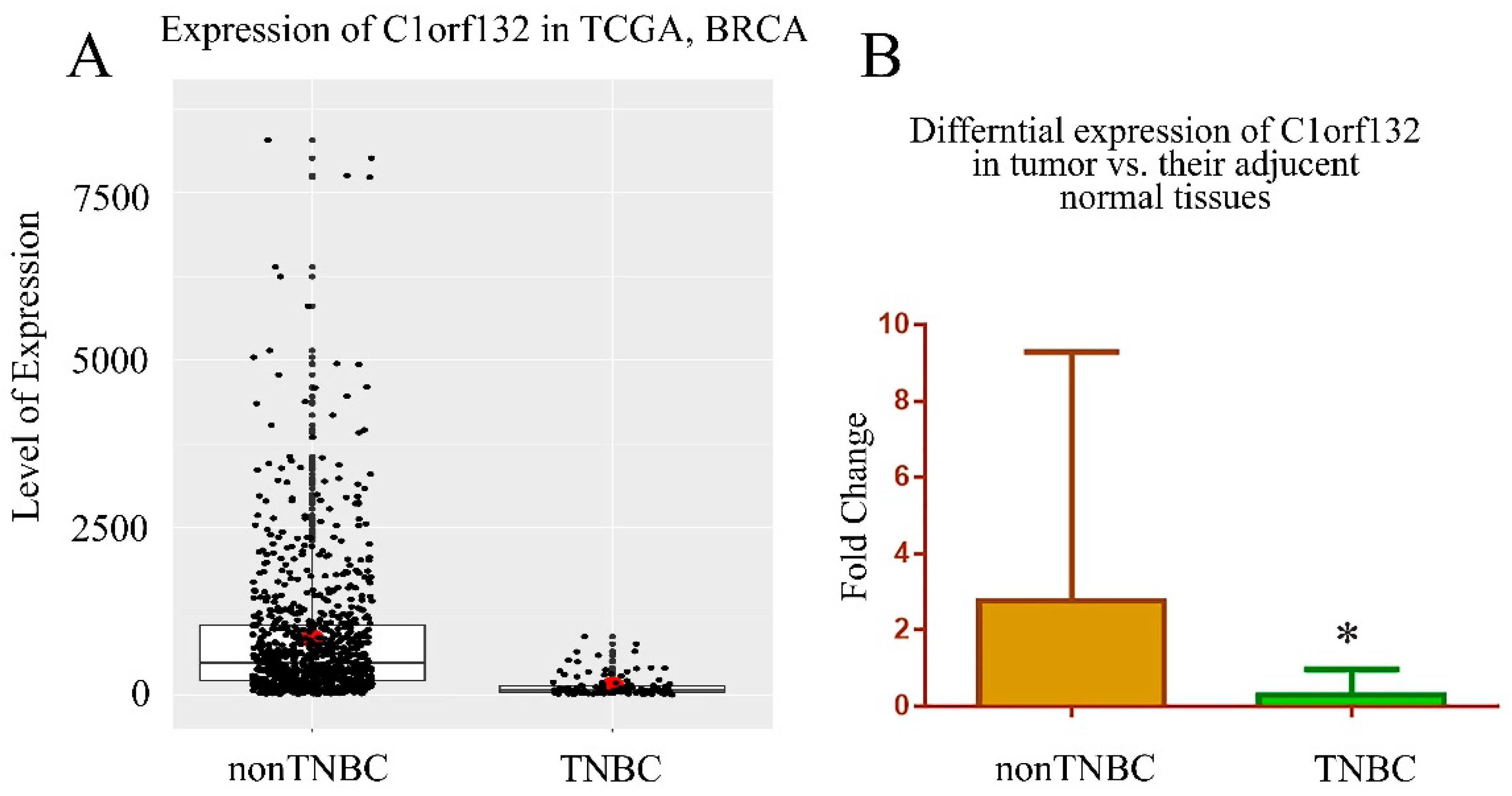
2.2. C1orf132 Was Significantly Downregulated in TNBC Tissue Samples
2.3. C1orf132 Transcribed from the p2 Promoter Localizes in the Nucleus
2.4. Promoter Deletion Enhanced the Migration Ability of MCF12A Cells
2.5. Gene Expression Profiling after C1orf132 Knock-Down
3. Discussion
4. Materials and Methods
4.1. Ethical Statement
4.2. Clinical Tissue Samples
4.3. Bioinformatics Analysis
4.4. Cloning the Putative Promoters of C1orf132
4.5. Cell Culture and Transfection
4.6. Fractionation Assay
4.7. Promoter Activity Reporter Assay
4.8. Promoter Deletion Using CRISPR/Cas9 System
4.9. Wound Healing Assay
4.10. Cell Cycle Analysis by Flow Cytometry
4.11. RNA Extraction and qRT-PCR
4.12. TaqMan miR Assay for miR-29c-3p Detection
4.13. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miller, K.D.; Siegel, R.L.; Khan, R.; Jemal, A. Cancer Statistics. J. Cancer Rehabil. 2018, 70, 7–30. [Google Scholar] [CrossRef]
- Sharifian, A.; Pourhoseingholi, M.A.; Emadedin, M.; Nejad, M.R.; Ashtari, S.; Hajizadeh, N.; Firouzei, S.A.; Hosseini, S.J. Burden of Breast Cancer in Iranian Women is Increasing. Asian Pac. J. Cancer Prev. 2015, 16, 5049–5052. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- The Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumors. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vuong, D.; Simpson, P.; Green, B.; Cummings, M.; Lakhani, S.R. Molecular classification of breast cancer. Virchows Arch. 2014, 465, 1–14. [Google Scholar] [CrossRef]
- Hon, J.D.C.; Singh, B.; Sahin, A.; Du, G.; Wang, J.; Wang, V.Y.; Deng, F.-M.; Zhang, D.Y.; Monaco, M.E.; Lee, P. Breast cancer molecular subtypes: From TNBC to QNBC. Am. J. Cancer Res. 2016, 6, 1864–1872. [Google Scholar]
- Nafissi, N.; Faraji, M.; Hosseini, M.; Shojaee, L.; Ziaei, F.; Akbari, M.E.; Mousavie, S.H. Relationships between Reproductive Risk Factors for Breast Cancer and Tumor Molecular Subtypes. Asian Pac. J. Cancer Prev. 2018, 19, 1767–1770. [Google Scholar] [CrossRef] [Green Version]
- Lesurf, R.; Aure, M.R.; Mørk, H.H.; Vitelli, V.; Lundgren, S.; Børresen-Dale, A.-L.; Kristensen, V.; Wärnberg, F.; Hallett, M.; Sørlie, T.; et al. Molecular Features of Subtype-Specific Progression from Ductal Carcinoma In Situ to Invasive Breast Cancer. Cell Rep. 2016, 16, 1166–1179. [Google Scholar] [CrossRef] [Green Version]
- Wang, K.C.; Chang, H.Y. Molecular mechanisms of long noncoding RNAs. Mol. Cell 2011, 43, 904–914. [Google Scholar] [CrossRef] [Green Version]
- Ling, H.; Vincent, K.; Pichler, M.; Fodde, R.; Berindan-Neagoe, I.; Slack, F.; Calin, G. Junk DNA and the long non-coding RNA twist in cancer genetics. Oncogene 2015, 34, 5003–5011. [Google Scholar] [CrossRef] [Green Version]
- Wu, Y.; Shao, A.; Wang, L.; Hu, K.; Yu, C.; Pan, C.; Zhang, S. The Role of lncRNAs in the Distant Metastasis of Breast Cancer. Front. Oncol. 2019, 9, 407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poli, E.; Zhang, J.; Nwachukwu, C.; Zheng, Y.; O Adedokun, B.; Olopade, O.I.; Han, Y.-J. Molecular Subtype-Specific Expression of MicroRNA-29c in Breast Cancer Is Associated with CpG Dinucleotide Methylation of the Promoter. PLoS ONE 2015, 10, e0142224. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhardwaj, A.; Singh, H.; Rajapakshe, K.; Tachibana, K.; Ganesan, N.; Pan, Y.; Gunaratne, P.H.; Coarfa, C.; Bedrosian, I. Regulation of miRNA-29c and its down-stream pathways in preneoplastic progression of triple-negative breast cancer. Oncotarget 2017, 8, 19645–19660. [Google Scholar] [CrossRef] [Green Version]
- Freire-Aradas, A.; Phillips, C.; Mosquera-Miguel, A.; Girón-Santamaría, L.; Gómez-Tato, A.; Casares De Cal, M.; Álvarez-Dios, J.; Ansede-Bermejo, J.; Torres-Español, M.; Schneider, P.M.; et al. Development of a methylation marker set for forensic age estimation using analysis of public methylation data and the Agena Bioscience EpiTYPER system. Forensic Sci. Int. Genet. 2016, 24, 65–74. [Google Scholar] [CrossRef]
- Zbieć-Piekarska, R.; Spólnicka, M.; Kupiec, T.; Parys-Proszek, A.; Makowska, Z.; Pałeczka, A.; Kucharczyk, K.; Płoski, R.; Branicki, W. Development of a foren-sically useful age prediction method based on DNA methylation analysis. Forensic Sci. Int. Genet. 2015, 17, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Jung, S.E.; Lim, S.M.; Hong, S.R.; Lee, E.H.; Shin, K.J.; Lee, H.Y. DNA methylation of the ELOVL2, FHL2, KLF14, C1orf132/MIR29B2C, and TRIM59 genes for age prediction from blood, saliva, and buccal swab samples. Forensic Sci. Int. Genet. 2019, 38, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Mirzadeh Azad, F.; Malakootian, M.; Mowla, S.J. lncRNA PSORS1C3 is regulated by glucocorticoids and fine-tunes OCT4 expression in non-pluripotent cells. Sci. Rep. 2019, 9, 8370. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Zhang, W.; Zhang, R.; Li, J.; Li, S.; Ma, Y.; Jin, W.; Wang, K. Overexpressed long noncoding RNA CRNDE with distinct alternatively spliced isoforms in multiple cancers. Front. Med. 2018, 13, 330–343. [Google Scholar] [CrossRef]
- Dennis, M.K.; Field, A.S.; Burai, R.; Ramesh, C.; Whitney, K.; Bologa, C.G. Long non-coding RNAs with enhancer-like function in humans. Cell 2012, 127, 358–366. [Google Scholar]
- Qi, Y.; Huang, Y.; Pang, L.; Gu, W.; Wang, N.; Hu, J.; Cui, X.; Zhang, J.; Zhao, J.; Liu, C.; et al. Prognostic value of the MicroRNA-29 family in multiple human cancers: A meta-analysis and systematic review. Clin. Exp. Pharmacol. Physiol. 2017, 44, 441–454. [Google Scholar] [CrossRef]
- Zhu, W.; He, J.; Chen, D.; Zhang, B.; Xu, L.; Ma, H.; Liu, X.; Zhang, Y.; Le, H. Expression of miR-29c, miR-93, and miR-429 as Potential Biomarkers for Detection of Early Stage Non-Small Lung Cancer. PLoS ONE 2014, 9, e87780. [Google Scholar] [CrossRef]
- Arechaga-Ocampo, E.; Lopez-Camarillo, C.; Villegas-Sepulveda, N.; Gonzalez-De la Rosa, C.H.; Perez-Añorve, I.X.; Roldan-Perez, R.; Flores-Perez, A.; Peña-Curiel, O.; Angeles-Zaragoza, O.; Corona, R.R.; et al. Tumor suppressor miR-29c regulates radioresistance in lung cancer cells. Tumor Biol. 2017, 39, 1010428317695010. [Google Scholar] [CrossRef] [Green Version]
- Zhang, N.-S.; Dai, G.-L.; Liu, S.-J. MicroRNA-29 family functions as a tumor suppressor by targeting RPS15A and regulating cell cycle in hepatocellular carcinoma. Int. J. Clin. Exp. Pathol. 2017, 10, 8031–8042. [Google Scholar]
- Jiang, J.; Yu, C.; Chen, M.; Zhang, H.; Tian, S.; Sun, C. Reduction of miR-29c enhances pancreatic cancer cell migration and stem cell-like phenotype. Oncotarget 2014, 6, 2767–2778. [Google Scholar] [CrossRef] [Green Version]
- Wheelock, M.J.; Shintani, Y.; Maeda, M.; Fukumoto, Y.; Johnson, K.R. Cadherin switching. J. Cell Sci. 2008, 121, 727–735. [Google Scholar] [CrossRef] [Green Version]
- Afzal Ashaie, M.; Hoque Chowdhury, E. Cadherins: The Superfamily Critically Involved in Breast Cancer. Curr. Pharm. Des. 2016, 22, 616–638. [Google Scholar] [CrossRef]
- Zhang, Y.; Xu, L.; Li, A.; Han, X. The roles of ZEB1 in tumorigenic progression and epigenetic modifications. Biomed. Pharmacother. 2019, 110, 400–408. [Google Scholar] [CrossRef] [PubMed]
- Wellner, U.; Schubert, J.; Burk, U.C.; Schmalhofer, O.; Zhu, F.; Sonntag, A.; Waldvogel, B.; Vannier, C.; Darling, D.; zur Hausen, A.; et al. The EMT-activator ZEB1 promotes tumor-igenicity by repressing stemness-inhibiting microRNAs. Nat. Cell Biol. 2009, 11, 1487–1495. [Google Scholar] [CrossRef] [PubMed]
- Chaffer, C.L.; Marjanovic, N.D.; Lee, T.; Bell, G.; Kleer, C.G.; Reinhardt, F.; D’Alessio, A.C.; Young, R.A.; Weinberg, R.A. Poised Chromatin at the ZEB1 Promoter Enables Breast Cancer Cell Plasticity and Enhances Tumorigenicity. Cell 2013, 154, 61–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giulianelli, S.; Riggio, M.; Guillardoy, T.; Pérez Piñero, C.; Gorostiaga, M.A.; Sequeira, G.; Pataccini, G.; Abascal, M.F.; Toledo, M.F.; Jacobsen, B.M.; et al. FGF2 induces breast cancer growth through ligand-independent activation and recruitment of ERα and PRBΔ4 isoform to MYC regulatory sequences. Int. J. Cancer 2019, 145, 1874–1888. [Google Scholar] [CrossRef]
- Yu, P.-J.; Ferrari, G.; Galloway, A.C.; Mignatti, P.; Pintucci, G. Basic fibroblast growth factor (FGF-2): The high molecular weight forms come of age. J. Cell Biochem. 2007, 100, 1100–1108. [Google Scholar] [CrossRef] [PubMed]
- Carausu, M.; Bidard, F.-C.; Callens, C.; Melaabi, S.; Jeannot, E.; Pierga, J.-Y.; Cabel, L. ESR1 mutations: A new biomarker in breast cancer. Expert Rev. Mol. Diagn. 2019, 19, 599–611. [Google Scholar] [CrossRef]
- Tomita, S.; Zhang, Z.; Nakano, M.; Ibusuki, M.; Kawazoe, T.; Yamamoto, Y.; Iwase, H. Estrogen receptor a gene ESR1 amplification may predict endocrine therapy responsiveness in breast. Cancer Sci. 2009, 100, 1012–1017. [Google Scholar] [CrossRef]
- Teleki, I.; Szasz, A.M.; Maros, M.E.; Gyorffy, B.; Kulka, J.; Meggyeshazi, N.; Kiszner, G.; Balla, P.; Samu, A.; Krenacs, T.; et al. Correlations of Differentially Expressed Gap Junction Connexins Cx26, Cx30, Cx32, Cx43 and Cx46 with Breast Cancer Progression and Prognosis. PLoS ONE 2014, 9, e112541. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shettar, A.; Damineni, S.; Mukherjee, G.; Kondaiah, P. Gap junction β-2 expression is negatively associated with the estrogen receptor status in breast cancer tissues and is a regulator of breast tumorigenesis. Oncol. Rep. 2018, 40, 3645–3653. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saitoh, A.; Hansen, L.A.; Vogel, J.C.; Udey, M.C. Characterization of Wnt gene expression in murine skin: Possible involvement of epidermis-derived Wnt-4 in cutaneous epithelial-mesenchymal interactions. Exp. Cell Res. 1998, 243, 150–160. [Google Scholar] [CrossRef]
- Reddy, S.; Andl, T.; Bagasra, A.; Lu, M.M.; Epstein, D.J.; Morrisey, E.E.; Millara, S.E. Characterization of Wnt gene expression in developing and postnatal hair follicles and identification of Wnt5a as a target of Sonic hedgehog in hair follicle morphogenesis. Mech Dev. 2001, 107, 69–82. [Google Scholar] [CrossRef]
- Heikkil, M.; Peltoketo, H.; Vainio, S. Wnts and the female reproductive system. J. Exp. Zool. 2001, 290, 616–623. [Google Scholar] [CrossRef]
- Stark, K.; Vainio, S.; Vassileva, G.; McMahon, A.P. Epithelial transformation of metanephric mesenchyme in the developing kidney regulated by Wnt-4. Nat. Cell Biol. 1994, 372, 679–683. [Google Scholar] [CrossRef] [PubMed]
- Vouyovitch, C.M.; Perry, J.K.; Liu, D.-X.; Bezin, L.; Vilain, E.; Diaz, J.-J.; Lobie, P.E.; Mertani, H.C. WNT4 mediates the autocrine effects of growth hormone in mammary carcinoma cells. Endocr. Relat. Cancer 2016, 23, 571–585. [Google Scholar] [CrossRef] [Green Version]
- Peng, F.; Wang, R.; Zhang, Y.; Zhao, Z.; Zhou, W.; Chang, Z.; Liang, H.; Zhao, W.; Qi, L.; Guo, Z.; et al. Differential expression analysis at the individual level reveals a lncRNA prognostic signature for lung adenocarcinoma. Mol. Cancer 2017, 16, 1–12. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Shafaroudi, A.M.; Sharifi-Zarchi, A.; Rahmani, S.; Nafissi, N.; Mowla, S.J.; Lauria, A.; Oliviero, S.; Matin, M.M. Expression and Function of C1orf132 Long-Noncoding RNA in Breast Cancer Cell Lines and Tissues. Int. J. Mol. Sci. 2021, 22, 6768. https://doi.org/10.3390/ijms22136768
Shafaroudi AM, Sharifi-Zarchi A, Rahmani S, Nafissi N, Mowla SJ, Lauria A, Oliviero S, Matin MM. Expression and Function of C1orf132 Long-Noncoding RNA in Breast Cancer Cell Lines and Tissues. International Journal of Molecular Sciences. 2021; 22(13):6768. https://doi.org/10.3390/ijms22136768
Chicago/Turabian StyleShafaroudi, Afsaneh Malekzadeh, Ali Sharifi-Zarchi, Saeid Rahmani, Nahid Nafissi, Seyed Javad Mowla, Andrea Lauria, Salvatore Oliviero, and Maryam M. Matin. 2021. "Expression and Function of C1orf132 Long-Noncoding RNA in Breast Cancer Cell Lines and Tissues" International Journal of Molecular Sciences 22, no. 13: 6768. https://doi.org/10.3390/ijms22136768
APA StyleShafaroudi, A. M., Sharifi-Zarchi, A., Rahmani, S., Nafissi, N., Mowla, S. J., Lauria, A., Oliviero, S., & Matin, M. M. (2021). Expression and Function of C1orf132 Long-Noncoding RNA in Breast Cancer Cell Lines and Tissues. International Journal of Molecular Sciences, 22(13), 6768. https://doi.org/10.3390/ijms22136768