
Plumbagin, a Natural Product with Potent Anticancer Activities, Binds to and Inhibits Dihydroorotase, a Key Enzyme in Pyrimidine Biosynthesis

 , , , and
, , , and
Abstract
:1. Introduction
2. Results
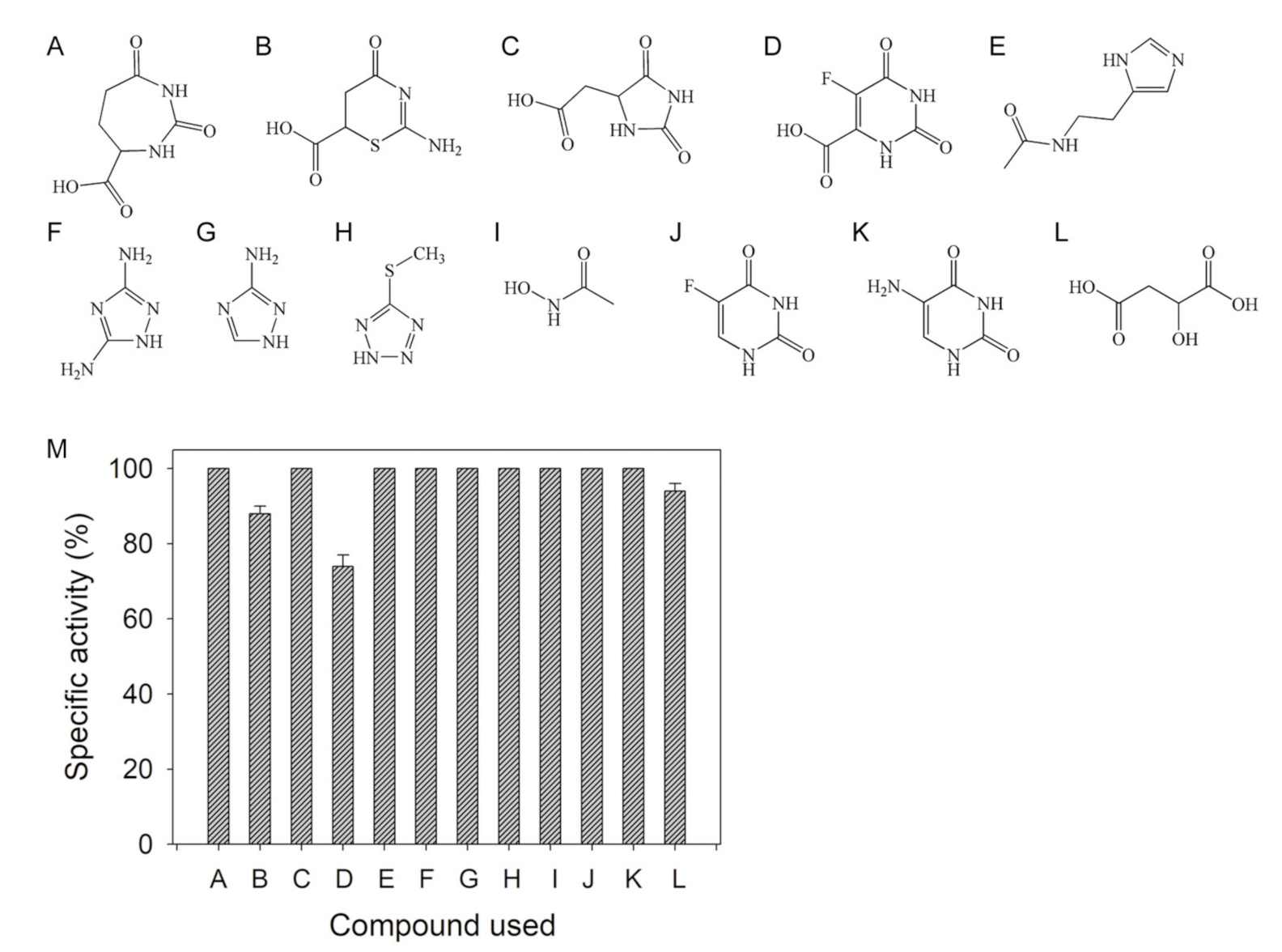
2.1. Inhibition of DHOase by Using Substrate Analogs
2.2. Inhibition of DHOase by Plant Extracts
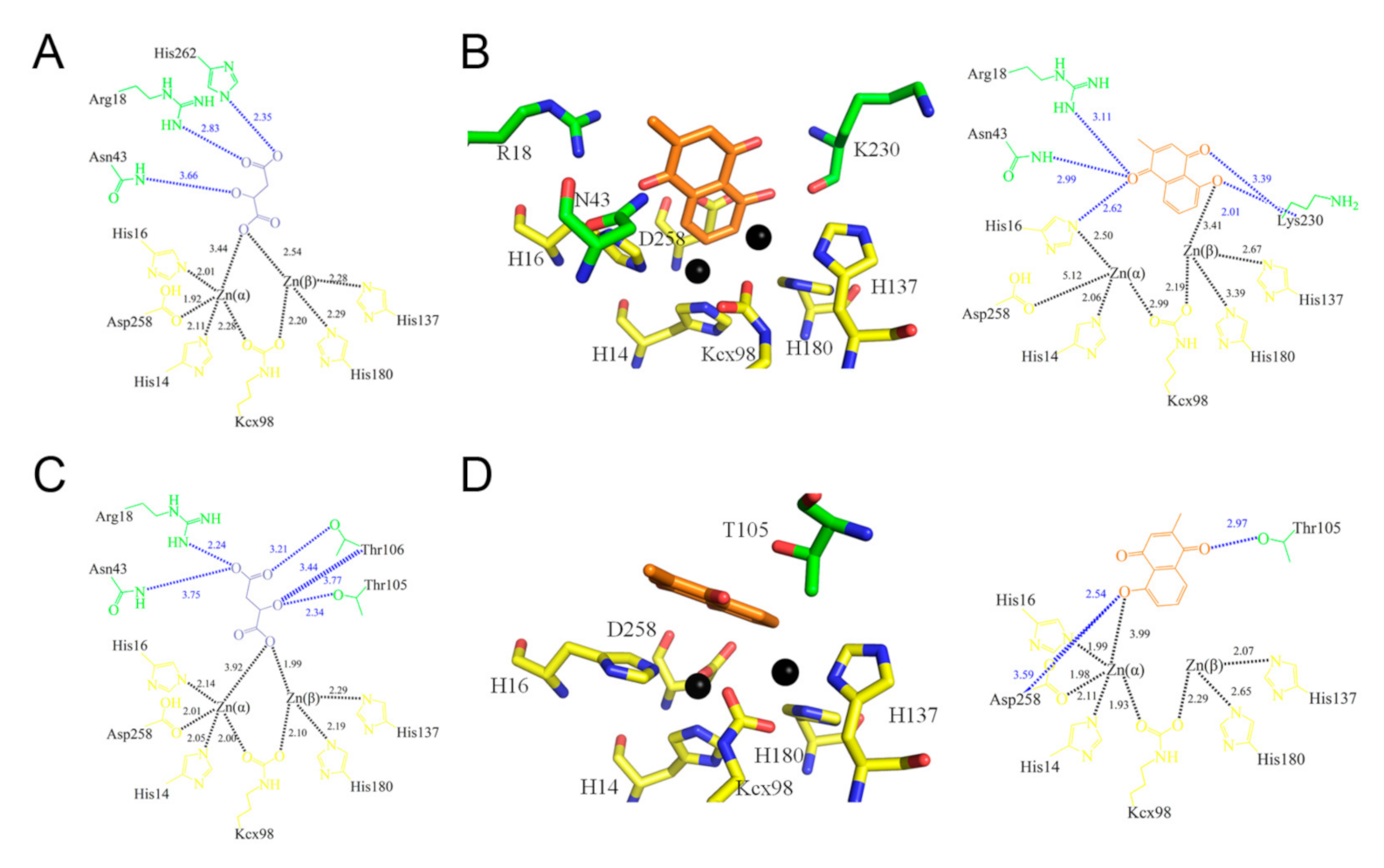
2.3. Crystal Structure of ScDHOase Complexed with PLU
2.4. Identification of PLU as a Competitive Inhibitor of DHOase
2.5. Mutational Analysis of Residues within the Active Site
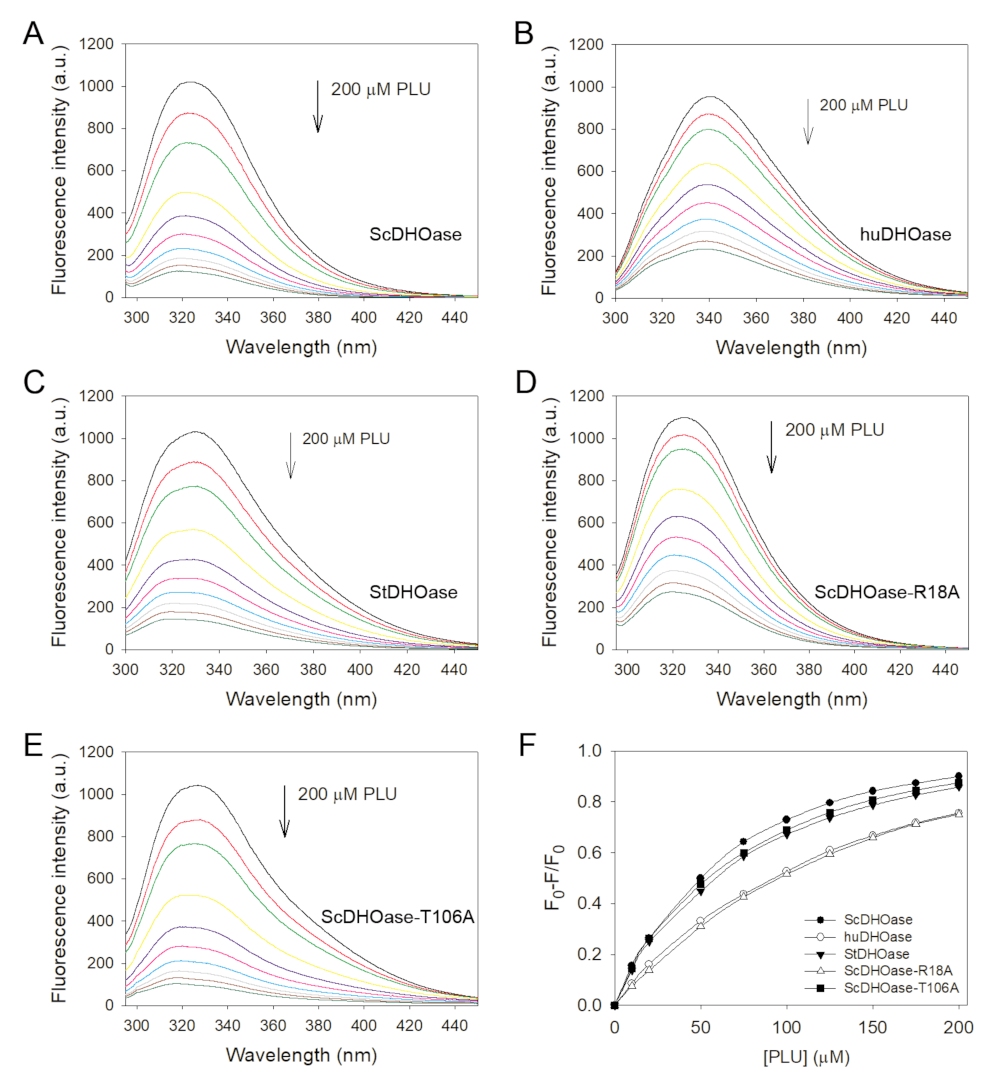
2.6. Binding Specificities of ScDHOase
2.7. Structure-Based Binding Analysis
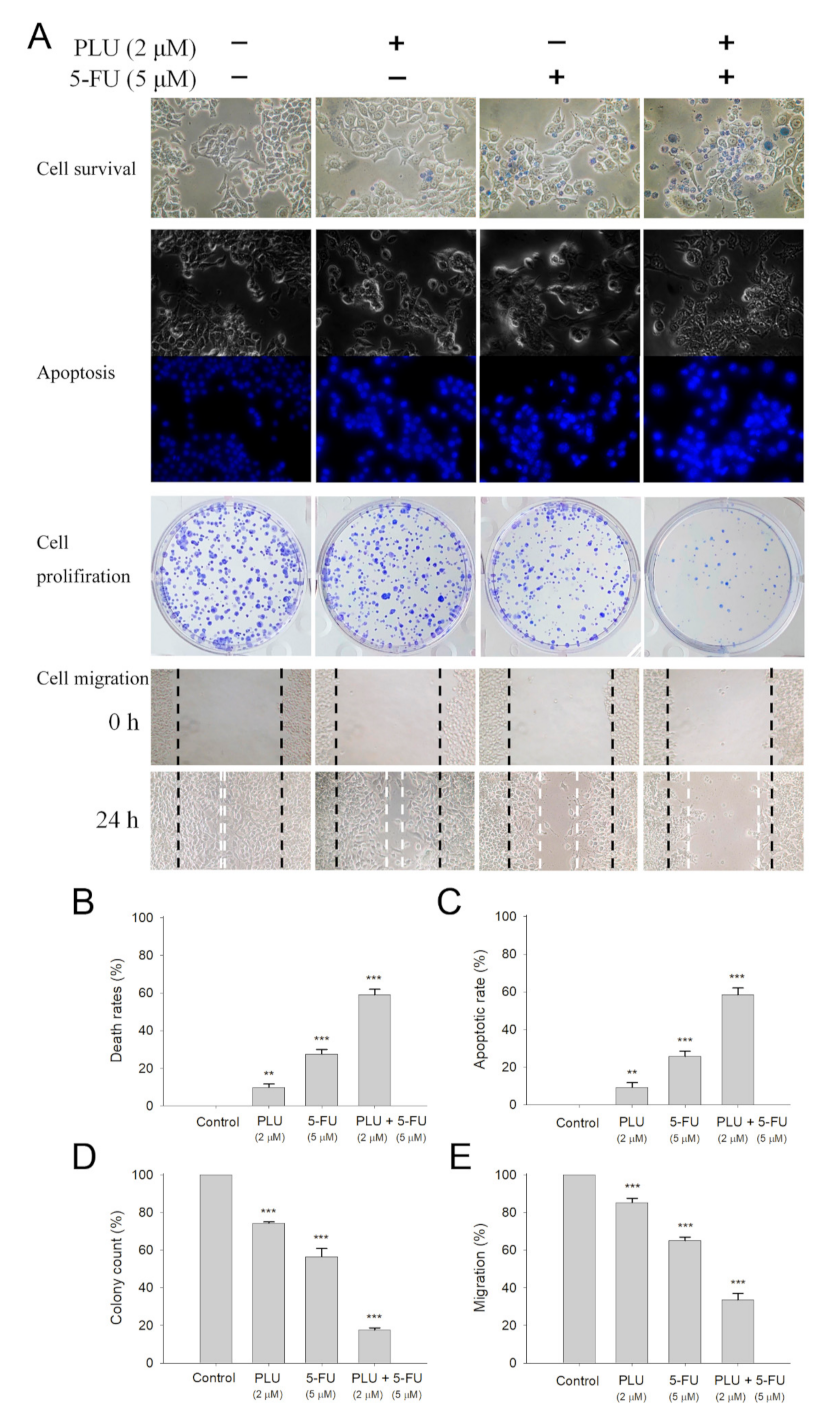
2.8. Anticancer Activity of PLU against 4T1 Cells
2.9. PLU Inhibited Bacterial Growth
2.10. PLU Could Act with 5-FU for Anti-4T1 Cancer Cells
3. Discussion
3.1. Identification of DHOase Inhibited by PLU
3.2. PLU as a Dirty Drug for Multiple Targets
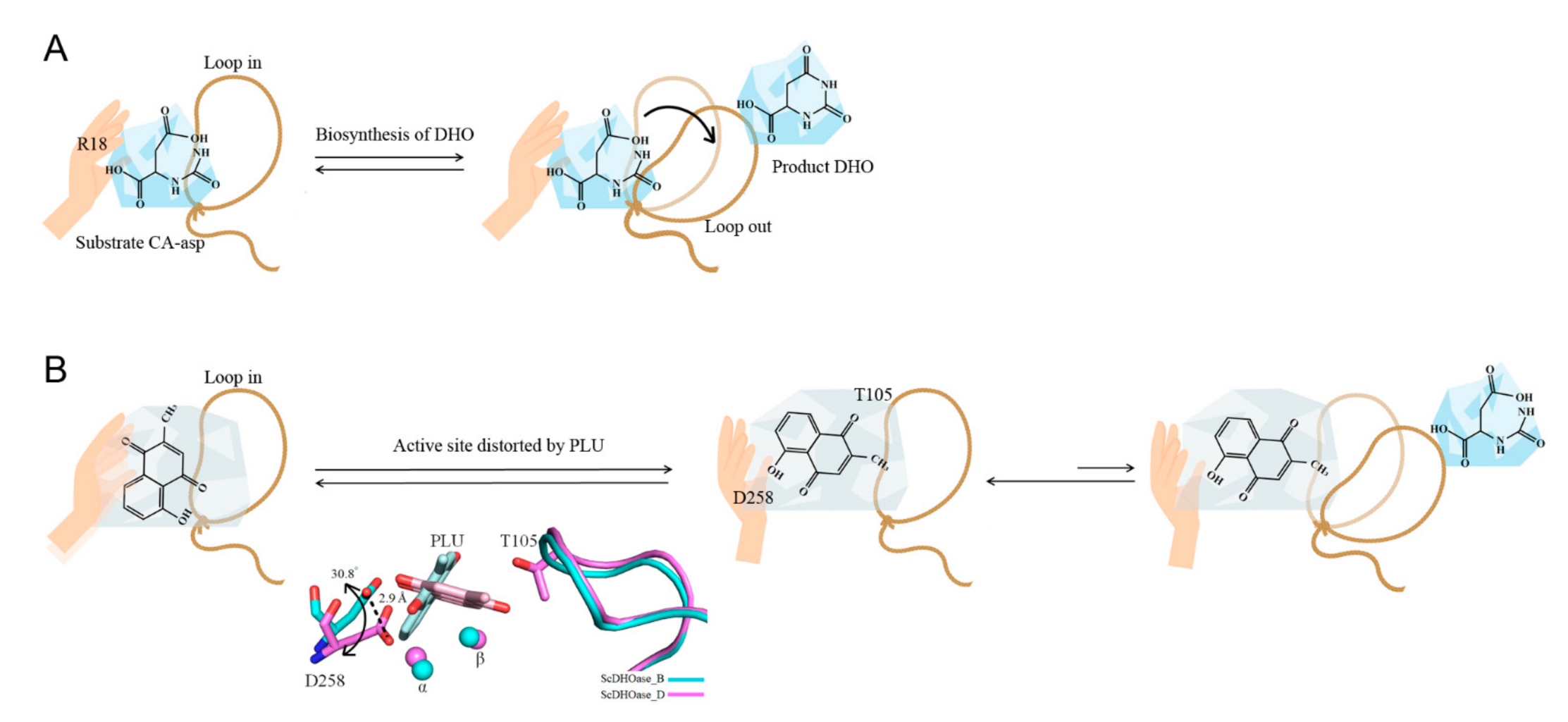
3.3. Loop-in Binding Mode of PLU and Malate
3.4. Dynamic Loop as a Part of the Catalytic Cycle in DHOase and DHPase and as a Drug Target
3.5. Active Site Distorted by PLU
4. Materials and Methods
4.1. Protein Expression and Purification
4.2. Site-Directed Mutagenesis
4.3. Crystallization Experiments
4.4. X-ray Diffraction Data and Structure Determination
4.5. Plant Materials and Extract Preparations
4.6. Gas Chromatography–Mass Spectrometry (GC–MS)
4.7. Enzyme Assay
4.8. Determination of the Dissociation Constant (Kd)
4.9. Trypan Blue Cytotoxicity Assay
4.10. Wound-Healing Assay
4.11. Clonogenic Formation Assay
4.12. Chromatin Condensation Assay
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Ridder, D.A.; Schindeldecker, M.; Weinmann, A.; Berndt, K.; Urbansky, L.; Witzel, H.R.; Heinrich, S.; Roth, W.; Straub, B.K. Key Enzymes in Pyrimidine Synthesis, CAD and CPS1, Predict Prognosis in Hepatocellular Carcinoma. Cancers 2021, 13, 744. [Google Scholar] [CrossRef] [PubMed]
- Verrier, E.R.; Weiss, A.; Bach, C.; Heydmann, L.; Turon-Lagot, V.; Kopp, A.; El Saghire, H.; Crouchet, E.; Pessaux, P.; Garcia, T.; et al. Combined Small Molecule and Loss-of-function Screen Uncovers Estrogen Receptor Alpha and CAD as Host Factors for HDV Infection and Antiviral Targets. Gut 2020, 69, 158–167. [Google Scholar] [CrossRef] [PubMed]
- Del Caño-Ochoa, F.; Ng, B.G.; Abedalthagafi, M.; Almannai, M.; Cohn, R.D.; Costain, G.; Elpeleg, O.; Houlden, H.; Karimiani, E.G.; Liu, P.; et al. Cell-based Analysis of CAD Variants Identifies Individuals Likely to Benefit from Uridine Therapy. Genet. Med. 2020, 22, 1598–1605. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Yang, K.; Wu, Q.; Kim, L.J.Y.; Morton, A.R.; Gimple, R.C.; Prager, B.C.; Shi, Y.; Zhou, W.; Bhargava, S.; et al. Targeting Pyrimidine Synthesis Accentuates Molecular Therapy Response in Glioblastoma Stem Cells. Sci. Transl. Med. 2019, 11, eaau4972. [Google Scholar] [CrossRef]
- Villa, E.; Ali, E.S.; Sahu, U.; Ben-Sahra, I. Cancer Cells Tune the Signaling Pathways to Empower de Novo Synthesis of Nucleotides. Cancers 2019, 11, 688. [Google Scholar] [CrossRef] [Green Version]
- Madak, J.T.; Bankhead, A., 3rd; Cuthbertson, C.R.; Showalter, H.D.; Neamati, N. Revisiting the Role of Dihydroorotate Dehydrogenase as a Therapeutic Target for Cancer. Pharmacol. Ther. 2019, 195, 111–131. [Google Scholar] [CrossRef]
- Garavito, M.F.; Narvaez-Ortiz, H.Y.; Zimmermann, B.H. Pyrimidine Metabolism: Dynamic and Versatile Pathways in Pathogens and Cellular Development. J. Genet. Genomics 2015, 42, 195–205. [Google Scholar] [CrossRef]
- Fung, S.K.; Lok, A.S. Drug insight: Nucleoside and nucleotide analog inhibitors for hepatitis B. Nat. Clin. Pract. Gastroenterol. Hepatol. 2004, 1, 90–97. [Google Scholar] [CrossRef]
- Longley, D.B.; Harkin, D.P.; Johnston, P.G. 5-Fluorouracil: Mechanisms of action and clinical strategies. Nat. Rev. Cancer 2003, 3, 330–338. [Google Scholar] [CrossRef]
- Del Cano-Ochoa, F.; Moreno-Morcillo, M.; Ramon-Maiques, S. CAD, A Multienzymatic Protein at the Head of de Novo Pyrimidine Biosynthesis. Subcell. Biochem. 2019, 93, 505–538. [Google Scholar]
- Samant, S.; Lee, H.; Ghassemi, M.; Chen, J.; Cook, J.L.; Mankin, A.S.; Neyfakh, A.A. Nucleotide biosynthesis is critical for growth of bacteria in human blood. PLoS Pathog. 2008, 4, e37. [Google Scholar] [CrossRef]
- Evans, D.R.; Guy, H.I. Mammalian pyrimidine biosynthesis: Fresh insights into an ancient pathway. J. Biol. Chem. 2004, 279, 33035–33038. [Google Scholar] [CrossRef] [Green Version]
- Christopherson, R.I.; Lyons, S.D.; Wilson, P.K. Inhibitors of de novo nucleotide biosynthesis as drugs. Acc. Chem. Res. 2002, 35, 961–971. [Google Scholar] [CrossRef]
- Zhu, J.; Thompson, C.B. Metabolic regulation of cell growth and proliferation. Nat. Rev. Mol. Cell. Biol. 2019, 20, 436–450. [Google Scholar] [CrossRef]
- Keshet, R.; Szlosarek, P.; Carracedo, A.; Erez, A. Rewiring urea cycle metabolism in cancer to support anabolism. Nat. Rev. Cancer 2018, 18, 634–645. [Google Scholar] [CrossRef]
- Lee, J.S.; Adler, L.; Karathia, H.; Carmel, N.; Rabinovich, S.; Auslander, N.; Keshet, R.; Stettner, N.; Silberman, A.; Agemy, L.; et al. Urea Cycle Dysregulation Generates Clinically Relevant Genomic and Biochemical Signatures. Cell 2018, 174, 1559–1570. [Google Scholar] [CrossRef] [Green Version]
- Lv, Y.; Wang, X.; Li, X.; Xu, G.; Bai, Y.; Wu, J.; Piao, Y.; Shi, Y.; Xiang, R.; Wang, L. Nucleotide de novo synthesis increases breast cancer stemness and metastasis via cGMP-PKG-MAPK signaling pathway. PLoS Biol. 2020, 18, e3000872. [Google Scholar] [CrossRef]
- Aoki, T.; Weber, G. Carbamoyl phosphate synthetase (glutamine-hydrolyzing): Increased activity in cancer cells. Science 1981, 212, 463–465. [Google Scholar] [CrossRef]
- Washabaugh, M.W.; Collins, K.D. Dihydroorotase from Escherichia coli. Purification and characterization. J. Biol. Chem. 1984, 259, 3293–3298. [Google Scholar] [CrossRef]
- Souciet, J.L.; Nagy, M.; Le Gouar, M.; Lacroute, F.; Potier, S. Organization of the yeast URA2 gene: Identification of a defective dihydroorotase-like domain in the multifunctional carbamoylphosphate synthetase-aspartate transcarbamylase complex. Gene 1989, 79, 59–70. [Google Scholar] [CrossRef]
- Rice, A.J.; Lei, H.; Santarsiero, B.D.; Lee, H.; Johnson, M.E. Ca-asp bound X-ray structure and inhibition of Bacillus anthracis dihydroorotase (DHOase). Bioorg. Med. Chem. 2016, 24, 4536–4543. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.; Chan, C.W.; Graham, S.C.; Christopherson, R.I.; Guss, J.M.; Maher, M.J. Structures of ligand-free and inhibitor complexes of dihydroorotase from Escherichia coli: Implications for loop movement in inhibitor design. J. Mol. Biol. 2007, 370, 812–825. [Google Scholar] [CrossRef]
- Del Cano-Ochoa, F.; Grande-Garcia, A.; Reverte-Lopez, M.; D’Abramo, M.; Ramon-Maiques, S. Characterization of the catalytic flexible loop in the dihydroorotase domain of the human multi-enzymatic protein CAD. J. Biol. Chem. 2018, 293, 18903–18913. [Google Scholar] [CrossRef] [Green Version]
- Grande-Garcia, A.; Lallous, N.; Diaz-Tejada, C.; Ramon-Maiques, S. Structure, functional characterization, and evolution of the dihydroorotase domain of human CAD. Structure 2014, 22, 185–198. [Google Scholar] [CrossRef] [Green Version]
- Krishnaswamy, M.; Purushothaman, K.K. Plumbagin: A study of its anticancer, antibacterial & antifungal properties. Indian J. Exp. Biol. 1980, 18, 876–877. [Google Scholar]
- Tripathi, S.K.; Panda, M.; Biswal, B.K. Emerging role of plumbagin: Cytotoxic potential and pharmaceutical relevance towards cancer therapy. Food Chem. Toxicol. 2019, 125, 566–582. [Google Scholar] [CrossRef]
- Kang, C.G.; Im, E.; Lee, H.J.; Lee, E.O. Plumbagin reduces osteopontin-induced invasion through inhibiting the Rho-associated kinase signaling pathway in A549 cells and suppresses osteopontin-induced lung metastasis in BalB/c mice. Bioorg. Med. Chem. Lett. 2017, 27, 1914–1918. [Google Scholar] [CrossRef]
- Guan, H.H.; Huang, Y.H.; Lin, E.S.; Chen, C.J.; Huang, C.Y. Structural basis for the interaction modes of dihydroorotase with the anticancer drugs 5-fluorouracil and 5-aminouracil. Biochem. Biophys. Res. Commun. 2021, 551, 33–37. [Google Scholar] [CrossRef]
- Rathod, P.K.; Khatri, A.; Hubbert, T.; Milhous, W.K. Selective activity of 5-fluoroorotic acid against Plasmodium falciparum in vitro. Antimicrob. Agents Chemother. 1989, 33, 1090–1094. [Google Scholar] [CrossRef] [Green Version]
- Huang, K.F.; Hsu, H.L.; Karim, S.; Wang, A.H. Structural and functional analyses of a glutaminyl cyclase from Ixodes scapularis reveal metal-independent catalysis and inhibitor binding. Acta Crystallogr. D Biol. Crystallogr. 2014, 70, 789–801. [Google Scholar] [CrossRef]
- Hill, A. Optimizing HIV treatment. Curr. Opin. HIV AIDS 2013, 8, 34–40. [Google Scholar] [CrossRef] [PubMed]
- De, U.; Son, J.Y.; Jeon, Y.; Ha, S.Y.; Park, Y.J.; Yoon, S.; Ha, K.T.; Choi, W.S.; Lee, B.M.; Kim, I.S.; et al. Plumbagin from a tropical pitcher plant (Nepenthes alata Blanco) induces apoptotic cell death via a p53-dependent pathway in MCF-7 human breast cancer cells. Food Chem. Toxicol. 2019, 123, 492–500. [Google Scholar] [CrossRef] [PubMed]
- Pulaski, B.A.; Ostrand-Rosenberg, S. Mouse 4T1 breast tumor model. Curr. Protoc. Immunol. 2001. Chapter 20, Unit 20.22. [Google Scholar] [CrossRef] [PubMed]
- Yoneda, T.; Michigami, T.; Yi, B.; Williams, P.J.; Niewolna, M.; Hiraga, T. Actions of bisphosphonate on bone metastasis in animal models of breast carcinoma. Cancer 2000, 88, 2979–2988. [Google Scholar] [CrossRef]
- Heppner, G.H.; Miller, F.R.; Shekhar, P.M. Nontransgenic models of breast cancer. Breast Cancer Res. 2000, 2, 331–334. [Google Scholar] [CrossRef] [Green Version]
- Sultanli, S.; Ghumnani, S.; Ashma, R.; Kubatzky, K.F. Plumbagin, a Biomolecule with (Anti)Osteoclastic Properties. Int. J. Mol. Sci. 2021, 22, 2779. [Google Scholar] [CrossRef]
- Roy, A. Plumbagin: A Potential Anti-cancer Compound. Mini Rev. Med. Chem. 2021, 21, 731–737. [Google Scholar] [CrossRef]
- Huang, C.Y. Structure, catalytic mechanism, posttranslational lysine carbamylation, and inhibition of dihydropyrimidinases. Adv. Protein Chem. Struct. Biol. 2020, 122, 63–96. [Google Scholar]
- Huang, Y.H.; Lien, Y.; Chen, J.H.; Lin, E.S.; Huang, C.Y. Identification and characterization of dihydropyrimidinase inhibited by plumbagin isolated from Nepenthes miranda extract. Biochimie 2020, 171–172, 124–135. [Google Scholar] [CrossRef]
- Thoden, J.B.; Phillips, G.N., Jr.; Neal, T.M.; Raushel, F.M.; Holden, H.M. Molecular structure of dihydroorotase: A paradigm for catalysis through the use of a binuclear metal center. Biochemistry 2001, 40, 6989–6997. [Google Scholar] [CrossRef]
- Gerlt, J.A.; Babbitt, P.C. Divergent evolution of enzymatic function: Mechanistically diverse superfamilies and functionally distinct suprafamilies. Annu. Rev. Biochem. 2001, 70, 209–246. [Google Scholar] [CrossRef]
- Cheng, J.H.; Huang, C.C.; Huang, Y.H.; Huang, C.Y. Structural basis for pH-dependent oligomerization of dihydropyrimidinase from Pseudomonas aeruginosa PAO1. Bioinorg. Chem. Appl. 2018, 2018, 9564391. [Google Scholar] [CrossRef] [Green Version]
- Tzeng, C.T.; Huang, Y.H.; Huang, C.Y. Crystal structure of dihydropyrimidinase from Pseudomonas aeruginosa PAO1: Insights into the molecular basis of formation of a dimer. Biochem. Biophys. Res. Commun. 2016, 478, 1449–1455. [Google Scholar] [CrossRef]
- Huang, C.Y. Inhibition of a putative dihydropyrimidinase from Pseudomonas aeruginosa PAO1 by flavonoids and substrates of cyclic amidohydrolases. PLoS ONE 2015, 10, e0127634. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, Y.C.; Chen, M.C.; Hsu, C.C.; Chan, S.I.; Yang, Y.S.; Chen, C.J. Crystal structures of vertebrate dihydropyrimidinase and complexes from Tetraodon nigroviridis with lysine carbamylation: Metal and structural requirements for post-translational modification and function. J. Biol. Chem. 2013, 288, 30645–30658. [Google Scholar] [CrossRef] [Green Version]
- Abendroth, J.; Niefind, K.; Schomburg, D. X-ray structure of a dihydropyrimidinase from Thermus sp. at 1.3 A resolution. J. Mol. Biol. 2002, 320, 143–156. [Google Scholar] [CrossRef]
- Ho, Y.Y.; Huang, Y.H.; Huang, C.Y. Chemical rescue of the post-translationally carboxylated lysine mutant of allantoinase and dihydroorotase by metal ions and short-chain carboxylic acids. Amino Acids 2013, 44, 1181–1191. [Google Scholar] [CrossRef]
- Ho, Y.Y.; Hsieh, H.C.; Huang, C.Y. Biochemical characterization of allantoinase from Escherichia coli BL21. Protein J. 2011, 30, 384–394. [Google Scholar] [CrossRef]
- Kim, K.; Kim, M.I.; Chung, J.; Ahn, J.H.; Rhee, S. Crystal structure of metal-dependent allantoinase from Escherichia coli. J. Mol. Biol. 2009, 387, 1067–1074. [Google Scholar] [CrossRef]
- Huang, C.Y.; Hsu, C.C.; Chen, M.C.; Yang, Y.S. Effect of metal binding and posttranslational lysine carboxylation on the activity of recombinant hydantoinase. J. Biol. Inorg. Chem. 2009, 14, 111–121. [Google Scholar] [CrossRef]
- Xu, Z.; Liu, Y.; Yang, Y.; Jiang, W.; Arnold, E.; Ding, J. Crystal structure of D-hydantoinase from Burkholderia pickettii at a resolution of 2.7 Angstroms: Insights into the molecular basis of enzyme thermostability. J. Bacteriol. 2003, 185, 4038–4049. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.Y.; Yang, Y.S. A novel cold-adapted imidase from fish Oreochromis niloticus that catalyzes hydrolysis of maleimide. Biochem. Biophys. Res. Commun. 2003, 312, 467–472. [Google Scholar] [CrossRef]
- Huang, C.Y.; Yang, Y.S. The role of metal on imide hydrolysis: Metal content and pH profiles of metal ion-replaced mammalian imidase. Biochem. Biophys. Res. Commun. 2002, 297, 1027–1032. [Google Scholar] [CrossRef]
- Yang, Y.S.; Ramaswamy, S.; Jakoby, W.B. Rat liver imidase. J. Biol. Chem. 1993, 268, 10870–10875. [Google Scholar] [CrossRef]
- Peng, W.F.; Huang, C.Y. Allantoinase and dihydroorotase binding and inhibition by flavonols and the substrates of cyclic amidohydrolases. Biochimie 2014, 101, 113–122. [Google Scholar] [CrossRef]
- Cheng, J.H.; Huang, Y.H.; Lin, J.J.; Huang, C.Y. Crystal structures of monometallic dihydropyrimidinase and the human dihydroorotase domain K1556A mutant reveal no lysine carbamylation within the active site. Biochem. Biophys. Res. Commun. 2018, 505, 439–444. [Google Scholar] [CrossRef]
- Cheon, Y.H.; Kim, H.S.; Han, K.H.; Abendroth, J.; Niefind, K.; Schomburg, D.; Wang, J.; Kim, Y. Crystal structure of D-hydantoinase from Bacillus stearothermophilus: Insight into the stereochemistry of enantioselectivity. Biochemistry 2002, 41, 9410–9417. [Google Scholar] [CrossRef]
- Gojkovic, Z.; Rislund, L.; Andersen, B.; Sandrini, M.P.; Cook, P.F.; Schnackerz, K.D.; Piskur, J. Dihydropyrimidine amidohydrolases and dihydroorotases share the same origin and several enzymatic properties. Nucleic Acids Res. 2003, 31, 1683–1692. [Google Scholar] [CrossRef] [Green Version]
- Lohkamp, B.; Andersen, B.; Piskur, J.; Dobritzsch, D. The crystal structures of dihydropyrimidinases reaffirm the close relationship between cyclic amidohydrolases and explain their substrate specificity. J. Biol. Chem. 2006, 281, 13762–13776. [Google Scholar] [CrossRef] [Green Version]
- Porter, T.N.; Li, Y.; Raushel, F.M. Mechanism of the dihydroorotase reaction. Biochemistry 2004, 43, 16285–16292. [Google Scholar] [CrossRef]
- Huang, Y.H.; Huang, C.Y. Creation of a putative third metal binding site in type II dihydroorotases significantly enhances enzyme activity. Protein Pept. Lett. 2015, 22, 1117–1122. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.C.; Tsau, H.W.; Chen, W.T.; Huang, C.Y. Identification and characterization of a putative dihydroorotase, KPN01074, from Klebsiella pneumoniae. Protein J. 2010, 29, 445–452. [Google Scholar] [CrossRef] [PubMed]
- Guyonvarch, A.; Nguyen-Juilleret, M.; Hubert, J.C.; Lacroute, F. Structure of the Saccharomyces cerevisiae URA4 gene encoding dihydroorotase. Mol. Gen. Genet. 1988, 212, 134–141. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.H.; Chen, I.C.; Huang, C.Y. Characterization of an SSB-dT25 complex: Structural insights into the S-shaped ssDNA binding conformation. RSC Adv. 2019, 9, 40388–40396. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.H.; Huang, C.Y. SAAV2152 is a single-stranded DNA binding protein: The third SSB in Staphylococcus aureus. Oncotarget 2018, 9, 20239–20254. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.H.; Guan, H.H.; Chen, C.J.; Huang, C.Y. Staphylococcus aureus single-stranded DNA-binding protein SsbA can bind but cannot stimulate PriA helicase. PLoS ONE 2017, 12, e0182060. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.H.; Lien, Y.; Huang, C.C.; Huang, C.Y. Characterization of Staphylococcus aureus primosomal DnaD protein: Highly conserved C-terminal region is crucial for ssDNA and PriA helicase binding but not for DnaA protein-binding and self-tetramerization. PLoS ONE 2016, 11, e0157593. [Google Scholar] [CrossRef]
- Huang, C.Y.; Hsu, C.H.; Sun, Y.J.; Wu, H.N.; Hsiao, C.D. Complexed crystal structure of replication restart primosome protein PriB reveals a novel single-stranded DNA-binding mode. Nucleic Acids Res. 2006, 34, 3878–3886. [Google Scholar] [CrossRef] [Green Version]
- Otwinowski, Z.; Minor, W. Processing of X-ray Diffraction Data Collected in Oscillation Mode. Methods Enzymol. 1997, 276, 307–326. [Google Scholar]
- Kabsch, W. XDS. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 125–132. [Google Scholar] [CrossRef] [Green Version]
- Terwilliger, T.C.; Adams, P.D.; Read, R.J.; McCoy, A.J.; Moriarty, N.W.; Grosse-Kunstleve, R.W.; Afonine, P.V.; Zwart, P.H.; Hung, L.W. Decision-making in structure solution using Bayesian estimates of map quality: The PHENIX AutoSol wizard. Acta Crystallogr. D Biol. Crystallogr. 2009, 65, 582–601. [Google Scholar] [CrossRef] [Green Version]
- Lebedev, A.A.; Young, P.; Isupov, M.N.; Moroz, O.V.; Vagin, A.A.; Murshudov, G.N. JLigand: A graphical tool for the CCP4 template-restraint library. Acta Crystallogr. D Biol. Crystallogr. 2012, 68, 431–440. [Google Scholar] [CrossRef] [Green Version]
- Terwilliger, T.C.; Grosse-Kunstleve, R.W.; Afonine, P.V.; Moriarty, N.W.; Zwart, P.H.; Hung, L.W.; Read, R.J.; Adams, P.D. Iterative model building, structure refinement and density modification with the PHENIX AutoBuild wizard. Acta Crystallogr. D Biol. Crystallogr. 2008, 64, 61–69. [Google Scholar] [CrossRef] [Green Version]
- Winn, M.D.; Ballard, C.C.; Cowtan, K.D.; Dodson, E.J.; Emsley, P.; Evans, P.R.; Keegan, R.M.; Krissinel, E.B.; Leslie, A.G.; McCoy, A.; et al. Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 2011, 67, 235–242. [Google Scholar] [CrossRef] [Green Version]
- Chen, V.B.; Arendall, W.B., 3rd; Headd, J.J.; Keedy, D.A.; Immormino, R.M.; Kapral, G.J.; Murray, L.W.; Richardson, J.S.; Richardson, D.C. MolProbity: All-atom structure validation for macromolecular crystallography. Acta Crystallogr. D Biol. Crystallogr. 2010, 66, 12–21. [Google Scholar] [CrossRef] [Green Version]
- Huang, Y.H.; Ning, Z.J.; Huang, C.Y. Crystal structure of dihydropyrimidinase in complex with anticancer drug 5-fluorouracil. Biochem. Biophys. Res. Commun. 2019, 519, 160–165. [Google Scholar] [CrossRef]
- Lin, H.H.; Huang, C.Y. Characterization of flavonol inhibition of DnaB helicase: Real-time monitoring, structural modeling, and proposed mechanism. J. Biomed. Biotechnol. 2012, 2012, 735368. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.C.; Huang, C.Y. Inhibition of Klebsiella pneumoniae DnaB helicase by the flavonol galangin. Protein J. 2011, 30, 59–65. [Google Scholar] [CrossRef]
- Strober, W. Trypan blue exclusion test of cell viability. Curr. Protoc. Immunol. 2001, 111, A3.B.1–A3.B.3. [Google Scholar]
- Liang, C.C.; Park, A.Y.; Guan, J.L. In vitro scratch assay: A convenient and inexpensive method for analysis of cell migration in vitro. Nat. Protoc. 2007, 2, 329–333. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.H.; Yang, W.L.; Lin, K.T.; Liu, C.H.; Liu, Y.W.; Huang, K.W.; Chang, P.M.; Lai, J.M.; Hsu, C.N.; Chao, K.M.; et al. Gene expression-based chemical genomics identifies potential therapeutic drugs in hepatocellular carcinoma. PLoS ONE 2011, 6, e27186. [Google Scholar] [CrossRef] [Green Version]
- Larsson, R.; Nygren, P. A rapid fluorometric method for semiautomated determination of cytotoxicity and cellular proliferation of human tumor cell lines in microculture. Anticancer Res. 1989, 9, 1111–1119. [Google Scholar]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data collection | |
| Crystal | PLU–ScDHOase |
| Wavelength (Å) | 0.9 |
| Resolution (Å) | 45.9–3.60 |
| Space group | P21 |
| Cell dimension a, b, c (Å) β (°) | 85.24, 88.36, 103.42 95.16 |
| Redundancy | 1.2 (1.2) |
| Completeness (%) | 94.54 (83.76) |
| <I/σI> | 5.4 (1.3) |
| CC1/2 | 0.942 (0.78) |
| Refinement | |
| Resolution (Å) | 45.9–3.60 |
| No. reflections | 16975 |
| Rwork/Rfree | 0.26/0.31 |
| No. atoms | |
| Ligands | 54 |
| Protein | 1460 |
| Zinc | 8 |
| Water | 0 |
| r.m.s deviations | |
| Bond lengths (Å) | 0.002 |
| Bond angles (°) | 0.54 |
| Ramachandran plot | |
| Favored (%) | 93.94 |
| Allowed (%) | 5.71 |
| Outliers (%) | 0.35 |
| PDB entry | 7CA1 |
| ScDHOase | Specific Activity (μmol/mg/min) | Relative Activity (100%) | Corresponding Residue in CAD |
|---|---|---|---|
| Wild type | 68 ± 4 | 100 | huDHOase |
| H14A | <10−3 | 0 | H1471 (metal binding) |
| H16A | <10−3 | 0 | H1473 (metal binding) |
| R18A | <10−3 | 0 | R1475 (substrate binding) |
| N43A | <10−3 | 0 | N1505 (substrate binding) |
| K98A | <10−3 | 0 | K1556 (metal binding) |
| T105A | 0.57 ± 0.04 | 0.8 | T1562 (dynamic loop) |
| T106A | 0.15 ± 0.01 | 0.2 | F1563 (dynamic loop) |
| H137A | <10−3 | 0 | H1590 (metal binding) |
| H180A | <10−3 | 0 | H1614 (metal binding) |
| D258A | <10−3 | 0 | D1686 (metal binding and catalysis) |
| D258E | 0.015 ± 0.003 | 0.02 | D1686 (metal binding and catalysis) |
| H262A | <10−3 | 0 | H1690 (substrate binding) |
| DHOase | λmax (nm) | λem Shift (nm) | Quenching (%) | Kd Value (μM) |
|---|---|---|---|---|
| ScDHOase | from 324 to 317.5 | 6.5 | 87.7 | 64.8 ± 1.6 |
| ScDHOase–R18A | from 324.5 to 320.5 | 4.0 | 75.1 | 181.0 ± 5.9 |
| ScDHOase–T106A | from 328 to 321.5 | 6.5 | 90.0 | 70.8 ± 2.6 |
| huDHOase | from 340 to 338.5 | 1.5 | 75.6 | 150.9 ± 4.1 |
| StDHOase | from 330.5 to 319.5 | 11.0 | 85.9 | 80.3 ± 2.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Guan, H.-H.; Huang, Y.-H.; Lin, E.-S.; Chen, C.-J.; Huang, C.-Y. Plumbagin, a Natural Product with Potent Anticancer Activities, Binds to and Inhibits Dihydroorotase, a Key Enzyme in Pyrimidine Biosynthesis. Int. J. Mol. Sci. 2021, 22, 6861. https://doi.org/10.3390/ijms22136861
Guan H-H, Huang Y-H, Lin E-S, Chen C-J, Huang C-Y. Plumbagin, a Natural Product with Potent Anticancer Activities, Binds to and Inhibits Dihydroorotase, a Key Enzyme in Pyrimidine Biosynthesis. International Journal of Molecular Sciences. 2021; 22(13):6861. https://doi.org/10.3390/ijms22136861
Chicago/Turabian StyleGuan, Hong-Hsiang, Yen-Hua Huang, En-Shyh Lin, Chun-Jung Chen, and Cheng-Yang Huang. 2021. "Plumbagin, a Natural Product with Potent Anticancer Activities, Binds to and Inhibits Dihydroorotase, a Key Enzyme in Pyrimidine Biosynthesis" International Journal of Molecular Sciences 22, no. 13: 6861. https://doi.org/10.3390/ijms22136861