
Pharmacological Characterisation of Pseudocerastes and Eristicophis Viper Venoms Reveal Anticancer (Melanoma) Properties and a Potentially Novel Mode of Fibrinogenolysis

 ,
,  , , , ,
, , , ,  ,
,  and
and
Abstract
:1. Introduction
2. Results and Discussion
2.1. Venom Profile
2.1.1. Proteomics
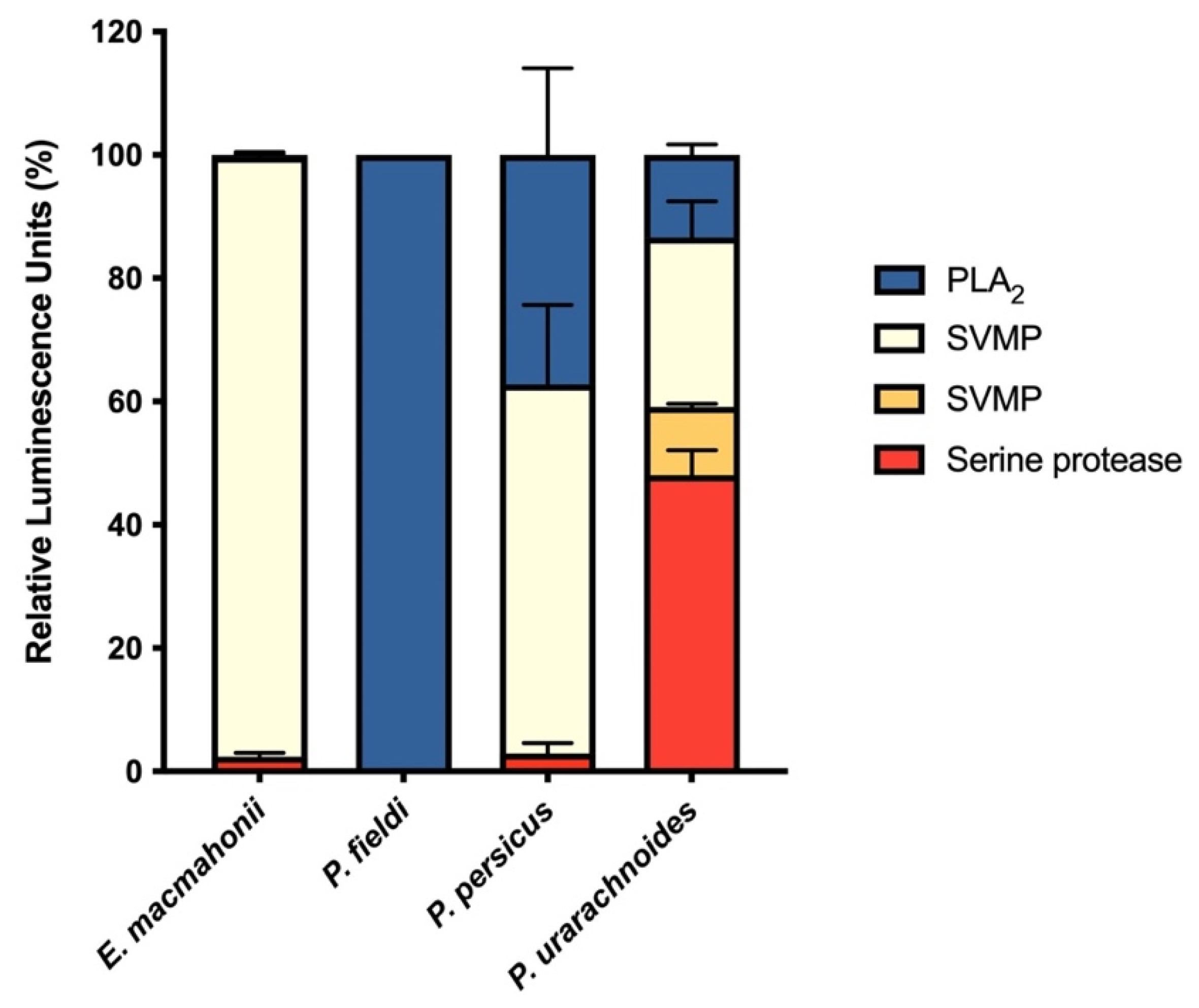
2.1.2. Protease Activity
2.2. Cytotoxicity
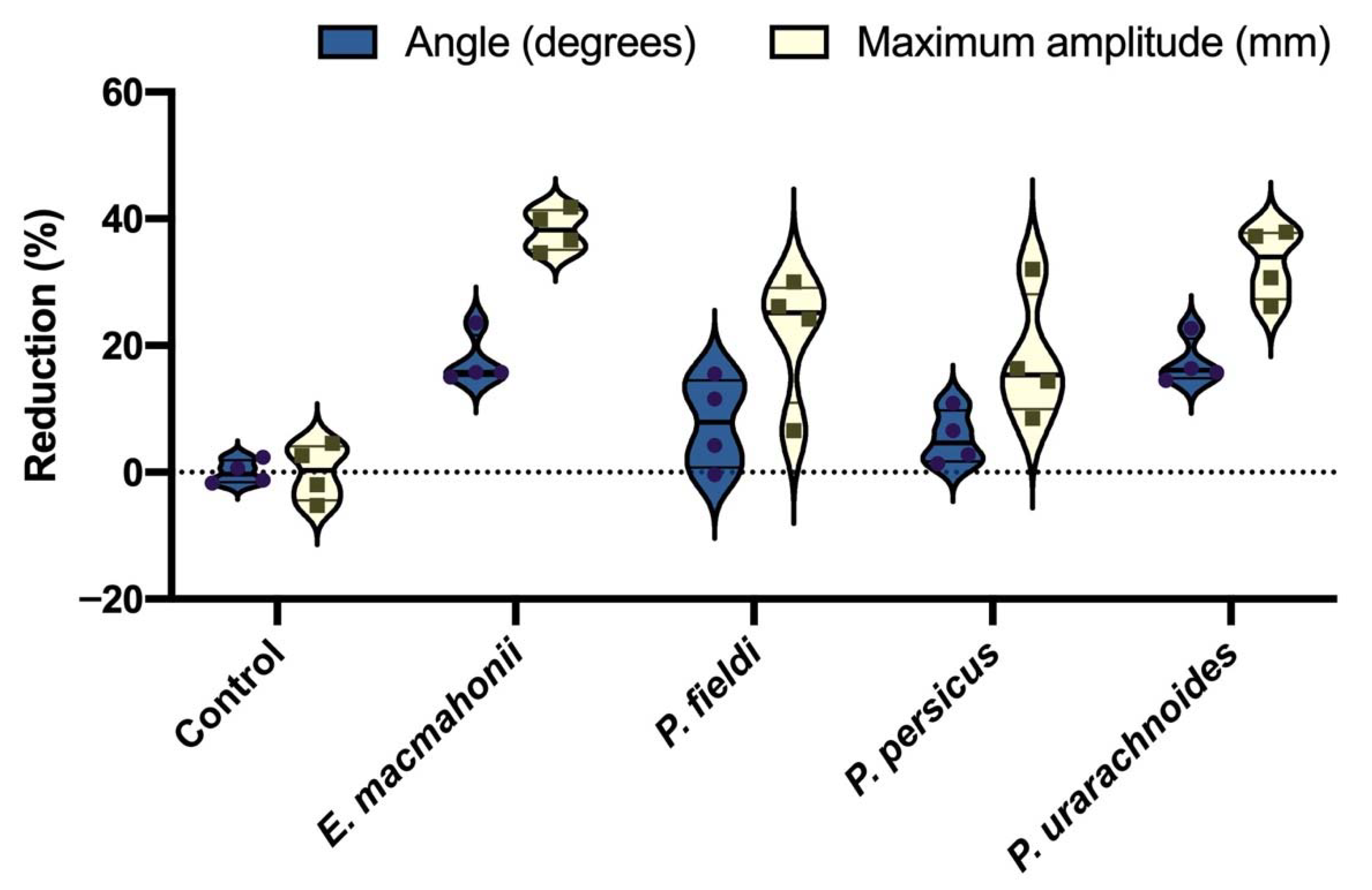
2.3. Coagulant Activity
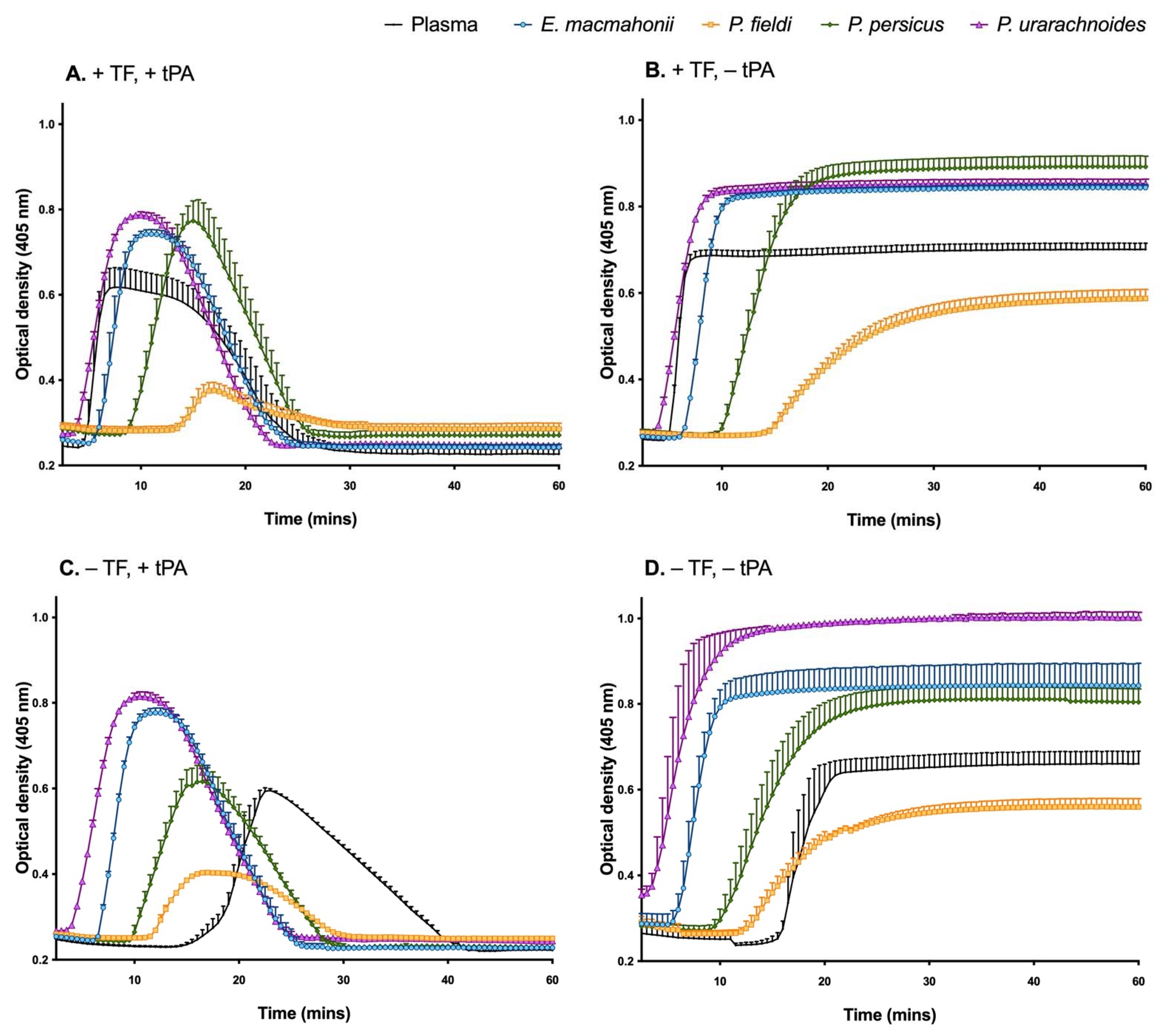
2.3.1. Coagulation and Fibrinolysis
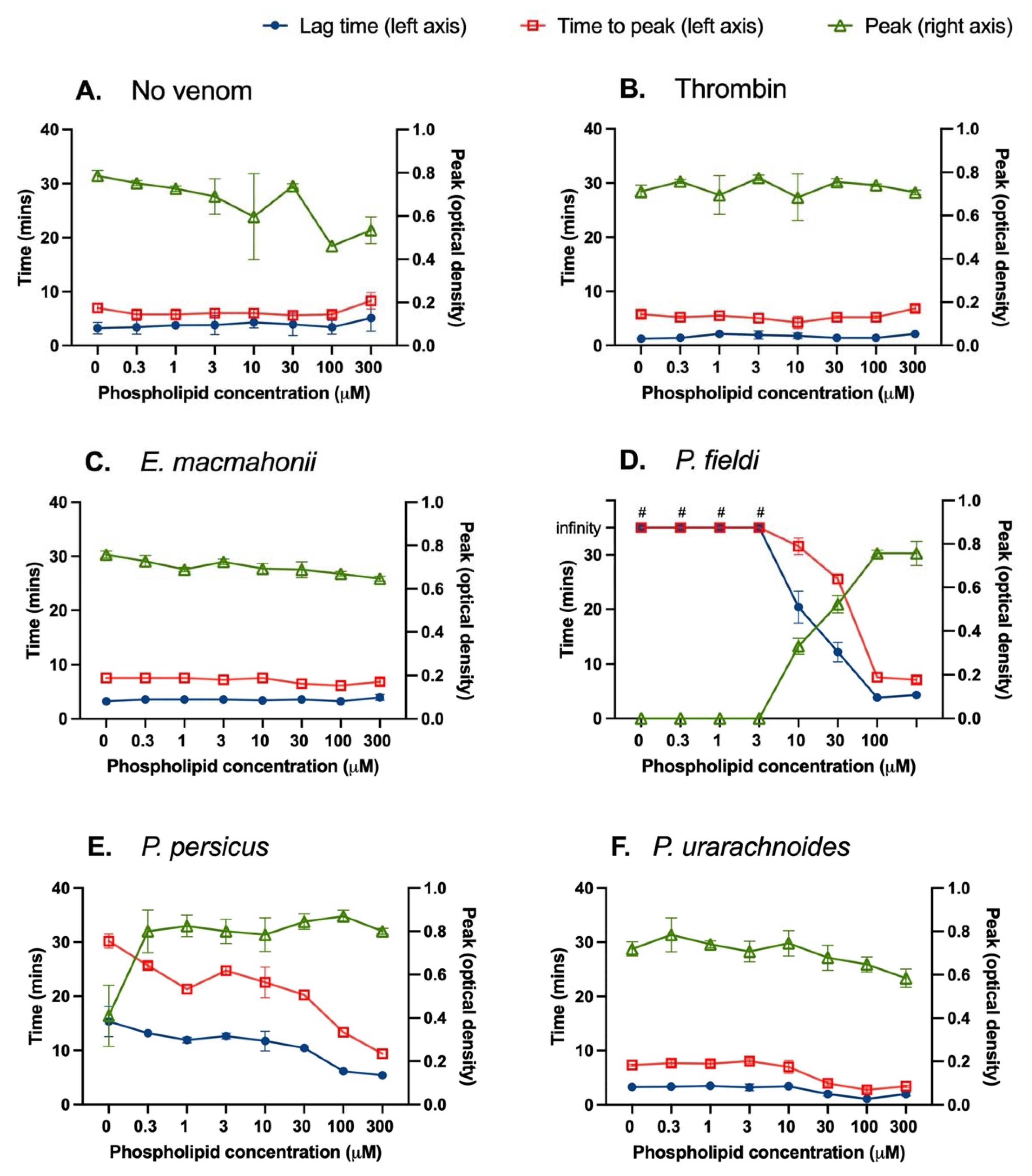
2.3.2. Phospholipid Interactions
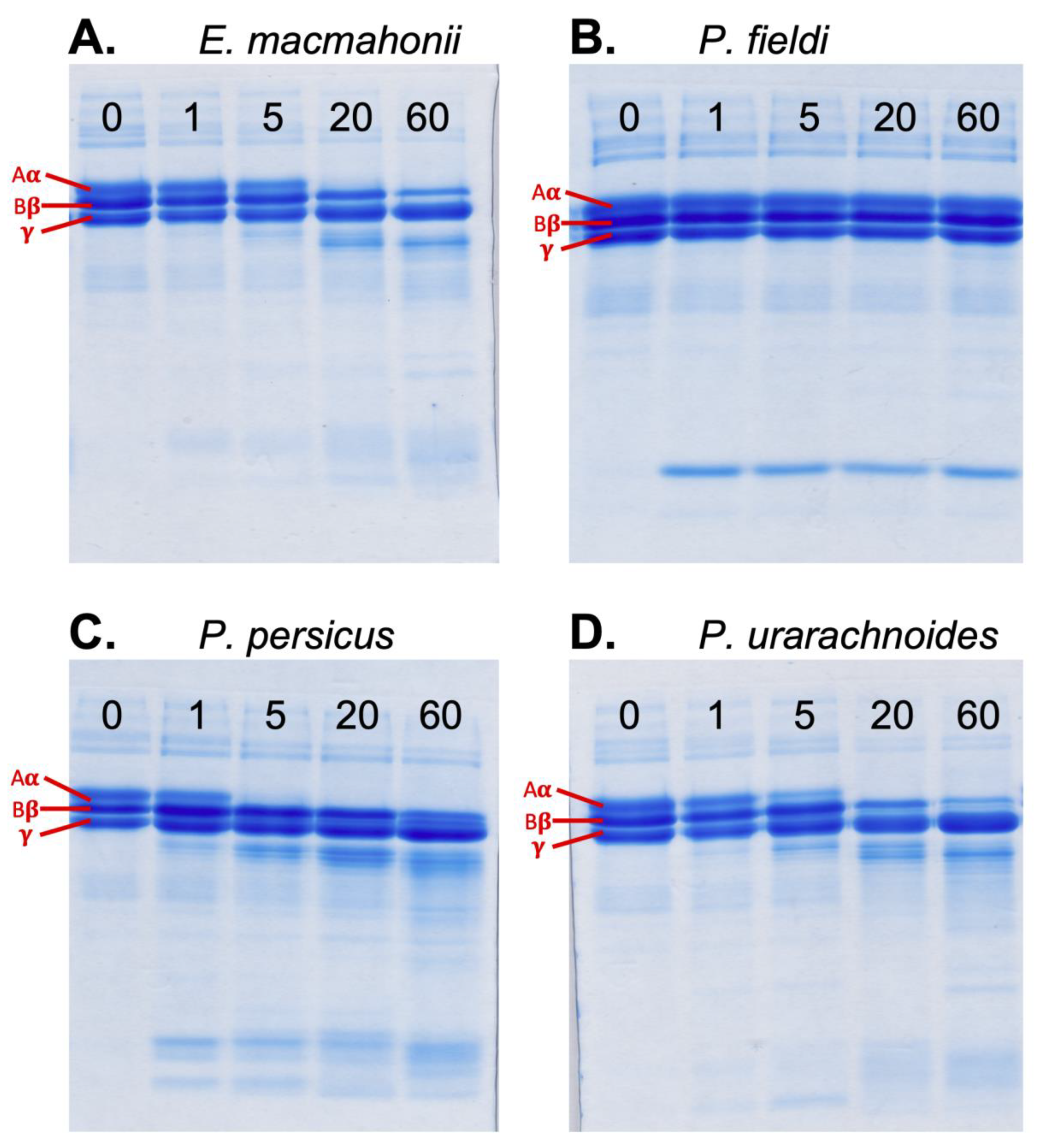
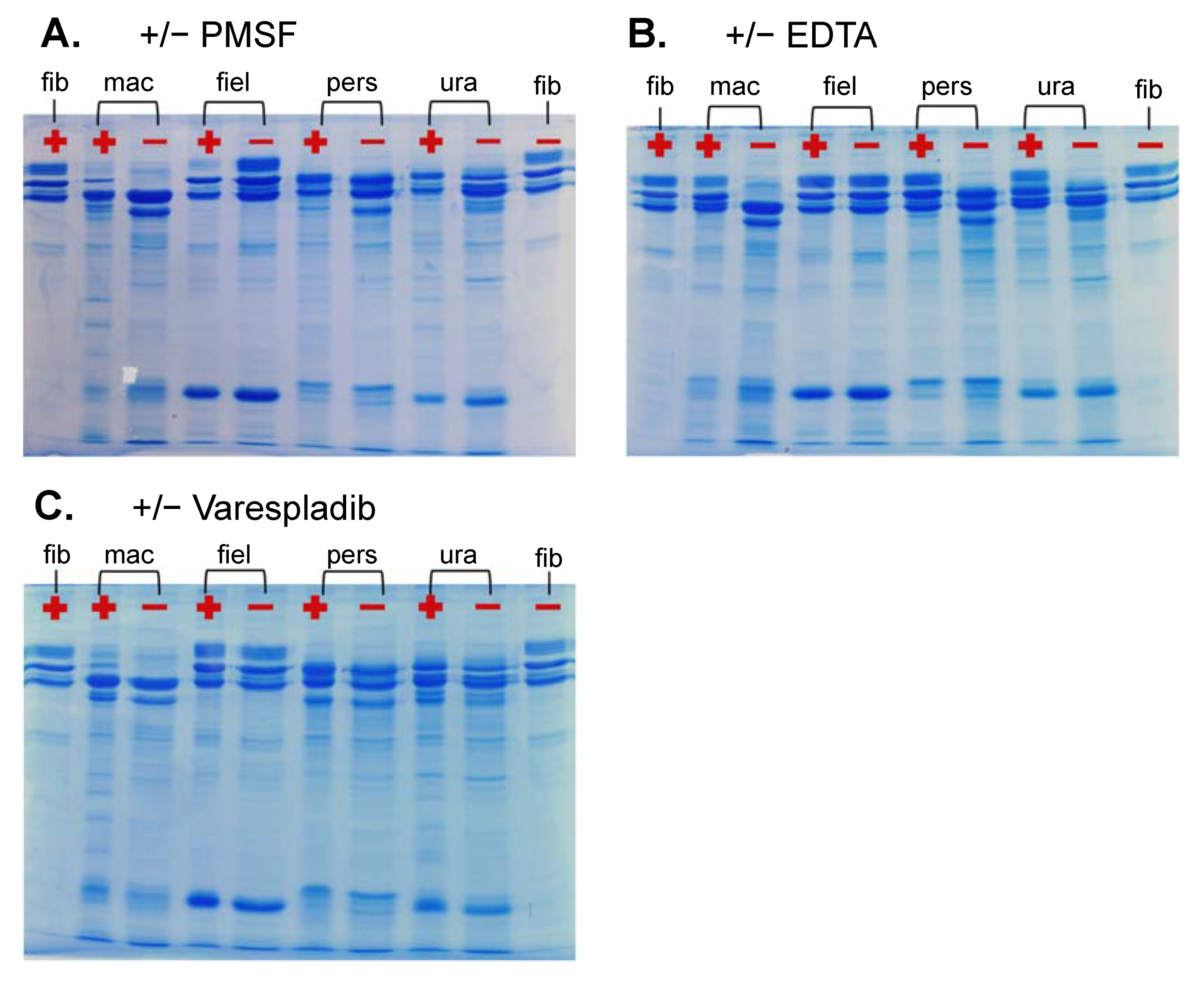
2.3.3. Fibrinogenolytic Activity
3. Materials and Methods
3.1. Venom Samples
3.2. Fluorometric Enzyme Activity Assays
3.2.1. Snake Venom Phospholipase A2 Activity (PLA2)
3.2.2. Snake Venom Metalloprotease (SVMP) Activity
3.2.3. Snake Venom Serine Protease (SVSP) Activity
3.3. Cytotoxicity
MTT Assays
3.4. Venom-Induced Coagulant Activity
3.4.1. Fibrinolysis
3.4.2. Phospholipid Interactions
3.4.3. Fibrinogenolysis
3.5. Statistics
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Casewell, N.R.; Wüster, W.; Vonk, F.J.; Harrison, R.A.; Fry, B.G. Complex cocktails: The evolutionary novelty of venoms. Trends Ecol. Evol. 2013, 28, 219–229. [Google Scholar] [CrossRef] [PubMed]
- Junqueira-de-Azevedo, I.L.; Bastos, C.M.V.; Ho, P.L.; Luna, M.S.; Yamanouye, N.; Casewell, N.R. Venom-related transcripts from Bothrops jararaca tissues provide novel molecular insights into the production and evolution of snake venom. Mol. Biol. Evol. 2015, 32, 754–766. [Google Scholar] [CrossRef] [Green Version]
- Kini, R.M. Excitement ahead: Structure, function and mechanism of snake venom phospholipase A2 enzymes. Toxicon 2003, 42, 827–840. [Google Scholar] [CrossRef] [PubMed]
- Serrano, S.M. The long road of research on snake venom serine proteinases. Toxicon 2013, 62, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Simurda, T.; Zolkova, J.; Kolkova, Z.; Loderer, D.; Dobrotova, M.; Skornova, I.; Brunclíkova, M.; Grendar, M.; Lasabova, Z.; Stasko, J. Comparison of clinical phenotype with genetic and laboratory results in 31 patients with congenital dysfibrinogenemia in northern Slovakia. Int. J. Hematol. 2020, 111, 795–802. [Google Scholar] [CrossRef]
- Mohamed Abd El-Aziz, T.; Garcia Soares, A.; Stockand, J.D. Snake Venoms in Drug Discovery: Valuable Therapeutic Tools for Life Saving. Toxins 2019, 11, 564. [Google Scholar] [CrossRef] [Green Version]
- Ali, S.A.; Hamid, F.; Abbasi, A.; Zaidi, Z.H.; Shehnaz, D. Pharmacological effects of the leaf-nosed viper snake (Eristocophis macmahoni) venom and its HPLC fractions. Toxicon 1999, 37, 1095–1107. [Google Scholar] [CrossRef]
- Scarborough, R.M.; Rose, J.W.; Hsu, M.A.; Phillips, D.R.; Fried, V.A.; Campbell, A.M.; Nannizzi, L.; Charo, I.F. Barbourin. A GPIIb-IIIa-specific integrin antagonist from the venom of Sistrurus m. barbouri. J. Biol. Chem 1991, 266, 9359–9362. [Google Scholar] [CrossRef]
- Siddiqi, A.R.; Persson, B.; Zaidi, Z.H.; Jörnvall, H. Characterization of two platelet aggregation inhibitor-like polypeptides from viper venom. Peptides 1992, 13, 1033–1037. [Google Scholar] [CrossRef]
- Alam, J.M.; Ali, S.A. Age-dependent variability of biochemical and biological properties in venoms of Leaf-Nose Viper snakes, Eristicophis macmahoni. Pak. J. Zool. 2001, 33, 173–177. [Google Scholar]
- Coimbra, F.C.; Bourke, L.A.; Huynh, T.M.; Vlecken, D.H.; Ghezellou, P.; Visser, J.C.; Dobson, J.S.; Fernandez-Rojo, M.A.; Ikonomopoulou, M.P.; Casewell, N.R.; et al. Extensive Variation in the Activities of Pseudocerastes and Eristicophis Viper Venoms Suggests Divergent Envenoming Strategies Are Used for Prey Capture. Toxins 2021, 13, 112. [Google Scholar]
- Bdolah, A. Comparison of venoms from two subspecies of the false horned viper (Pseudocerastes persicus). Toxicon 1986, 24, 726–729. [Google Scholar] [CrossRef]
- Oyama, E.; Takahashi, H. Distribution of low molecular weight platelet aggregation inhibitors from snake venoms. Toxicon 2007, 49, 293–298. [Google Scholar] [CrossRef]
- Shahbazi, B.; Najafabadi, Z.S.; Goudarzi, H.; Sajadi, M.; Tahoori, F.; Bagheri, M. Cytotoxic effects of Pseudocerastes persicus venom and its HPLC fractions on lung cancer cells. J. Venom. Anim. Toxins Incl. Trop. Dis. 2019, 25, e20190009. [Google Scholar] [CrossRef] [PubMed]
- Batzri-Izraeli, R.; Bdolah, A. Isolation and characterization of the main toxic fraction from the venom of the false horned viper (Pseudocerastes fieldi). Toxicon 1982, 20, 867–875. [Google Scholar] [CrossRef]
- Tsai, M.; Lee, C.; Bdolah, A. Mode of neuromuscular blocking action of a toxic phospholipase A2 from Pseudocerastes fieldi (Field’s horned viper) snake venom. Toxicon 1983, 21, 527–534. [Google Scholar] [CrossRef]
- Mallow, D.; Ludwig, D.; Nilson, G. True Vipers: Natural History and Toxinology of Old World Vipers; Krieger Publishing Company: Malabar, FL, USA, 2003. [Google Scholar]
- Ali, S.A.; Jackson, T.N.; Casewell, N.R.; Low, D.H.; Rossi, S.; Baumann, K.; Fathinia, B.; Visser, J.; Nouwens, A.; Hendrikx, I. Extreme venom variation in Middle Eastern vipers: A proteomics comparison of Eristicophis macmahonii, Pseudocerastes fieldi and Pseudocerastes persicus. J. Proteom. 2015, 116, 106–113. [Google Scholar] [CrossRef] [PubMed]
- Nget-Hong, T.; Ponnudurai, G. A comparative study of the biological properties of venoms of some old world vipers (Subfamily viperinae). Int. J. Biochem. 1992, 24, 331–336. [Google Scholar] [CrossRef]
- Soengas, M.S.; Lowe, S.W. Apoptosis and melanoma chemoresistance. Oncogene 2003, 22, 3138–3151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Moral-Sanz, J.; Fernandez-Rojo, M.A.; Potriquet, J.; Mukhopadhyay, P.; Brust, A.; Wilhelm, P.; Smallwood, T.B.; Clark, R.J.; Fry, B.G.; Alewood, P.F. ERK and mTORC1 Inhibitors Enhance the Anti-Cancer Capacity of the Octpep-1 Venom-Derived Peptide in Melanoma BRAF (V600E) Mutations. Toxins 2021, 13, 146. [Google Scholar] [CrossRef]
- Ikonomopoulou, M.P.; Fernandez-Rojo, M.A.; Pineda, S.S.; Cabezas-Sainz, P.; Winnen, B.; Morales, R.A.; Brust, A.; Sánchez, L.; Alewood, P.F.; Ramm, G.A. Gomesin inhibits melanoma growth by manipulating key signaling cascades that control cell death and proliferation. Sci. Rep. 2018, 8, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Lassen, A.; Atefi, M.; Robert, L.; Wong, D.J.; Cerniglia, M.; Comin-Anduix, B.; Ribas, A. Effects of AKT inhibitor therapy in response and resistance to BRAF inhibition in melanoma. Mol. Cancer 2014, 13, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Swenson, S.; Markland, F., Jr. Snake venom fibrin (ogen) olytic enzymes. Toxicon 2005, 45, 1021–1039. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, E.F.; Flores-Ortiz, R.J.; Alvarenga, V.G.; Eble, J.A. Direct Fibrinolytic Snake Venom Metalloproteinases Affecting Hemostasis: Structural, Biochemical Features and Therapeutic Potential. Toxins 2017, 9, 392. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Boffa, M.-C.; Boffa, G.A. A phospholipase A2 with anticoagulant activity II. Inhibition of the phospholipid activity in coagulation. Biochim. Et Biophys. Acta (BBA) Enzymol. 1976, 429, 839–852. [Google Scholar] [CrossRef]
- Saikia, D.; Thakur, R.; Mukherjee, A.K. An acidic phospholipase A2 (RVVA-PLA2-I) purified from Daboia russelli venom exerts its anticoagulant activity by enzymatic hydrolysis of plasma phospholipids and by non-enzymatic inhibition of factor Xa in a phospholipids/Ca2+ independent manner. Toxicon 2011, 57, 841–850. [Google Scholar] [CrossRef] [PubMed]
- Wolberg, A.S.; Campbell, R.A. Thrombin generation, fibrin clot formation and hemostasis. Transfus. Apher. Sci. 2008, 38, 15–23. [Google Scholar] [CrossRef] [Green Version]
- Cavigiolio, G.; Jayaraman, S. Proteolysis of apolipoprotein A-I by secretory phospholipase A2: A new link between inflammation and atherosclerosis. J. Biol. Chem. 2014, 289, 10011–10023. [Google Scholar] [CrossRef] [Green Version]
- Panagides, N.; Jackson, T.N.; Ikonomopoulou, M.P.; Arbuckle, K.; Pretzler, R.; Yang, D.C.; Ali, S.A.; Koludarov, I.; Dobson, J.; Sanker, B. How the cobra got its flesh-eating venom: Cytotoxicity as a defensive innovation and its co-evolution with hooding, aposematic marking, and spitting. Toxins 2017, 9, 103. [Google Scholar] [CrossRef] [Green Version]
- Higgins, D.; Mann, K. The interaction of bovine factor V and factor V-derived peptides with phospholipid vesicles. J. Biol. Chem. 1983, 258, 6503–6508. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Toxin Type | E. macmahonii | P. fieldi | P. persicus | P. urarachnoides |
|---|---|---|---|---|
| CRiSP | X | X | X | n.d. |
| Disintegrin | X | n.d. | n.d. | n.d. |
| SVSP | X | X | X | X |
| Kunitz peptide | n.d. | X | X | n.d. |
| LAAO | X | n.d. | n.d. | X |
| Lectin | X | X | X | X |
| Natriuretic peptides | n.d. | n.d. | n.d. | n.d. |
| NGF | n.d. | X | X | n.d. |
| Nucleotidase | n.d. | X | n.d. | n.d. |
| PLA2 | X | X | X | X |
| SVMP | X | n.d. | X | X |
| VEGF | X | X | X | n.d. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
op den Brouw, B.; Ghezellou, P.; Casewell, N.R.; Ali, S.A.; Fathinia, B.; Fry, B.G.; Bos, M.H.A.; Ikonomopoulou, M.P. Pharmacological Characterisation of Pseudocerastes and Eristicophis Viper Venoms Reveal Anticancer (Melanoma) Properties and a Potentially Novel Mode of Fibrinogenolysis. Int. J. Mol. Sci. 2021, 22, 6896. https://doi.org/10.3390/ijms22136896
op den Brouw B, Ghezellou P, Casewell NR, Ali SA, Fathinia B, Fry BG, Bos MHA, Ikonomopoulou MP. Pharmacological Characterisation of Pseudocerastes and Eristicophis Viper Venoms Reveal Anticancer (Melanoma) Properties and a Potentially Novel Mode of Fibrinogenolysis. International Journal of Molecular Sciences. 2021; 22(13):6896. https://doi.org/10.3390/ijms22136896
Chicago/Turabian Styleop den Brouw, Bianca, Parviz Ghezellou, Nicholas R. Casewell, Syed Abid Ali, Behzad Fathinia, Bryan G. Fry, Mettine H.A. Bos, and Maria P. Ikonomopoulou. 2021. "Pharmacological Characterisation of Pseudocerastes and Eristicophis Viper Venoms Reveal Anticancer (Melanoma) Properties and a Potentially Novel Mode of Fibrinogenolysis" International Journal of Molecular Sciences 22, no. 13: 6896. https://doi.org/10.3390/ijms22136896
APA Styleop den Brouw, B., Ghezellou, P., Casewell, N. R., Ali, S. A., Fathinia, B., Fry, B. G., Bos, M. H. A., & Ikonomopoulou, M. P. (2021). Pharmacological Characterisation of Pseudocerastes and Eristicophis Viper Venoms Reveal Anticancer (Melanoma) Properties and a Potentially Novel Mode of Fibrinogenolysis. International Journal of Molecular Sciences, 22(13), 6896. https://doi.org/10.3390/ijms22136896