
β-Ketophosphonates with Pentalenofuran Scaffolds Linked to the Ketone Group for the Synthesis of Prostaglandin Analogs †

 , ,
, , 
Abstract
:
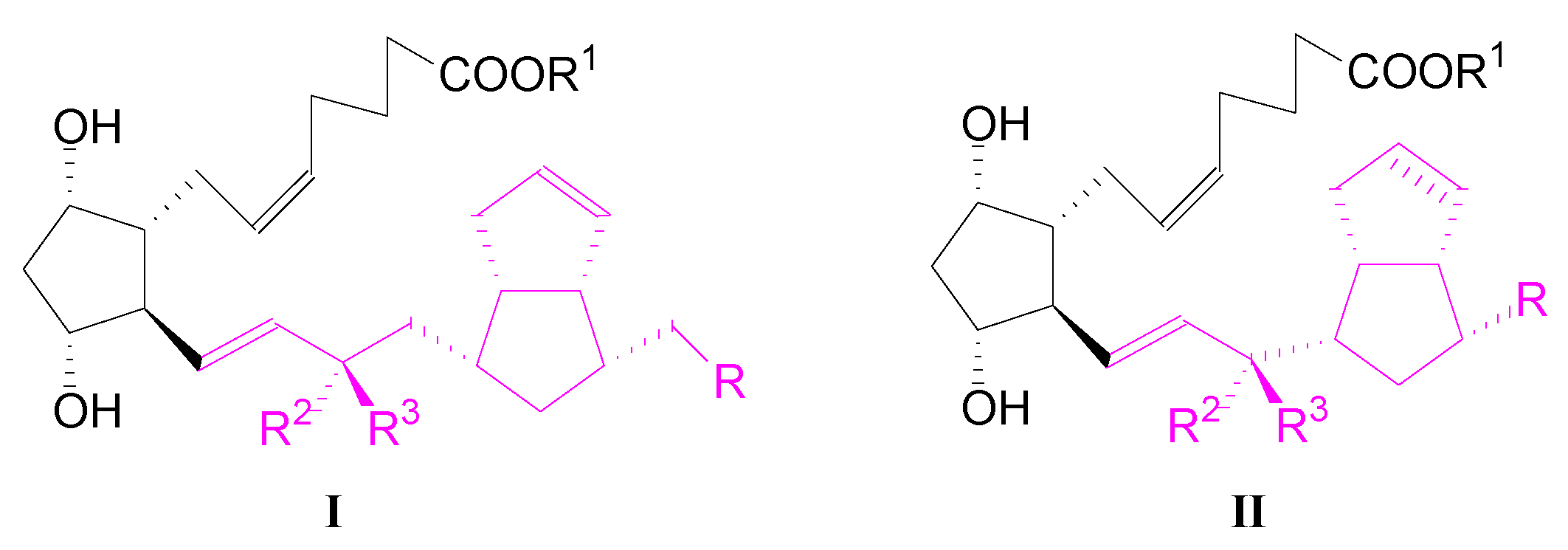
1. Introduction
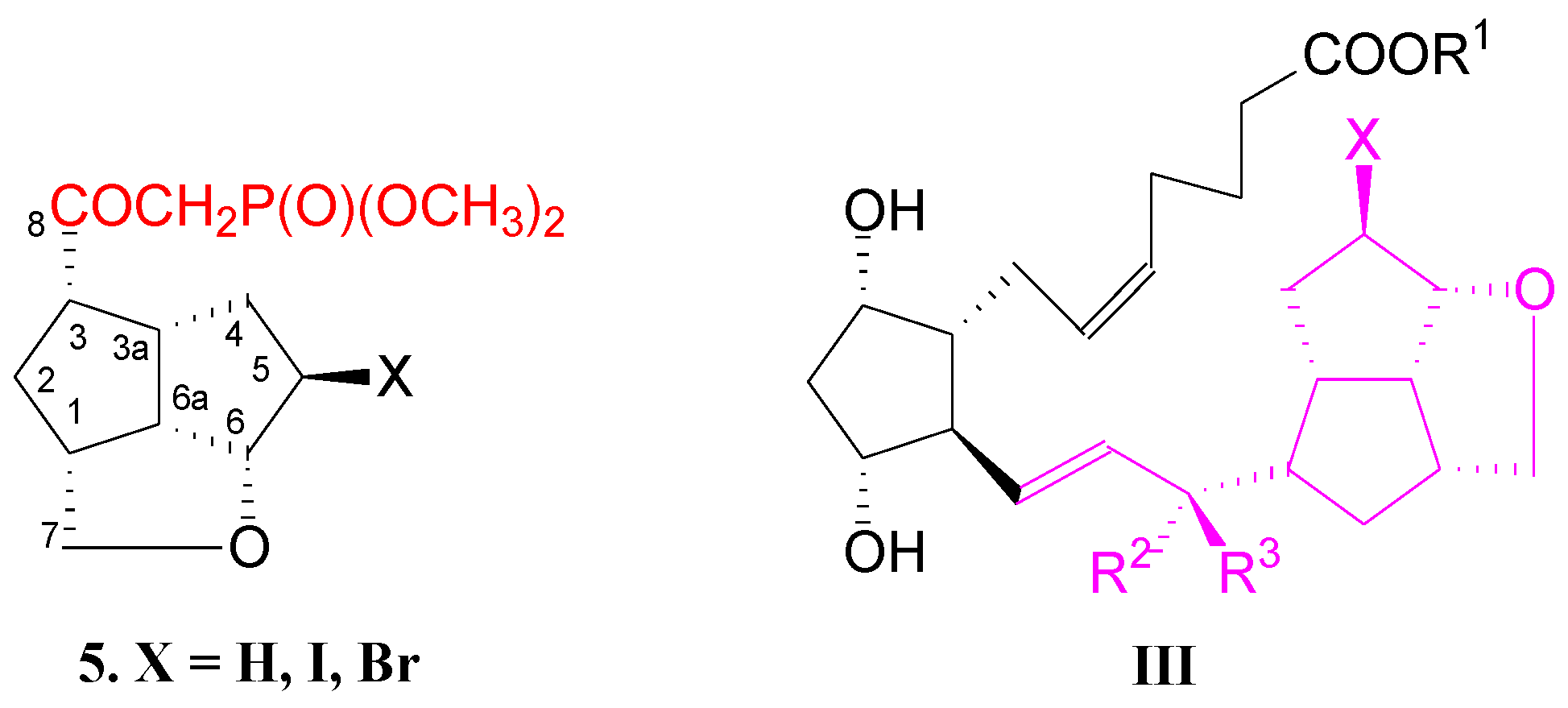
2. Results and Discussions
- -
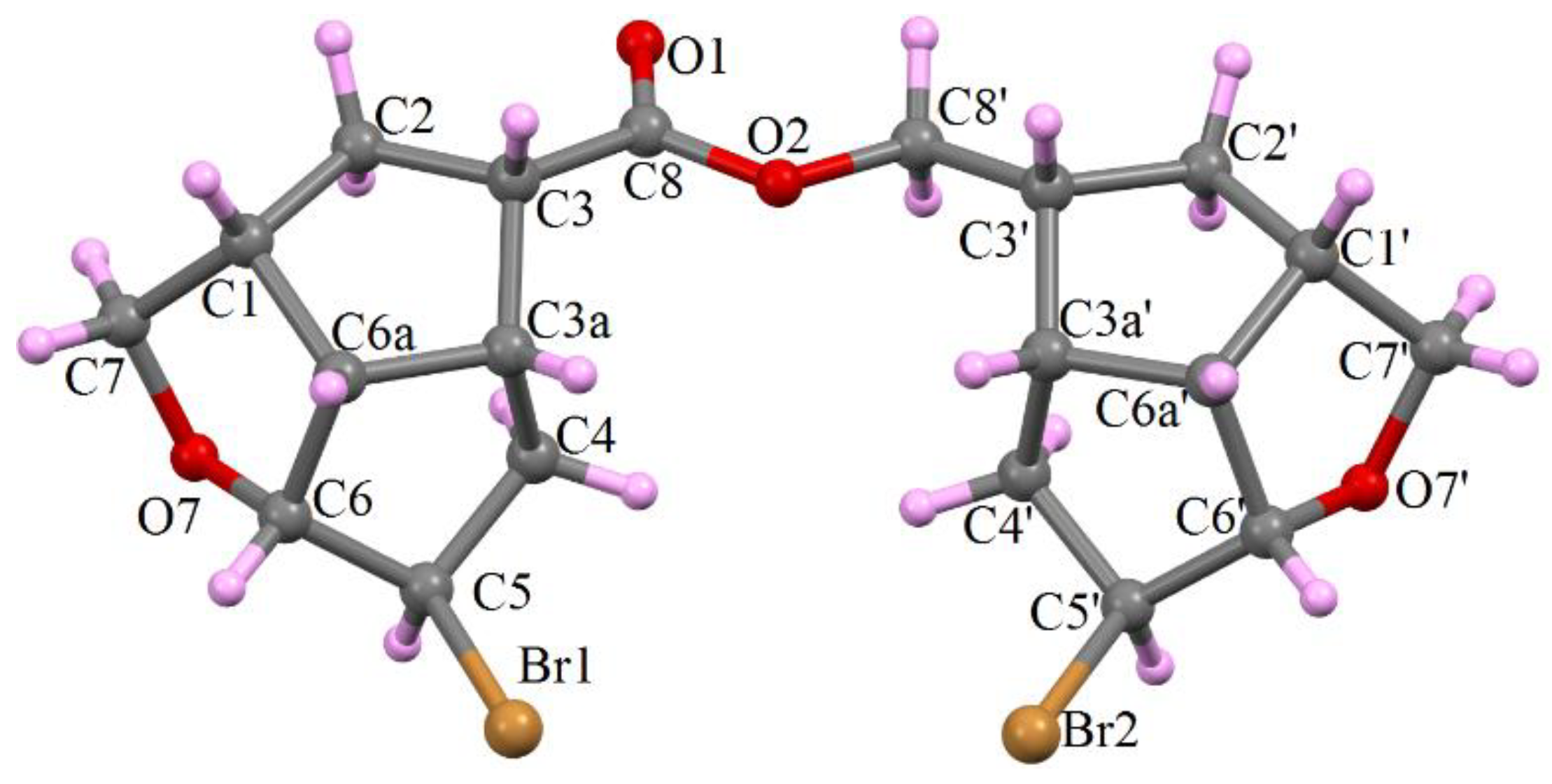
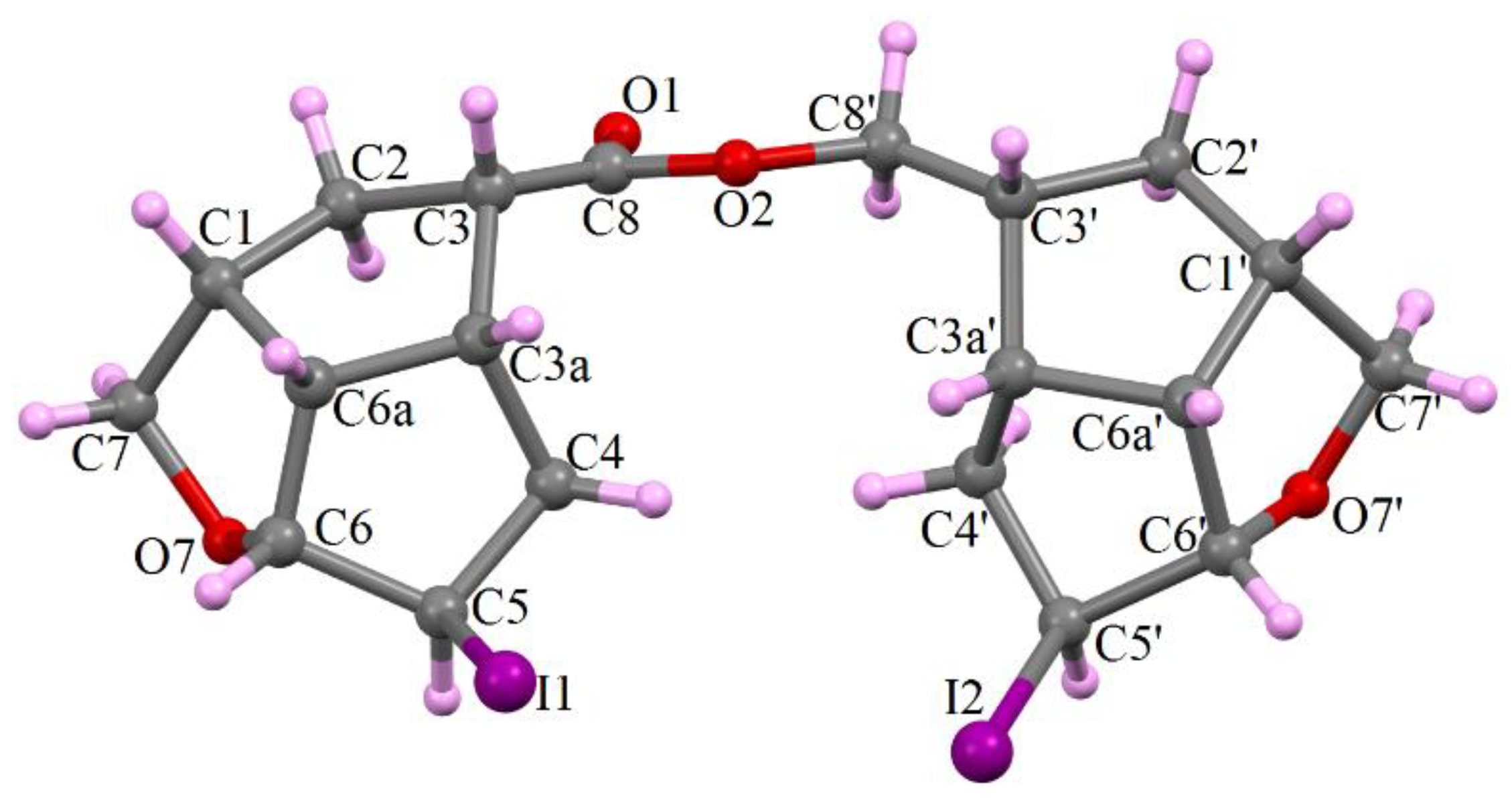
- Compound 6c provided suitable crystals for single crystal X-ray determination and its structural configuration is presented in Figure 3. As we observed in NMR spectra, the molecule 6c is an ester of the acid 3c, formed in Jones oxidation of the alcohol 2c, with the starting un-oxidized alcohol 2c, presented in the reaction mixture until its complete oxidation.
- -
- By contrast with compound 6c, compound 6b had no suitable single crystals and the structure was determined by the XRPD method, presented in detail at Experimental Section 3.2.
3. Experimental
3.1. General Information
3.1.1. Chemistry: Synthesis of Compound 3a
3.1.2. Synthesis of Compound 3b
3.1.3. Synthesis of Compound 3c
3.1.4. Synthesis of Compound 4a
3.1.5. Synthesis of Compound 4b
3.1.6. Synthesis of Compound 4c
3.1.7. Synthesis of β-Ketophosphonate 5a
3.1.8. Synthesis of β-Ketophosphonate 5b
3.1.9. Synthesis of β-Ketophosphonate 5c
3.2. X-ray Crystallography
4. Conclusions
5. Patents
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Collins, P.W.; Djuric, S.W. Synthesis of Therapeutically Useful Prostaglandin and Prostacyclin Analogs. Chem. Rev. 1993, 93, 1533–1564. [Google Scholar] [CrossRef]
- Yankee, E.W.; Axen, U.; Bundy, G.L. Total synthesis of 15-methylprostaglandins. J. Am. Chem. Soc. 1974, 96, 5865–5876. [Google Scholar] [CrossRef]
- Crabbe, P.; Fried, J.H. 4,5,13-Prostatrienoic Acid Derivatives. U.S. Patent 3,879,438(A), 25 April 1975. [Google Scholar]
- Simons, B.; Dammann, H.-G.; Muller, P.; Orth, D.; Raduntz, H.-E. 13-Thiaprostaglandins Having Cytoprotective Activity. U.S. Patent 4,622,316, 11 November 1986. [Google Scholar]
- Kolb, M.; Van Hijfte, L.; Ireland, R.E. A highly convergent synthesis of mexiprestil: 16(R) 16-methoxy 16-methyl PGE1 methyl ester. Tetrahedron Lett. 1988, 29, 6769–6772. [Google Scholar] [CrossRef]
- Skuballa, W.; Raduechel, B.; Vorbrueggen, H.; Elger, W.; Loge, O.; Schillinger, E. Pharmaceutically Active 9-Chloroprostaglandins. U.S. Patent 4,444,788, 24 April 1984. [Google Scholar]
- Suga, H.; Konishi, Y.; Wakatsuka, H.; Miyake, H.; Kori, S.; Hayashi, M. Synthesis of 16,16-Dimethyl-Trans-Δ2-PGE1 Methyl Ester (ONO-802). Prostaglandins 1978, 15, 907–912. [Google Scholar] [CrossRef]
- Baird, D.T.; Rodger, M.; Cameron, I.; Roberts, I. Prostaglandins and antigestagens for the interruption of early pregnancy. J. Reprod. Fertil. Suppl. 1988, 36, 173–179. [Google Scholar]
- Hayashi, M.; Konishi, Y.; Arai, Y. 6,9-Methano-PGI2 Analogues. U.S. Patent 4,479,966, 30 October 1984. [Google Scholar]
- Sodeoke, Y.M.; Ogawa, Y.; Kirio, M. Shibasaki, Stereocontrolled Synthesis of Exocyclic Olefins Using Arene Tricarbonyl Chromium Complex-Catalyzed Hydrogenation. I. Efficient Synthesis of Carbacyclin and Its Analogs. Chem. Pharm. Bull. Tokyo 1991, 39, 309–322. [Google Scholar] [CrossRef] [Green Version]
- Kovacs, G.; Simonidesz, V.; Tomoskozi, I.; Kormoczy, P.; Szekely, I.; Papp-Behr, A.; Stadler, I.; Szekeres, L.; Pap, G. A new stable prostacyclin mimic, 7-oxo-PGI2. J. Med. Chem. 1982, 25, 105–107. [Google Scholar] [CrossRef] [PubMed]
- Djuric, S.W.; Miyano, M.; Clare, M.; Rydzewski, R.M. A stereocontrolled synthesis of a novel prostacyclin analog “allene-carbacyclin”. Application of molecular mechanics calculations to organic synthesis. Tetrahedron Lett. 1987, 28, 299–302. [Google Scholar] [CrossRef]
- Schneider, J.; Friderichs, E.; Koegel, B.; Seipp, U.; Stahlberg, H.-J.; Terlinden, R.; Heintze, K. Taprostene sodium. Cardiovasc. Drug Rev. 1993, 11, 479–500. [Google Scholar] [CrossRef]
- Aristoff, P.A.; Johnson, P.D.; Harrison, A.W. Total synthesis of a novel antiulcer agent via a modification of the intramolecular Wadsworth-Emmons-Wittig reaction. J. Am. Chem. Soc. 1985, 107, 7967–7974. [Google Scholar] [CrossRef]
- Kluge, A.F.; Kertesz, D.J.; O’Yang, C.; Wu, H. Potent prostacyclin analogs based on the bicyclo[4.2.0]octane ring system. J. Org. Chem. 1987, 52, 2860–2868. [Google Scholar] [CrossRef]
- Vorbrueggen, H.; Nieuweber, B.; Stuerzebecher, C.S. New Carbacyclines, Preparation thereof and Drug Containing them. WO Patent 86/00895, 13 February 1986. [Google Scholar]
- Faustini, F.; Panzeri, A.; Orzi, F.; Di Sale, E.; Ceserani, R. Furyl Derivatives of 16-Substituted Prostaglandins. U.S. Patent 4,585,791, 29 April 1986. [Google Scholar]
- Tamura, T.; Inukai, N.; Iwamoto, H.; Yanagisawa, I.; Ishii, Y.; Takagi, T.; Tomioka, K.; Murakami, M. Studies on prostaglandins. VII. Synthesis of prostaglandin derivatives possessing ylidene group on C-16. Yamanouchi Seiyaku Kenkyu Hokoku 1980, 4, 16–29. [Google Scholar]
- Hess, H.J.E.; Schaff, T.K. Neue 15-Substituierte-Omega-Pentanorprostaglandine. DE Patent 2,355,731, 22 May 1974. [Google Scholar]
- Ngo, V.X.; Old, D.W.; Burk, R.M. 15-Aryl Prostaglandins as EP4 Agonists, and Methods of Use Thereof. U.S. Patent 9,540,357, 10 January 2017. [Google Scholar]
- Tănase, C.; Drăghici, C.; Căproiu, M.T. New β-ketophosphonates for the synthesis of prostaglandin analogues. 1. Phosphonates with a bicyclo[3.3.0]octene scaffold spaced by a methylene group from the β-ketone. Prostaglandins Leukot. Essent. Fat. Acids 2021. under revision. [Google Scholar] [CrossRef]
- Tănase, C.; Căproiu, M.T.; Drăghici, C. New ß-ketophosphonates for the synthesis of prostaglandin analogues. 2. Phosphonates with a bicyclo[3.3.0]octene and bicyclo[3.3.0]octane scaffolds linked to the ß-ketone group. New J. Chem. 2020, 44, 20405–20410. [Google Scholar] [CrossRef]
- Tănase, C.; Căproiu, M.T.; Drăghici, C. β-Ketophosphonates with a Hexahydro- and Octahydropentalene Functionalized Fragment for Obtaining New Prostaglandin Analogs. Patent Pending A/00517, 14 August 2020. [Google Scholar]
- Tănase, C.; Cocu, F.; Drăghici, C.; Hanganu, A.; Pintilie, L.; Maganu, M.; Munteanu, C.V.A.; Shova, S. Secondary Compounds in the Catalytic Hydrogenation of Enone and Allylic Alcohol Prostaglandin Intermediates: Isolation, Characterization, X-ray Crystallography. New J. Chem. 2019, 43, 7582–7599. [Google Scholar] [CrossRef]
- Tănase, C.; Cocu, F.; Drăghici, C.; Căproiu, M.T. 1,2,3,3a,4,6a-Hexahydro-1,3-pentalenedimethanol, mono and bis OH-Protected Derivatives, Useful Intermediates for Fine Organic Synthesis. Rev. Roum. Chim. 2008, 53, 195–202. [Google Scholar]
- Tănase, C.; Drăghici, C.; Shova, S.; Maganu, M.; Cojocaru, A.; Munteanu, C.V.A.; Cocu, F. Regioselective reactions on a 1,3-disubstituted dihydroxymethyl or dicarboxyl hexahydropentalene skeleton F. Tetrahedron 2015, 71, 6852–6859. [Google Scholar] [CrossRef]
- Tojo, G.; Fernández, M. Oxidation of Primary Alcohols to Carboxylic Acids, A Guide to Current Common Practice. In Basic Reactions in Organic Synthesis; Springer Science + Business Media, LLC: Berlin/Heidelberg, Germany, 2007; pp. 13–31. [Google Scholar]
- Tănase, C.; Drăghici, C.; Hanganu, A.; Pintilie, L.; Maganu, M.; Gal, E.; Borodi, G. The First Pentalenofurane Scaffold in the ω-side Chain of a PGF1 Analog. 2021; submitted. [Google Scholar]
- CrysAlisPro; Rigaku Oxford Diffraction: Oxfordshire, UK, 2015.
- Sheldrick, G.M. A Short History of SHELX. Acta Crystallogr. 2008, A64, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Sheldrick, G.M. Crystal structure refinement with SHELXL. Acta Crystallogr. 2015, A71, 3–8. [Google Scholar] [CrossRef] [Green Version]
- Dolomanov, O.V.; Bourhis, L.J.; Gildea, R.J.; Howard, J.A.K.; Puschmann, H. OLEX2: A complete structure solution refinement and analysis program. J. Appl. Cryst. 2009, 42, 339–341. [Google Scholar] [CrossRef]
- Griffin, J.F.; Duax, W.L.; Weeks, C.M. Atlas of Steroid Structure, 2nd ed.; IFI/PLENUM: New York, NY, USA; Washington, DC, USA; London, UK, 1984; p. 8. [Google Scholar]
- Tănase, C.; Drăghici, C.; Căproiu, M.T. β-Ketophosphonates with a Functionalized Pentalenofurane Fragment for Obtaining New Prostaglandin Analogs. Patent Pending A/00516, 14 August 2020. [Google Scholar]
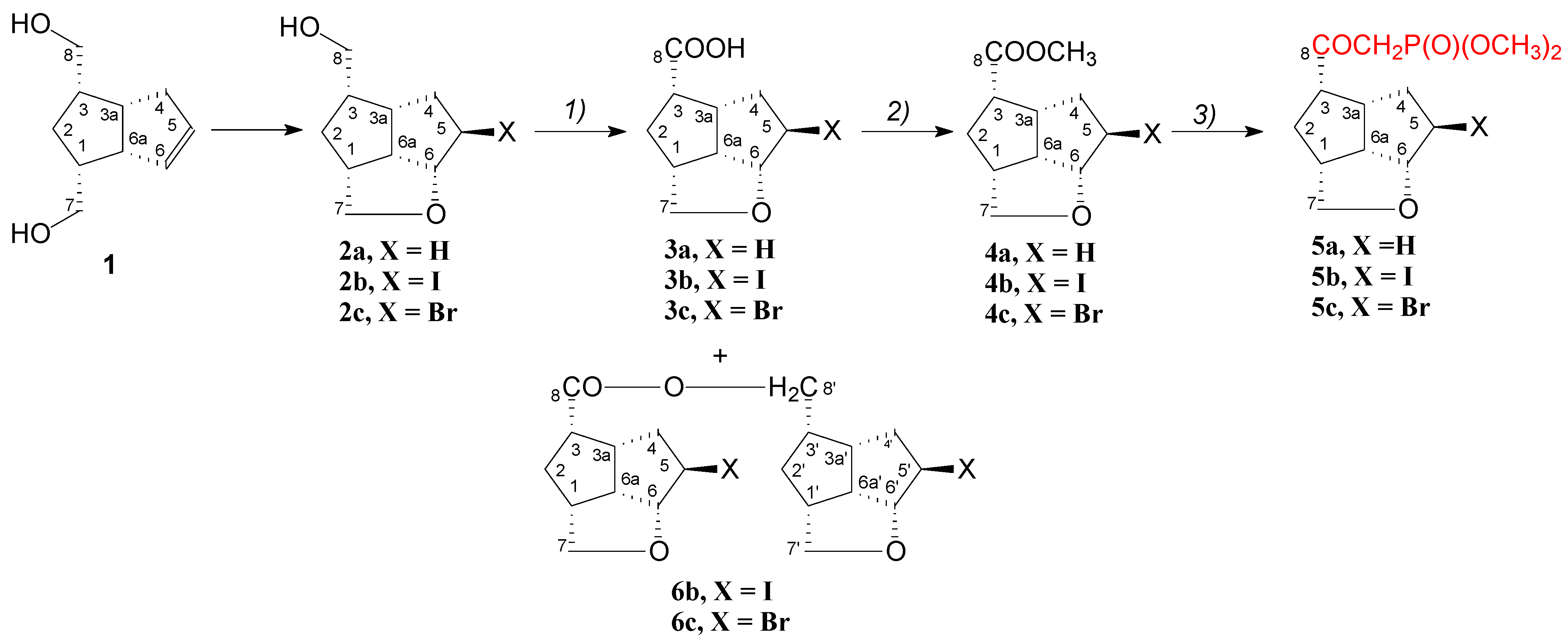


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 6c | ΔCs | ΔCs | ΔCs | ΔCs | ΔCs | ΔCs |
| (C5) | (O7) | (C3) | (C4′) | (C7′) | (C3′) | |
| 12.36 | 15.43 | 5.91 | 14.38 | 14.92 | 5.55 | |
| 6b | ΔCs | ΔCs | ΔCs | ΔCs | ΔCs | ΔCs |
| (C5) | (O7) | (C3) | (C4′) | (O7′) | (C3′) | |
| 5.81 | 9.55 | 3.35 | 9.31 | 15.46 | 1.96 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tănase, C.I.; Drăghici, C.; Căproiu, M.T.; Hanganu, A.; Borodi, G.; Maganu, M.; Gal, E.; Pintilie, L. β-Ketophosphonates with Pentalenofuran Scaffolds Linked to the Ketone Group for the Synthesis of Prostaglandin Analogs. Int. J. Mol. Sci. 2021, 22, 6787. https://doi.org/10.3390/ijms22136787
Tănase CI, Drăghici C, Căproiu MT, Hanganu A, Borodi G, Maganu M, Gal E, Pintilie L. β-Ketophosphonates with Pentalenofuran Scaffolds Linked to the Ketone Group for the Synthesis of Prostaglandin Analogs. International Journal of Molecular Sciences. 2021; 22(13):6787. https://doi.org/10.3390/ijms22136787
Chicago/Turabian StyleTănase, Constantin I., Constantin Drăghici, Miron Teodor Căproiu, Anamaria Hanganu, Gheorghe Borodi, Maria Maganu, Emese Gal, and Lucia Pintilie. 2021. "β-Ketophosphonates with Pentalenofuran Scaffolds Linked to the Ketone Group for the Synthesis of Prostaglandin Analogs" International Journal of Molecular Sciences 22, no. 13: 6787. https://doi.org/10.3390/ijms22136787
APA StyleTănase, C. I., Drăghici, C., Căproiu, M. T., Hanganu, A., Borodi, G., Maganu, M., Gal, E., & Pintilie, L. (2021). β-Ketophosphonates with Pentalenofuran Scaffolds Linked to the Ketone Group for the Synthesis of Prostaglandin Analogs. International Journal of Molecular Sciences, 22(13), 6787. https://doi.org/10.3390/ijms22136787