1. Introduction
Pancreatic ductal adenocarcinoma (PDAC) has a very poor prognosis with a 5-year overall survival of approximately 9% [
1]. Surgery remains the only means of cure. However, at the time of diagnosis, up to 30% of patients present with unresectable locally advanced pancreatic cancer. In a subset of these patients, failing to control the primary tumor may result in complications that can lead to death. Therefore, making unresectable tumors resectable may improve outcomes.
As gemcitabine has been shown to both be effective and to enhance radiosensitivity on PDAC cells, chemoradiotherapy (CRT) with gemcitabine is one of the current effective option for treating non-metastatic unresectable pancreatic cancer [
2]. However, gemcitabine-based CRT has a high of rate toxicity, leading to a reduction of gemcitabine dose or chemotherapy discontinuation. Using modern techniques of radiotherapy such as intensity-modulated radiotherapy (IMRT) or stereotactic body radiotherapy (SBRT) may simultaneously enhance dose to tumor and decrease dose to organ-at-risk, leading to a reduction of gemcitabine-radiation related toxicity. Besides targeted therapies that could be radiosensitizers and/or chemopotentiating agents may enhance this synergy.
Recently, DNA repair has become a major topic of investigation for the treatment of cancer. The key determinant of cellular radiosensitivity is the capacity of cells to repair highly lethal DNA double-strand breaks (DSBs). Therefore, targeting proteins implicated in the response to ionizing radiation-induced DNA damage, such as poly(ADP-ribose) polymerase-1 (PARP-1), may be an appropriate strategy. Indeed, PARP-1 is a major DNA damage sensor allowing the recruitment of DNA repair proteins involved in DNA single- and double-strand breaks repair [
3]. At the outset of PARP inhibitors (PARPi) development, targeting BRCA1/2 mutant tumors has been the main approach and is known as the concept of synthetic lethality. Ongoing phase III trials are testing olaparib in different tumor types with BRCA1/2 mutations such as breast cancer (clinical trial.gov NCT02032823, NCT02000622), ovarian cancer (NCT01844986, NCT01874363) and pancreatic cancer (NCT02184195). More recently, the use of PARPi has been investigated on BRCA1/2 wild-type tumors in combination with radiotherapy [
3]. Olaparib has been one of the most studied PARPi, which potentiated the effect of radiotherapy in different cell lines and tumor xenograft models [
4]. However, association of olaparib, gemcitabine and irradiation has never been assessed on BRCA wild type pancreatic cancer cells yet. The main purpose of this study is to evaluate the in vitro antitumor efficacy of the association of olaparib (PARPi) and gemcitabine, combined with different radiation doses, on four pancreatic cancer cell lines.
3. Discussion
Effective treatments for locally advanced pancreatic cancer are limited. Therefore, any new modality to replace or support current treatments for pancreatic cancer would be highly valuable.
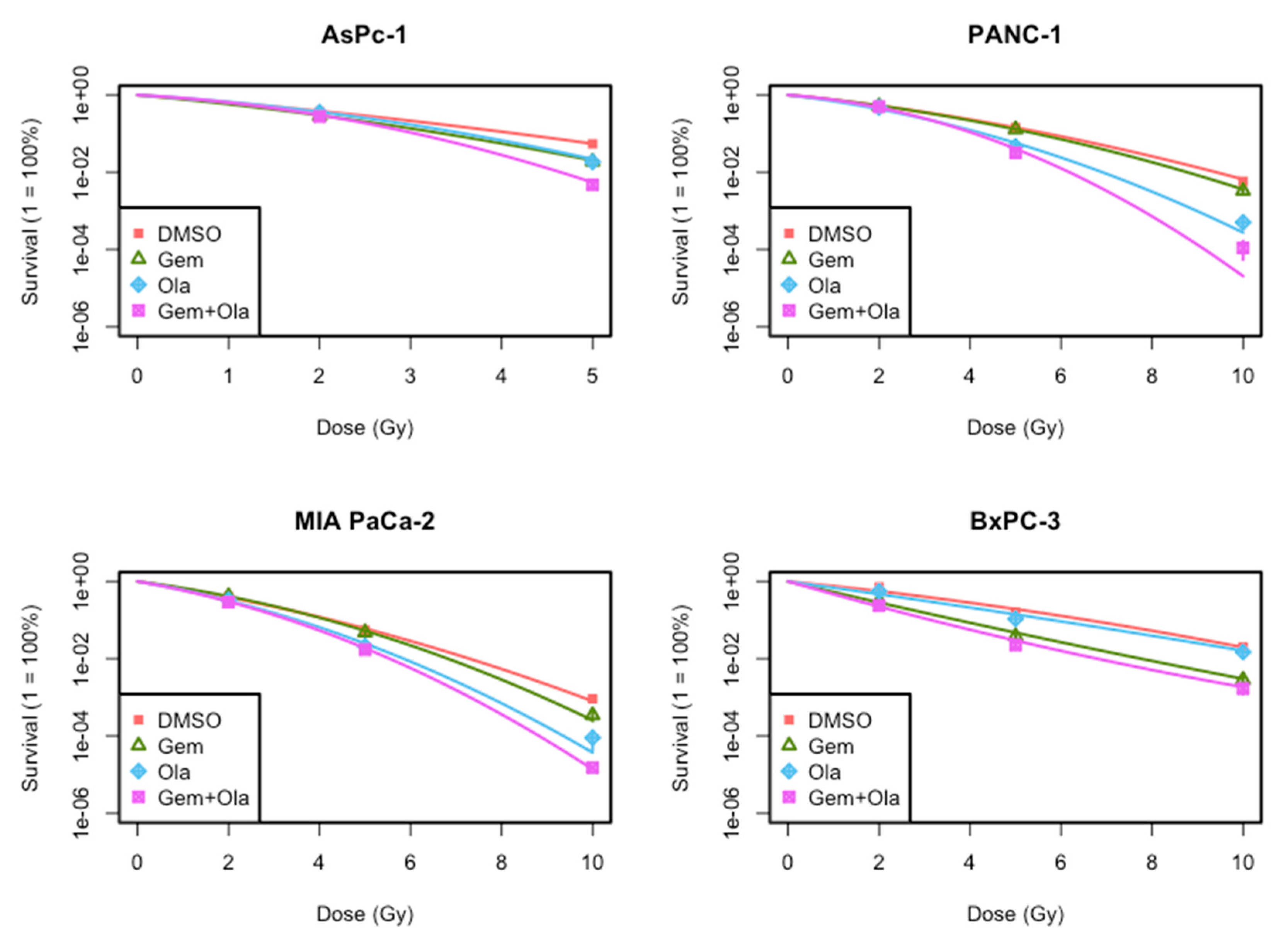
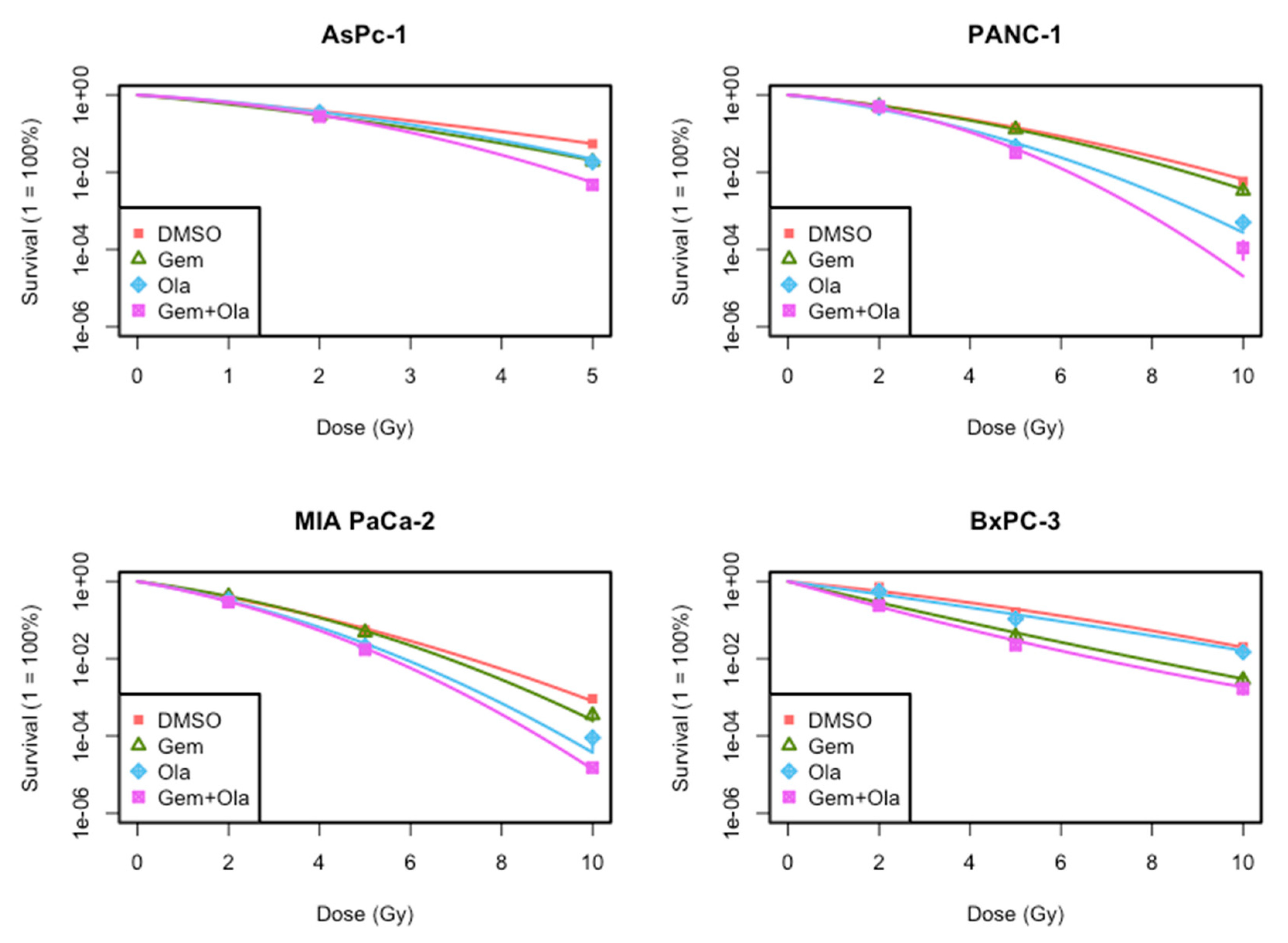
In this study, we evaluated in vitro efficacy of the association of gemcitabine, olaparib and irradiation in four locally advanced pancreatic cancer cell lines. First, our results indicated that this triple association reduced cell growth and clonogenic survival, compared to both gemcitabine/irradiation or olaparib/irradiation combinations and standard treatment combining chemoradiotherapy with gemcitabine. This effect was mainly due to the radiosensitizing effect of olaparib. Indeed, we depicted that although olaparib alone at a concentration of 1 µM was not toxic for all four cell lines as chosen for this project, it had a clear radiosensitizing effect and particularly with high dose of radiation (10 Gy). This is consistent with results obtained by Vance et al., who treated MIA PaCa-2 cells with 1 µM of olaparib and increasing doses of irradiation (ranged from 2 to 8 Gy) and found a radiation enhancement ratio of 1.5 ± 0.1, whereas cytotoxicity of olaparib alone was 1.1 ± 0.1 [
5]. It is also coherent with Karnak et al. who reported radiosensitization of AsPC-1 cell line with olaparib (SER = 1.2 ± 0.2) [
6]. We also studied two other pancreatic cell lines that have never been studied before with olaparib, PANC-1 and BxPC-3. We pointed out that the magnitude radiosensitization with olaparib was dependent of the cell line. As cell lines have different genomic background, the extent of radiosensitization could be due to their genomic alterations. Indeed, our data showed that olaparib and gemcitabine have a synergistic radiosensitization effect on BxPC-3 cell line but not in three other cell lines. The combination of olaparib with gemcitabine-based chemoradiotherapy should then be assessed in vivo.
Another aspect of the present study was the investigation of cell cycle perturbation with PARP inhibitors. We and others have shown that PARP-1 inhibition resulted in a greater accumulation of cells in the G2/M phase in response to radiation, likely due to persistent DNA damage, whereas gemcitabine blocked cells in early S-phase, likely due to the inhibition of replication [
7,
8,
9]. As G2 checkpoint activation is known to be a consequence of persistent DNA damage, we assessed DSBs repair through residual γ-H2Ax foci, 24 h after treatments and showed that olaparib combined with irradiations (10 Gy) increased γ-H2AX foci compared to control irradiated cells. To properly compare the radiosensitization effect of gemcitabine with olaparib, we assessed radiosensitization effect of a non-cytotoxic concentration of gemcitabine (10 nM). As Pauwels et al. presented in different types of cancer cell lines, radiosensitization with gemcitabine was time-exposure dependent and was higher with 24 h exposure before irradiation [
10], therefore we treated cell lines with 24 h of gemcitabine before irradiation. To display radiosensitization with CHK1/2 (AZD7762) inhibitor in pancreatic cancer, Morgan et al. irradiated and co-treated MIA PaCa-2 cells with gemcitabine/AZD7762 and showed a radiation enhancement ratio of 1.5 with gemcitabine treatment alone. In their treatment schedules, cells were treated 24 h before irradiation with gemcitabine and at time of irradiation, approximately 45% of cells were in S-phase [
11]. Indeed, Im et al. presented that radiosensitization with gemcitabine required a depletion in deoxynucleotide for approximately 4 h with accumulation of cells in S-phase [
12]. These observations are coherent with our hypothesis that gemcitabine could synchronize cell lines into S-phase, thus leading to enhanced sensitization after irradiation and olaparib treatment. As S-phase is known to be a radioresistant phase, olaparib treatment could overcome intrinsic radioresistance.
Ewald et al. showed that H2AX phosphorylation was not only associated with DSBs but also with agents that inhibits DNA synthesis, such as gemcitabine and was due to stalled replication forks during S-phase [
13]. Moreover, they displayed that some γ-H2AX foci co-localized with DNA damage response proteins (Mre11, Rad50 and Nbs1) at the site of stalled replication forks, shortly after nucleoside analogue exposure. More recently, Bryant et al. presented that treatment with hydroxyurea induced stalled replication forks, thus stimulating PARP-1 activation, leading to recruitment of Mre11 and Nbs1, thus promoting repair through homologous recombination [
14]. Thus, there is an interplay between gemcitabine inducing stalled replication forks, PARP activation and homologous recombination. The radiosensitizing effect of PARPi may be due to different mechanisms. First, PARP inhibitor inhibits enzymatic activity of PARP, thus decreasing protein PARylation. Besides, PARPi efficacy may also be attributed to PARP trapping, whereby it remains to be restrained at the site of DNA damage, hence preventing DNA repair. Recently, Parsels et al. showed that PARP trapping with olaparib and ATR inhibition could overcome intrinsic resistance of HR-proficient pancreatic cancer cells [
15].
We also reported that cell death was partly induced by autophagy, specifically for gemcitabine-base treatments, but apoptosis was clearly not involved. Our results are consistent with the literature. Indeed, Chen et al. showed that even with the highest concentration of olaparib (1 µM), there was at most 6% of apoptosis in PANC-1 and BxPC-3 pancreatic cancer cell lines [
16]. More recently, Pandita et al. displayed that in MIA PaCa-2 cell line, treatment with 660 nM of gemcitabine resulted in 2% of apoptosis [
17]. MIA PaCa-2 cells seemed to be more resistant to gemcitabine. This was partly explained by the fact that gemcitabine induced apoptosis through p53 pathway and mutant p53 cells [
18]. Autophagy is a conserved process by which cytoplasm and cellular organelles are degraded in lysosomes. There is much controversy concerning the role of treatment induced-autophagy in pancreatic cancer. Authors argued that gemcitabine-induced autophagy prevents pancreatic carcinoma entering in apoptotic pathway, thus allowing resistance to treatment [
19,
20]. Others claimed that autophagy constituted a death mechanism, when pancreatic cancer cells were treated with gemcitabine and/or ionizing radiation [
18,
20]. Indeed, Rosenfeldt et al. used a humanized genetically modified mouse model of PDAC and showed that autophagy’s role in tumor development was connected to the status of p53. Indeed, loss of autophagy in tumor lacking p53 accelerated tumor onset, whereas it blocked progression to high grade malignant tumors in p53 wild-type cells [
20]. These data are consistent with results published by Fiorini et al., showing in a mutant p53 cell line, that incubation of gemcitabine with p53 reactivating molecules, induced autophagy and apoptosis. However, inhibiting autophagy enhanced anti-proliferative activity of combined treatment, thus demonstrating that autophagy induced by gemcitabine in p53 wild-type cell line may have a pro-survival effect [
18]. Here, we presented on four p53 mutant cell lines, that gemcitabine induced autophagy compared to control, but this effect was clearly attenuated with increasing radiation doses, and did not seem to be synergic. On the other hand, surviving fraction and cell growth were decreased with irradiation and gemcitabine compared to control, and the addition of olaparib also decreased surviving fraction. These data suggest that some other types of cell death contribute to the effectiveness of the combined triple treatment with gemcitabine, olaparib and irradiation. Several studies have examined the mechanism of cell-death induced by PARP inhibition. Some authors explored senescence as a mode of cell death after irradiation and treatment with PARPi. A radiosensitization of tumor cells was observed via the promotion of senescence [
21]. Another induced cell death could be mitotic catastrophe [
8].
Given the lack of effective strategies to treat pancreatic cancer and based on these data showing that PARPi could radiosensitize PDAC cell lines, targeting DNA damage through PARP inhibition should be explored as therapeutic options to treat locally advanced PDAC.
4. Materials and Methods
4.1. Cell Culture
The human pancreatic cancer cell lines MIA PaCa-2, AsPC-1, PANC-1 and BxPC-3 were purchased from American Type Culture Collection (CRL-1420TM, CRL-1682, CRL-1469 and CRL-1687, respectively, ATCC; Rockville, MD, USA) and banked at Centre Paul Strauss. Cells were cultured at 37 °C in a humidified atmosphere containing 5% CO2 and 95% air. PANC-1 was cultured in Dulbecco’s modified Eagle’s medium (DMEM; PAN Biotech GmbH, Aidenbach, Germany) supplemented with 10% fetal bovine serum (FBS, PAN Biotech GmbH) and 1% of a solution of penicillin (10000 IU/mL) and streptomycin (10 mg/mL) (PAN Biotech GmbH). MIA PaCa-2 was cultured in the same conditions with 1% of 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES, 1 mM, PAN Biotech GmbH), 1% of sodium pyruvate (10 mM, PAN Biotech GmbH) and 1% of non-essential amino acids (NEAA, PAN Biotech GmbH). AsPC-1 and BxPC-3 were cultured in Roswell Park Memorial Institute medium (RPMI; PAN Biotech GmbH) supplemented with 10% FBS, and 1% of a solution of penicillin (10000 IU/mL) and streptomycin (10 mg/mL). Subconfluent cell monolayers were trypsinized once a week using 0.5% trypsin containing 2% EDTA (PAN Biotech GmbH) and plated at passage ratios between 0.25:10 to 1:10, according to the cell line or used directly for study after enumeration determined with a Countess® Cell Counter (Countess, Invitrogen, Carlsbad, CA, USA).
4.2. Drugs and Chemicals
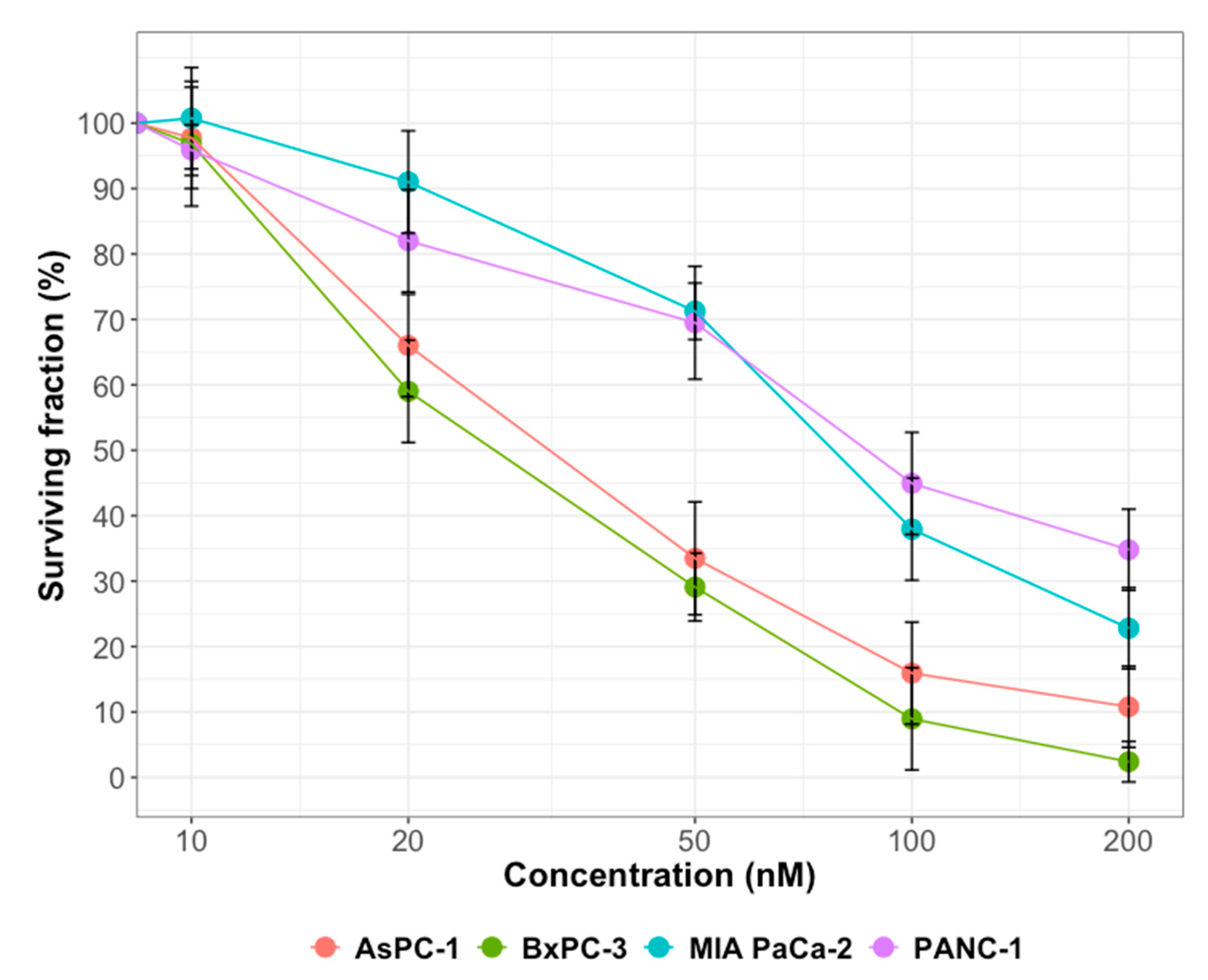
Olaparib and gemcitabine were provided by Selleck Chemicals (Houston, TX, USA). Stock solutions were prepared at 10 mM in DMSO and stored in aliquots at −20 °C. Olaparib was used at 1 µM and gemcitabine at 10 nM.
4.3. Irradiation Exposure
Cells seeded in 6-well plates were exposed, at room temperature, to photon irradiation, one hour after pharmacological treatment (olaparib and/or gemcitabine), at one fraction-doses of 2, 5 and 10 Gy. A 137Cs γ-irradiator (Biobeam GM 8000, GSM GmbH, Leipzig, Germany) was used in the Paul Strauss Center (Strasbourg, France) at a dose rate of 2.5 Gy/min. Control flasks were sham irradiated.
4.4. Clonogenic Survival Assay
Cells were seeded in 6–well plates at a density of 2 × 105 cells per well and allowed to adhere overnight in standard culture conditions. Cells were exposed to DMSO (control), olaparib and/or gemcitabine 1 h before irradiation with 2, 5 or 10 Gy. Following treatment for 24 h, cells were trypsinized, collected and numbered. Then, cells were seeded at optimized densities according to radiation dose and plated at two different dilutions into 6-well plates. Twelve to nineteen days later, depending on cell lines, the clones were stained using 0.05% crystal violet (Sigma-Aldrich, Lyon, France) in 5% ethanol solution and positive colonies (≥50 cells) were scored. The plating efficiency (PE) and then the surviving fractions were calculated. Survival curves were plotted using surviving fractions for different doses. To generate the radiation dose–response curves, the data was fitted to the linear quadratic (LQ) model, where S(D) is the fraction of cells surviving a dose of D and α/β are inactivation constants: S(D) = e^(−αD−βD^2). Semi-logarithmic survival curves were constructed for determination of survival ability of cells in response to various treatments. The linear-quadratic (LQ) cell survival curve parameters were calculated with CFAssay Package using R Software. SER were determined by calculating the ratio of doses in treated and control conditions for a given isoeffect (SF = 0.1).
4.5. PARP Inhibitor Activity
Cells were pre-incubated with olaparib (1 µM) during 1 h. The treated cells were then exposed to 500 µM of H2O2 for 10 min. Cells were fixed with a solution of methanol/acetone 1:1 at 4 °C for 20 min. Next, cells were permeabilized by three washes with PBS-tween. The poly(ADP-ribose) (PAR) signal was then detected by incubation with the anti-PAR antibody (1/1000; clone 10H Sugimura) in PBS-tween and BSA 0.1% for 2 h at 4 °C. Then, the cells were washed three times with PBS-tween. The cells were incubated with the secondary mouse antibody anti-IgG coupled with Alexa 594 (1/2000; Invitrogen) in PBS-tween and BSA 0.1% for 2 h at 4 °C. Subsequently, cells were washed with PBS-tween, next PBS and finally PBS-DAPI. Last, cell glass slides were prepared by fixation with Mowiol and antifading agent DABCO. Finally, detection of PAR was analyzed by immunofluorescence microscopic imaging using an Olympus BH-2 fluorescent microscope equipped with a digital camera.
4.6. Cell Cycle Distribution
Twenty-four hours after treatments, treated cells were trypsinized, harvested and washed twice in phosphate buffered saline (PBS, pH 7.2) fixed in 1 mL of ice-cold 70% ethanol and stored at −20 °C. The pellet was resuspended in 200 μL PBS containing 1 mg/mL RNase A, and the cellular DNA was stained with propidium iodide (PI; 0.1 mg/mL; Sigma, Lyon, France). Cells were incubated at 37 °C in the dark for 30 min prior to analysis. Cell cycle determination was performed using a BD AccuriTM C6 flow cytometer (Becton Dickinson, BD, San Jose, CA, USA) and fluorescence of at least 10,000 cells were analyzed using BD Accurri and the ModFit softwares, provided by the manufacturer.
4.7. Apoptotic and Necrotic Detection Assay
Apoptotic cells were quantified 24 and 48 h after irradiation. Harvested cells were washed with PBS and resuspended in 200 μL of 1X Annexin V Binding Buffer (FITC Annexin V Apoptosis Detection kit, BD Pharmingen). Then, 5 μL of Annexin V and 5 μL of propidium iodide (PI) were added to 100 μL of the solution. After incubation in the dark at room temperature for 15 min, 200 μL 1X Annexin V Binding Buffer was added to the solution and the fluorescence of 10,000 cells was analyzed using a BD AccuriTM C6 flow cytometer and software (BD). Annexin V+/PI− cells were recorded as being early apoptotic, whereas Annexin V+/PI+ were considered to be necrotic.
4.8. Autophagic Detection Assay
For autophagy determination 24 h after irradiation, we used the Cyto-IDTM Autophagy Detection kit (Enzo Life Sciences, Plymouth Meeting, PA, USA) according to the manufacturer’s instructions. Cells were washed in PBS (pH 7.2) and resuspended in 500 μL PBS containing Cyto-ID® Green Detection Reagent (0.1% v/v). Then, cells were incubated at 37 °C in the dark for 30 min and were resuspended in 200 μL PBS. The fluorescence emission of 10,000 cells was analyzed using a BD AccuriTM C6 cytometer and software (BD).
4.9. Determination of γ-H2AX Formation
For γ-H2AX foci visualization, cells grown on Lab-Tek chambers (Nunc, Thermofisher, Illkirch, France) were treated as indicated earlier and fixed with a 4% paraformaldehyde solution for 15 min. Then samples were permeabilized with PBS containing 0.5% Triton-X-100 and blocked with 10% BSA. Then, cells were incubated overnight at 4 °C, with blocking buffer containing primary antibody against γ-H2AX (clone JBW301; Merck Millipore, Darmstadt, Germany) followed by incubation with Alexa Fluor 488 goat anti-mouse IgG (Invitrogen) for 3 h at room temperature. Samples were counterstained with antifading agent containing DAPI. The formation of foci in nuclei were monitored by immunofluorescence using an Olympus BH-2 fluorescence microscope equipped with digital camera.
For γ-H2AX analysis in flow cytometry, 24 h after treatment, cells were harvested and washed in ice-cold PBS, fixed in 1 mL of 70% ethanol and stored at −20 °C for 24 h. Fixed cells were centrifuged and pellet was resuspended twice in 2 mL cold PBS containing 10% bovine serum albumin (BSA, Sigma-Aldrich, Lyon, France) and 0,2% Triton-X-100 (T-PBS-BSA). Then, labelling was performed using a solution of monoclonal mouse anti-phospho-histone-H2AX (ser 139) polyclonal antibody (clone JBW301; Merck Millipore, Darmstadt, Germany) diluted in 1/100 in T-PBS-BSA. Cells were stored at 4 °C overnight. Cells were washed twice with 2 mL T-PBS-BSA and resuspended in 100 µL of a solution containing an anti-mouse IgG fluorescein-conjugated antibody diluted at 1:200 (Invitrogen) and were stored 1 h in room temperature and gently shaken. Then, 2 mL T-PBS-BSA wad added, cells were centrifuged and pellet was resuspended in 200 µL of PBS containing PI (5 µg/mL) and RNAse A (100 µg/mL). Next, cells were stored at room temperature in the dark for 30 min and gently shaken. The fluorescence of 10,000 cells was analyzed using a BD Accuri
TM C6 flow cytometer and software (BD). For quantification of γ-H2AX positivity, a gate was arbitrarily set on the control, untreated sample to define a region of positive staining for γ-H2AX of approximately 5% [
22]. This gate was then overlaid on the treated samples.
4.10. Statistical Analysis
Experiments were performed at a minimum in triplicate and results were expressed as the mean ± standard deviation (SD) unless otherwise indicated. Statistical differences of data were assessed by the
t-test using R Software (ver. 3.4.0
http://cran.r-project.org/).
p-Values lower than 0.05 were considered as statistically significant. For clonogenic survival assay, results from 4 experiments were subjected to linear-quadratic regression analysis, using the maximum likelihood approach. Differences between curves were evaluated using a two-way ANOVA test.










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}