
The Double Mutation DSG2-p.S363X and TBX20-p.D278X Is Associated with Left Ventricular Non-Compaction Cardiomyopathy: Case Report

 , , , , , , , , , ,
, , , , , , , , , ,  ,
, 
Abstract
:1. Introduction
2. Results
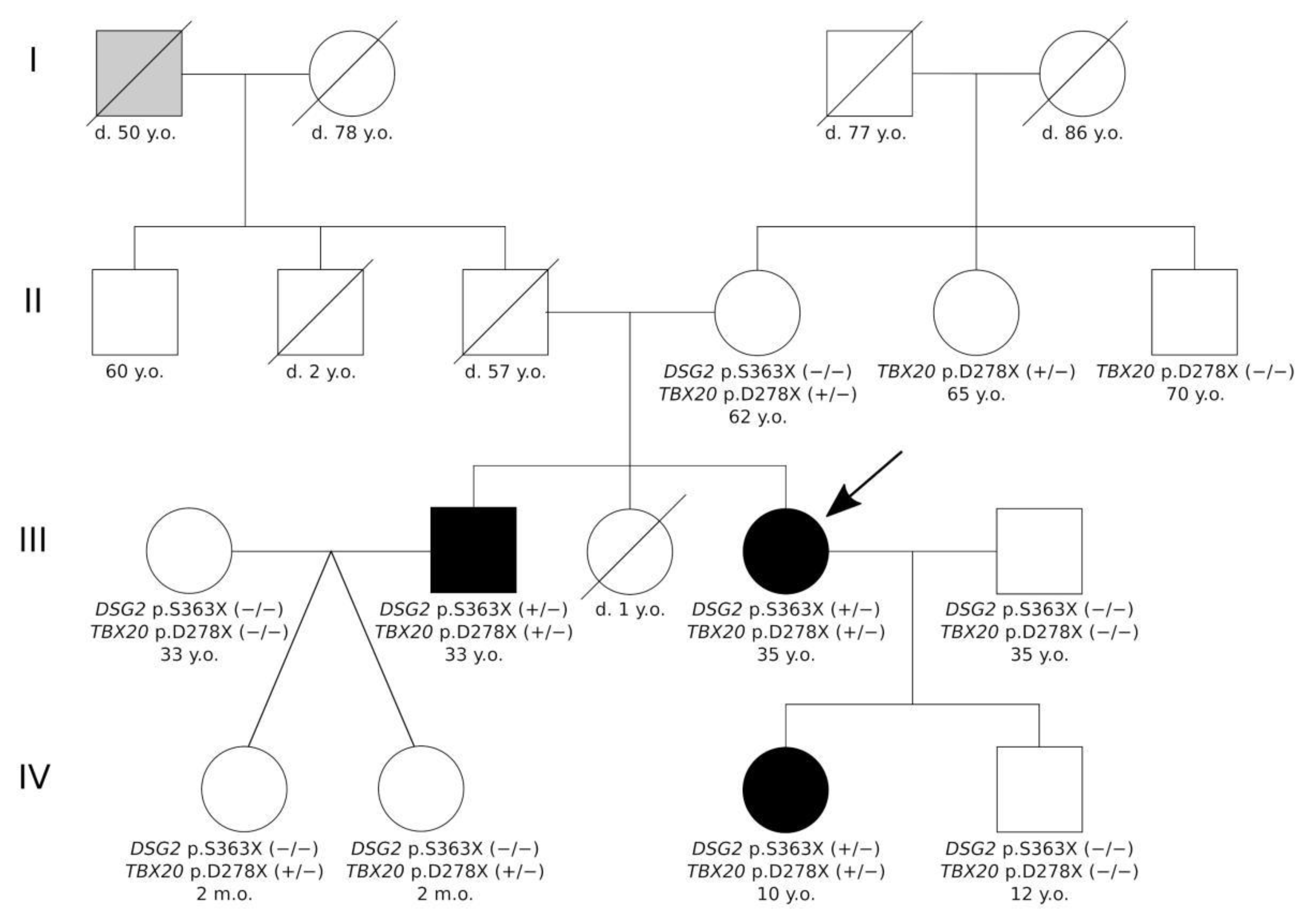
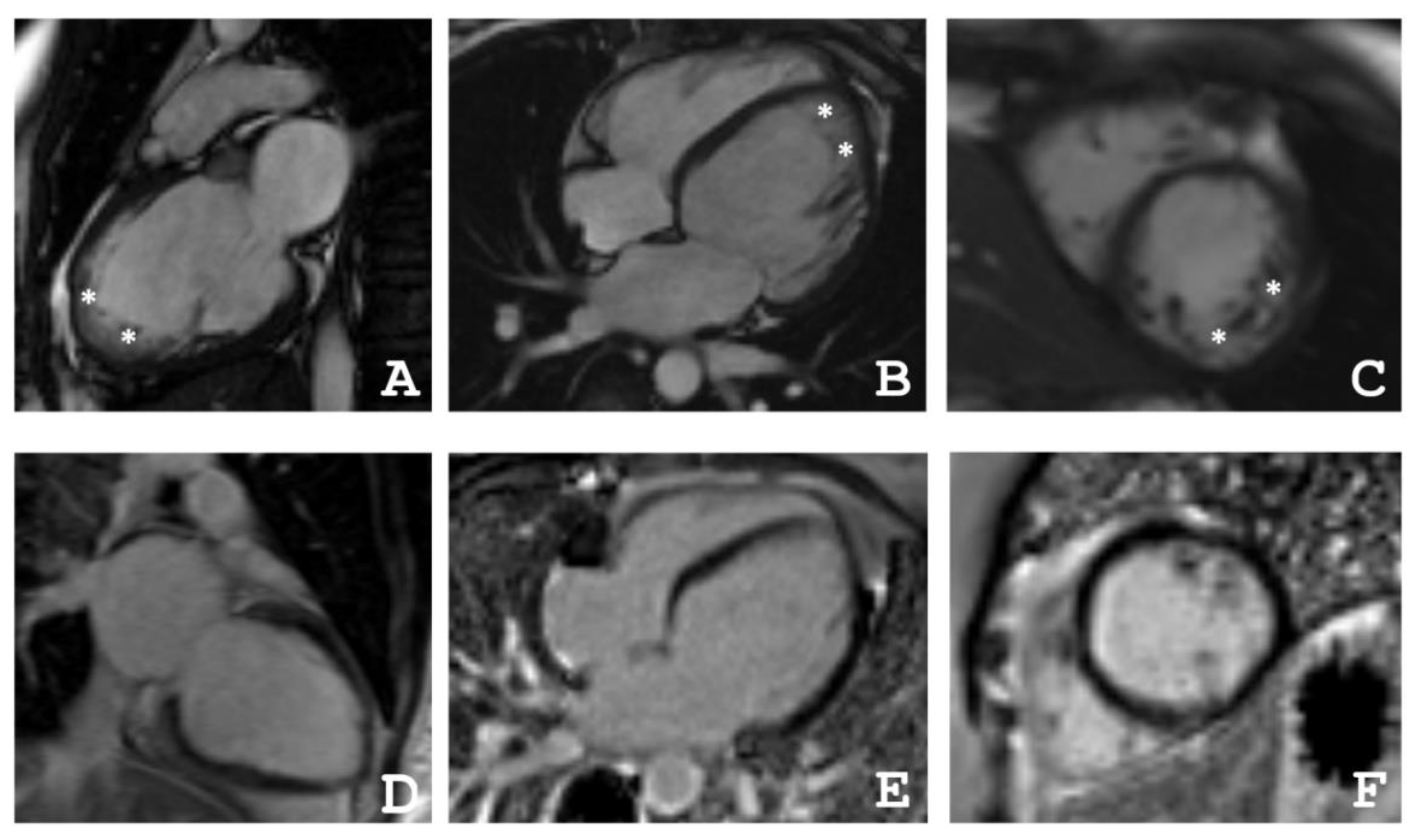
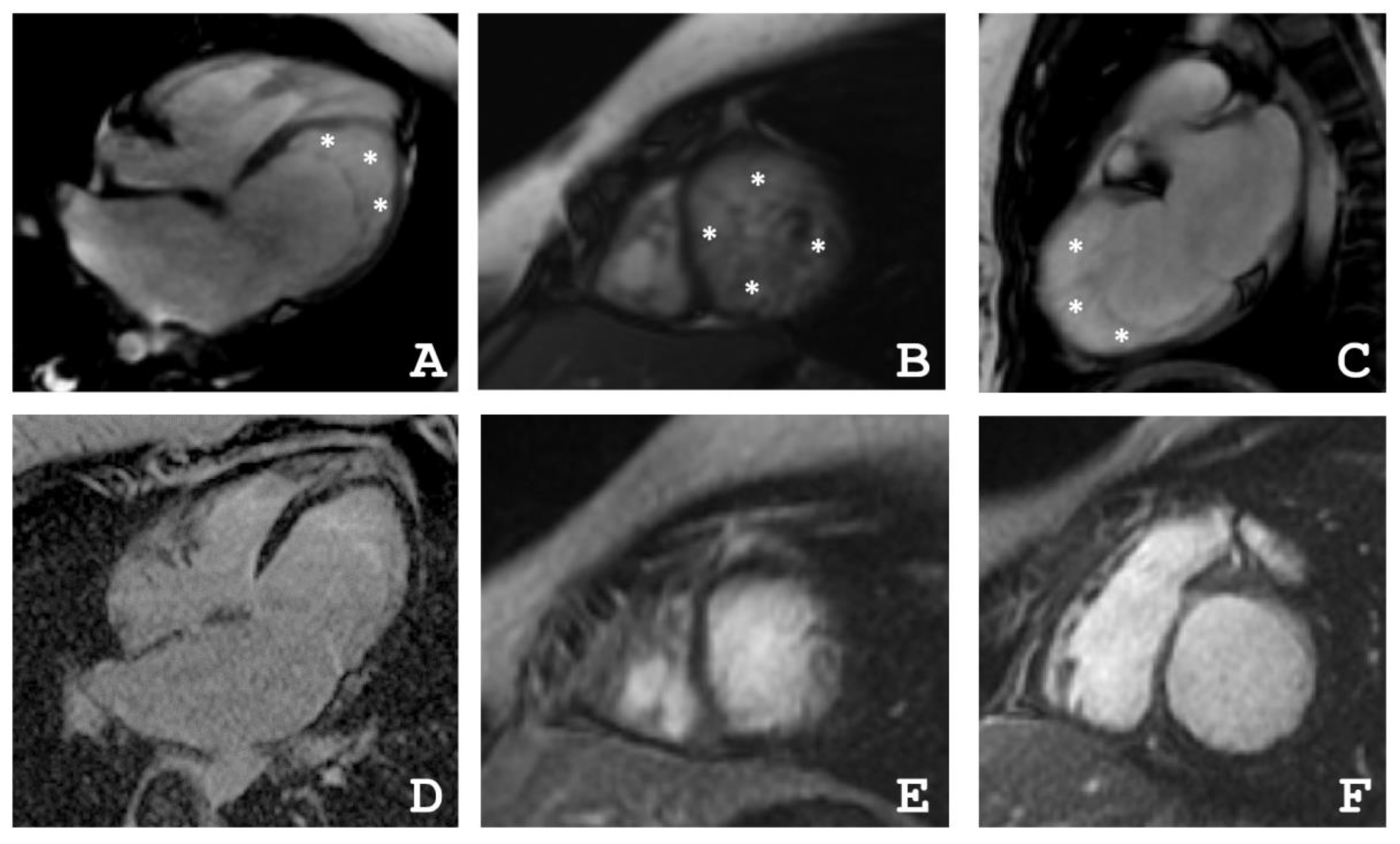
2.1. Clinical Investigations
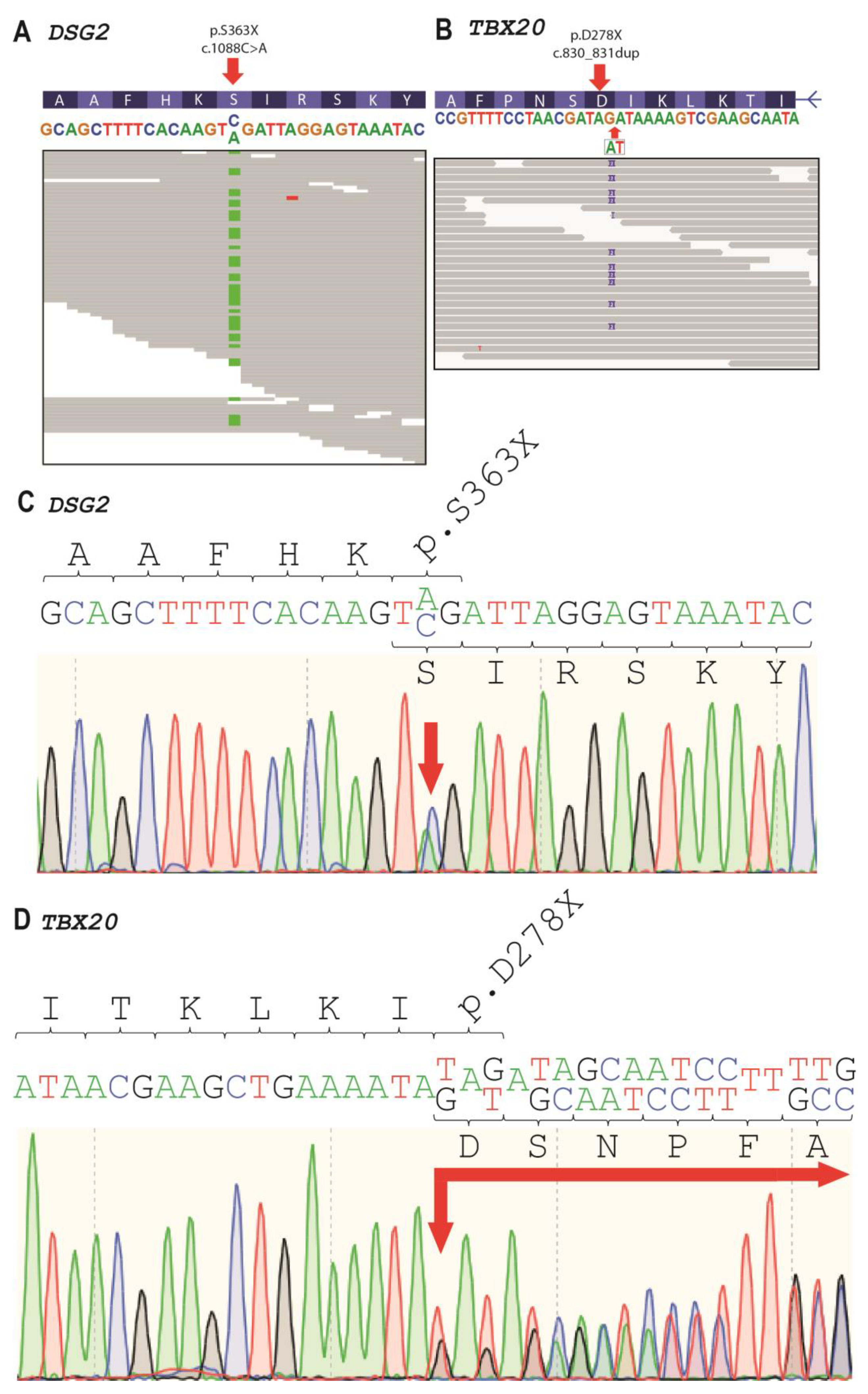
2.2. Genetic Analysis
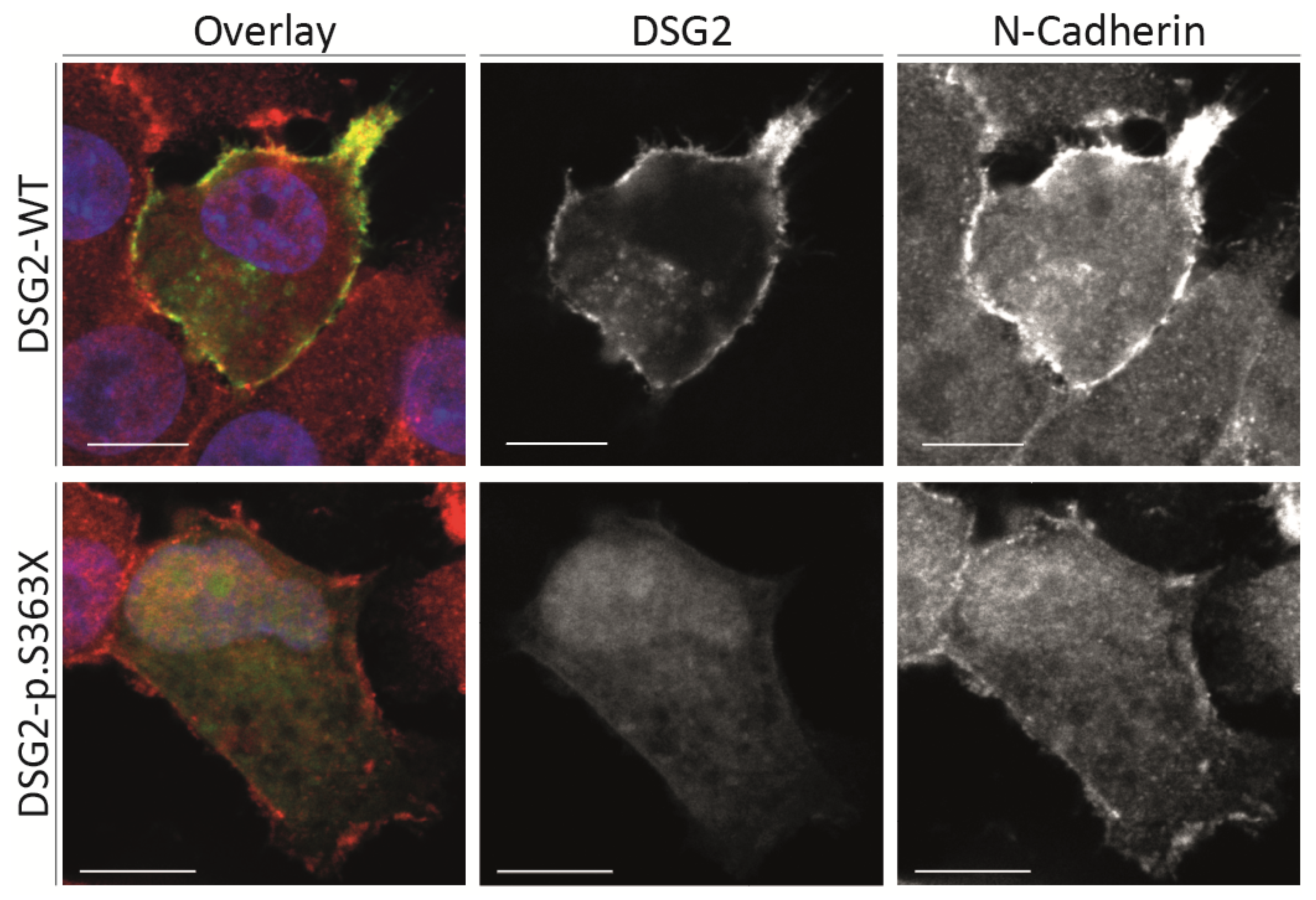
2.3. Cell Culture Experiments
3. Discussion
4. Materials and Methods
4.1. Clinical Description of the Patients
4.2. Cardiac Magnetic Resonance Imaging
4.3. Molecular Genetic Analysis
4.4. Plasmid Generation
4.5. Cell Culture, Immunocytochemistry and Confocal Microscopy
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Appendix A
References
- Stöllberger, C.; Wegner, C.; Finsterer, J. Left ventricular hypertrabeculation/noncompaction, cardiac phenotype, and neuromuscular disorders. Herz 2019, 44, 659–665. [Google Scholar] [CrossRef]
- Myasnikov, R.P.; Kulikova, O.V.; Meshkov, A.N.; Kiseleva, A.V.; Shumarina, A.O.; Koretskiy, S.N.; Zharikova, A.A.; Divashuk, M.G.; Kharlap, M.S.; Serduk, S.E.; et al. New variant of MYH7 gene nucleotide sequence in familial non-compaction cardiomyopathy with benign course. Ration. Pharmacother. Cardiol. 2020, 16, 383–391. [Google Scholar] [CrossRef]
- Myasnikov, R.P.; Blagova, O.V.; Kulikova, O.V.; Mershina, E.A.; Kharlap, M.S.; Andreenko, E.Y.; Koretsky, S.N.; Serdyuk, S.E.; Bazaeva, E.V.; Virabova, I.A.; et al. The specifics of noncompacted cardiomyopathy manifestation. Cardiovasc. Ther. Prev. 2015, 14, 78. [Google Scholar] [CrossRef]
- Li, S.; Zhang, C.; Liu, N.; Bai, H.; Hou, C.; Wang, J.; Song, L.; Pu, J. Genotype-positive status is associated with poor prognoses in patients with left ventricular noncompaction cardiomyopathy. J. Am. Heart Assoc. 2018, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kulikova, O.V.; Myasnikov, R.P.; Mershina, E.A.; Pilus, P.S.; Koretskiy, S.N.; Meshkov, A.N.; Kiseleva, A.V.; Kharlap, M.S.; Sinitsyn, V.E.; Sdvigova, N.A.; et al. The familial form of non-compaction cardiomyopathy: Types of myocardial remodeling, clinical course. Multicenter register results. Ther. Arch. 2021, 94. [Google Scholar] [CrossRef]
- Gerull, B.; Klaassen, S.; Brodehl, A. The Genetic Landscape of Cardiomyopathies. In Genetic Causes of Cardiac Disease; Springer: Cham, Switzerland, 2019; pp. 45–91. [Google Scholar]
- Sedaghat-Hamedani, F.; Haas, J.; Zhu, F.; Geier, C.; Kayvanpour, E.; Liss, M.; Lai, A.; Frese, K.; Pribe-Wolferts, R.; Amr, A.; et al. Clinical genetics and outcome of left ventricular non-compaction cardiomyopathy. Eur. Heart J. 2017, 38, 3449–3460. [Google Scholar] [CrossRef] [PubMed]
- Kayvanpour, E.; Sedaghat-Hamedani, F.; Gi, W.T.; Tugrul, O.F.; Amr, A.; Haas, J.; Zhu, F.; Ehlermann, P.; Uhlmann, L.; Katus, H.A.; et al. Clinical and genetic insights into non-compaction: A meta-analysis and systematic review on 7598 individuals. Clin. Res. Cardiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Posch, M.G.; Perrot, A.; Berger, F.; Özcelik, C. Molecular genetics of congenital atrial septal defects. Clin. Res. Cardiol. 2010, 99, 137–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gehmlich, K.; Syrris, P.; Reimann, M.; Asimaki, A.; Ehler, E.; Evans, A.; Quarta, G.; Pantazis, A.; Saffitz, J.E.; McKenna, W.J. Molecular changes in the heart of a severe case of arrhythmogenic right ventricular cardiomyopathy caused by a desmoglein-2 null allele. Cardiovasc. Pathol. 2012, 21, 275–282. [Google Scholar] [CrossRef]
- Kessler, E.L.; Nikkels, P.G.; van Veen, T.A. Disturbed Desmoglein-2 in the intercalated disc of pediatric patients with dilated cardiomyopathy. Hum. Pathol. 2017, 67, 101–108. [Google Scholar] [CrossRef]
- Brodehl, A.; Gaertner-Rommel, A.; Milting, H. Molecular insights into cardiomyopathies associated with desmin (DES) mutations. Biophys. Rev. 2018, 10, 983–1006. [Google Scholar] [CrossRef] [PubMed]
- Chin, T.K.; Perloff, J.K.; Williams, R.G.; Jue, K.; Mohrmann, R. Isolated noncompaction of left ventricular myocardium. A study of eight cases. Circ. J. 1990, 83, 507–517. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jenni, R.; Oechslin, E.; Schneider, J.; Attenhofer Jost, C.; Kaufmann, P.A. Echocardiographic and pathoanatomical characteristics of isolated left ventricular non-compaction: A step towards classification as a distinct cardiomyopathy. Heart 2001, 86, 666–671. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stöllberger, C.; Finsterer, J. Left ventricular hypertrabeculation/noncompaction. J. Am. Soc. Echocardiogr. 2004, 17, 91–100. [Google Scholar] [CrossRef]
- Grothoff, M.; Pachowsky, M.; Hoffmann, J.; Posch, M.; Klaassen, S.; Lehmkuhl, L.; Gutberlet, M. Value of cardiovascular MR in diagnosing left ventricular non-compaction cardiomyopathy and in discriminating between other cardiomyopathies. Eur. Radiol. 2012, 22, 2699–2709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jacquier, A.; Thuny, F.; Jop, B.; Giorgi, R.; Cohen, F.; Gaubert, J.Y.; Vidal, V.; Bartoli, J.M.; Habib, G.; Moulin, G. Measurement of trabeculated left ventricular mass using cardiac magnetic resonance imaging in the diagnosis of left ventricular non-compaction. Eur. Heart J. 2010, 31, 1098–1104. [Google Scholar] [CrossRef] [Green Version]
- Petersen, S.E.; Selvanayagam, J.B.; Wiesmann, F.; Robson, M.D.; Francis, J.M.; Anderson, R.H.; Watkins, H.; Neubauer, S. Left Ventricular Non-Compaction. J. Am. Coll. Cardiol. 2005, 46, 101–105. [Google Scholar] [CrossRef] [Green Version]
- Chitaev, N.A.; Troyanovsky, S.M. Direct Ca 2. Cell 1997, 138, 193–201. [Google Scholar] [CrossRef] [Green Version]
- Richard, P.; Ader, F.; Roux, M.; Donal, E.; Eicher, J.C.; Aoutil, N.; Huttin, O.; Selton-Suty, C.; Coisne, D.; Jondeau, G.; et al. Targeted panel sequencing in adult patients with left ventricular non-compaction reveals a large genetic heterogeneity. Clin. Genet. 2019, 95, 356–367. [Google Scholar] [CrossRef]
- Brodehl, A.; Meshkov, A.; Myasnikov, R.; Kiseleva, A.; Kulikova, O.; Klauke, B.; Sotnikova, E.; Stanasiuk, C.; Divashuk, M.; Pohl, G.M.; et al. Hemi- and homozygous loss-of-function mutations in dsg2 (Desmoglein-2) cause recessive arrhythmogenic cardiomyopathy with an early onset. Int. J. Mol. Sci. 2021, 22, 3786. [Google Scholar] [CrossRef]
- Kant, S.; Holthöfer, B.; Magin, T.M.; Krusche, C.A.; Leube, R.E. Desmoglein 2-Dependent Arrhythmogenic Cardiomyopathy Is Caused by a Loss of Adhesive Function. Circ. Cardiovasc. Genet. 2015, 8, 553–563. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef] [PubMed]
- Green, K.J.; Simpson, C.L. Desmosomes: New perspectives on a classic. J. Investig. Dermatol. 2007, 127, 2499–2515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Debus, J.D.; Milting, H.; Brodehl, A.; Kassner, A.; Anselmetti, D.; Gummert, J.; Gaertner-Rommel, A. In vitro analysis of arrhythmogenic cardiomyopathy associated desmoglein-2 (DSG2) mutations reveals diverse glycosylation patterns. J. Mol. Cell. Cardiol. 2019, 129, 303–313. [Google Scholar] [CrossRef] [Green Version]
- Harrison, O.J.; Brasch, J.; Lasso, G.; Katsamba, P.S.; Ahlsen, G.; Honig, B.; Shapiro, L. Structural basis of adhesive binding by desmocollins and desmogleins. Proc. Natl. Acad. Sci. USA 2016, 113, 7160–7165. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Nekrasova, O.E.; Patel, D.M.; Klessner, J.L.; Godsel, L.M.; Koetsier, J.L.; Amargo, E.V.; Desai, B.V.; Green, K.J. The C-terminal unique region of desmoglein 2 inhibits its internalization via tail-tail interactions. J. Cell Biol. 2012, 199, 699–711. [Google Scholar] [CrossRef] [Green Version]
- Costa, S.; Cerrone, M.; Saguner, A.M.; Brunckhorst, C.; Delmar, M.; Duru, F. Arrhythmogenic cardiomyopathy: An in-depth look at molecular mechanisms and clinical correlates. Trends Cardiovasc. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Van Der Zwaag, P.A.; Jongbloed, J.D.H.; Van Den Berg, M.P.; Van Der Smagt, J.J.; Jongbloed, R.; Bikker, H.; Hofstra, R.M.W.; Van Tintelen, J.P. A genetic variants database for arrhythmogenic right ventricular dysplasia/cardiomyopathy. Hum. Mutat. 2009, 30, 1278–1283. [Google Scholar] [CrossRef] [PubMed]
- Posch, M.G.; Posch, M.J.; Geier, C.; Erdmann, B.; Mueller, W.; Richter, A.; Ruppert, V.; Pankuweit, S.; Maisch, B.; Perrot, A.; et al. A missense variant in desmoglein-2 predisposes to dilated cardiomyopathy. Mol. Genet. Metab. 2008, 95, 74–80. [Google Scholar] [CrossRef]
- Ramond, F.; Janin, A.; Di Filippo, S.; Chanavat, V.; Chalabreysse, L.; Roux-Buisson, N.; Sanlaville, D.; Touraine, R.; Millat, G. Homozygous PKP2 deletion associated with neonatal left ventricle noncompaction. Clin. Genet. 2017, 91, 126–130. [Google Scholar] [CrossRef]
- Chen, Y.; Xiao, D.; Zhang, L.; Cai, C.L.; Li, B.Y.; Liu, Y. The Role of Tbx20 in Cardiovascular Development and Function. Front. Cell Dev. Biol. 2021, 9. [Google Scholar] [CrossRef]
- Kodo, K.; Ong, S.G.; Jahanbani, F.; Termglinchan, V.; Hirono, K.; Inanloorahatloo, K.; Ebert, A.D.; Shukla, P.; Abilez, O.J.; Churko, J.M.; et al. iPSC-derived cardiomyocytes reveal abnormal TGF-β signalling in left ventricular non-compaction cardiomyopathy. Nat. Cell Biol. 2016, 18, 1031–1042. [Google Scholar] [CrossRef] [Green Version]
- Beyder, A.; Mazzone, A.; Strege, P.R.; Tester, D.J.; Saito, Y.A.; Bernard, C.E.; Enders, F.T.; Ek, W.E.; Schmidt, P.T.; Dlugosz, A.; et al. Loss-of-function of the voltage-gated sodium channel NaV1.5 (channelopathies) in patients with irritable bowel syndrome. Gastroenterology 2014, 146, 1659–1668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- World Medical Association declaration of Helsinki. Ethical principles for medical research involving human subjects. JAMA J. Am. Med. Assoc. 2013, 310, 2191–2194. [CrossRef] [PubMed] [Green Version]
- Brodehl, A.; Stanasiuk, C.; Anselmetti, D.; Gummert, J.; Milting, H. Incorporation of desmocollin-2 into the plasma membrane requires N-glycosylation at multiple sites. FEBS Open Bio 2019, 9, 996–1007. [Google Scholar] [CrossRef] [PubMed]
- Brodehl, A.; Gaertner-Rommel, A.; Klauke, B.; Grewe, S.A.; Schirmer, I.; Peterschröder, A.; Faber, L.; Vorgerd, M.; Gummert, J.; Anselmetti, D.; et al. The novel αB-crystallin (CRYAB) mutation p.D109G causes restrictive cardiomyopathy. Hum. Mutat. 2017, 38, 947–952. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient | The Ratio NC/C in Segments | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | |
| III-2 | 0 | 0 | 0 | 0 | 0 | 1.1 | 4.0 | 0 | 0 | 1.6 | 2.1 | 3.5 | 0 | 0 | 1.4 | 0 | 0 |
| III-4 | 1.2 | 0 | 0 | 0 | 1.2 | 2.2 | 1.1 | 0 | 0 | 3.8 | 3.0 | 3.0 | 4.9 | 3.3 | 3.5 | 2.8 | 5.9 |
| IV-3 | 3.3 | 1 | 0 | 0 | 0 | 0 | 3.5 | 2.5 | 1.1 | 0 | 0 | 1 | 8.3 | 8.3 | 6.3 | 8.3 | 10.5 |
| Patient | EDV | EF | Grothoff, [16] | Jacquier, % [17] | Petersen, [18] | |||
|---|---|---|---|---|---|---|---|---|
| Mass Index of NC, g/m² | NC/Myocardial mass, % | NC/C ≥ 3:1 in One Segment(1–3, 7–16) | NC/C ≥ 2:1 in 4–6th Segments | |||||
| III-2 | 77 | 31 | 12 | 20 | + | − | 20 | + |
| III-4 | 82 | 55 | 8 | 14 | + | + | 16 | + |
| IV-3 | 64 | 48 | 21 | 36 | + | − | 36 | + |
| Gene | Genomic Coordinates (GRCh37) | cDNA Change | dbSNP | Kind of Mutation | Protein Change | MAF 1 | ACMG Classification | II-4 | III-2 | III-4 | IV-3 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| DSG2 | 18:29111023 | NM_001943.5:c.1088C>A | rs751527714 | nonsense | NP_001934.2:p.S363X | 0.000006577 | Pathogenic | − | + | + | + |
| SCN5A | 3:38597154 | NM_000335.5:c.4532G>T | rs368219299 | missense | NP_000326.2:p.R1511L | 0.00002630 | VUS | + | + | + | − |
| TTN | 2:179402474 | NM_003319.4:c.72265C>T | rs143556947 | missense | NP_003310.4:p.R24089C | 0.00009206 | VUS | − | − | + | − |
| TTN | 2:179442717 | NM_003319.4:c.41330T>C | rs368301580 | missense | NP_003310.4:p.I13777T | 0.00002630 | VUS | − | − | + | − |
| TGFB3 | 14:76429800 | NM_003239.5:c.785G>T | rs1595339233 | missense | NP_003230.1:p.G262V | 0.00001381 | VUS | − | + | + | + |
| TBX20 | 7:35271174 | NM_001077653.2: c.830_831dup | NA | nonsense | NP_001071121.1:p.D278X | Novel | Likely pathogenic | + | + | + | + |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Myasnikov, R.; Brodehl, A.; Meshkov, A.; Kulikova, O.; Kiseleva, A.; Pohl, G.M.; Sotnikova, E.; Divashuk, M.; Klimushina, M.; Zharikova, A.; et al. The Double Mutation DSG2-p.S363X and TBX20-p.D278X Is Associated with Left Ventricular Non-Compaction Cardiomyopathy: Case Report. Int. J. Mol. Sci. 2021, 22, 6775. https://doi.org/10.3390/ijms22136775
Myasnikov R, Brodehl A, Meshkov A, Kulikova O, Kiseleva A, Pohl GM, Sotnikova E, Divashuk M, Klimushina M, Zharikova A, et al. The Double Mutation DSG2-p.S363X and TBX20-p.D278X Is Associated with Left Ventricular Non-Compaction Cardiomyopathy: Case Report. International Journal of Molecular Sciences. 2021; 22(13):6775. https://doi.org/10.3390/ijms22136775
Chicago/Turabian StyleMyasnikov, Roman, Andreas Brodehl, Alexey Meshkov, Olga Kulikova, Anna Kiseleva, Greta Marie Pohl, Evgeniia Sotnikova, Mikhail Divashuk, Marina Klimushina, Anastasia Zharikova, and et al. 2021. "The Double Mutation DSG2-p.S363X and TBX20-p.D278X Is Associated with Left Ventricular Non-Compaction Cardiomyopathy: Case Report" International Journal of Molecular Sciences 22, no. 13: 6775. https://doi.org/10.3390/ijms22136775