
Disruption of O-Linked N-Acetylglucosamine Signaling in Placenta Induces Insulin Sensitivity in Female Offspring


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
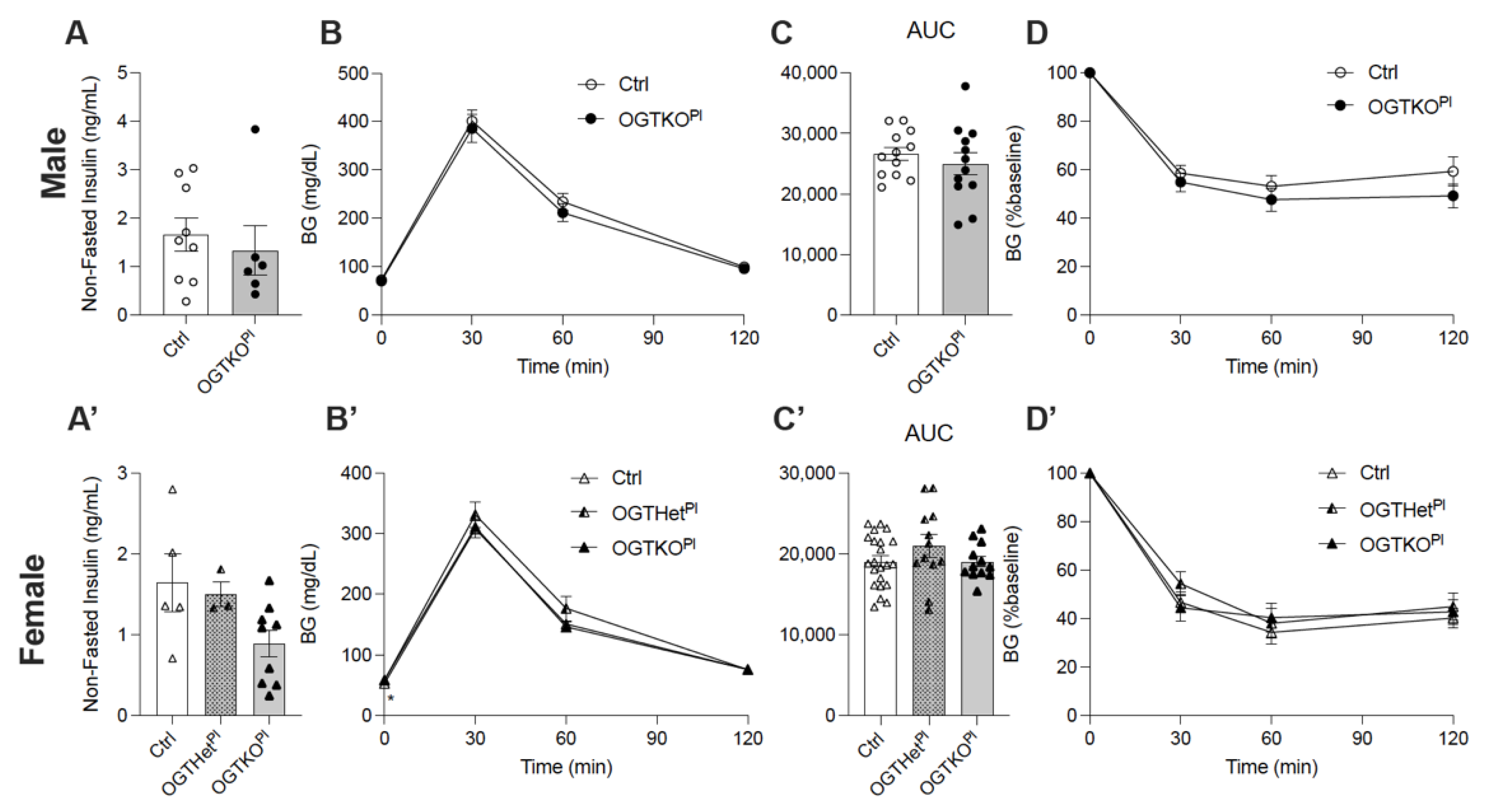
2.1. Reduced Placental OGT in Female Offspring on a Normal Chow Diet Led to an Increase in Fasting Blood Glucose
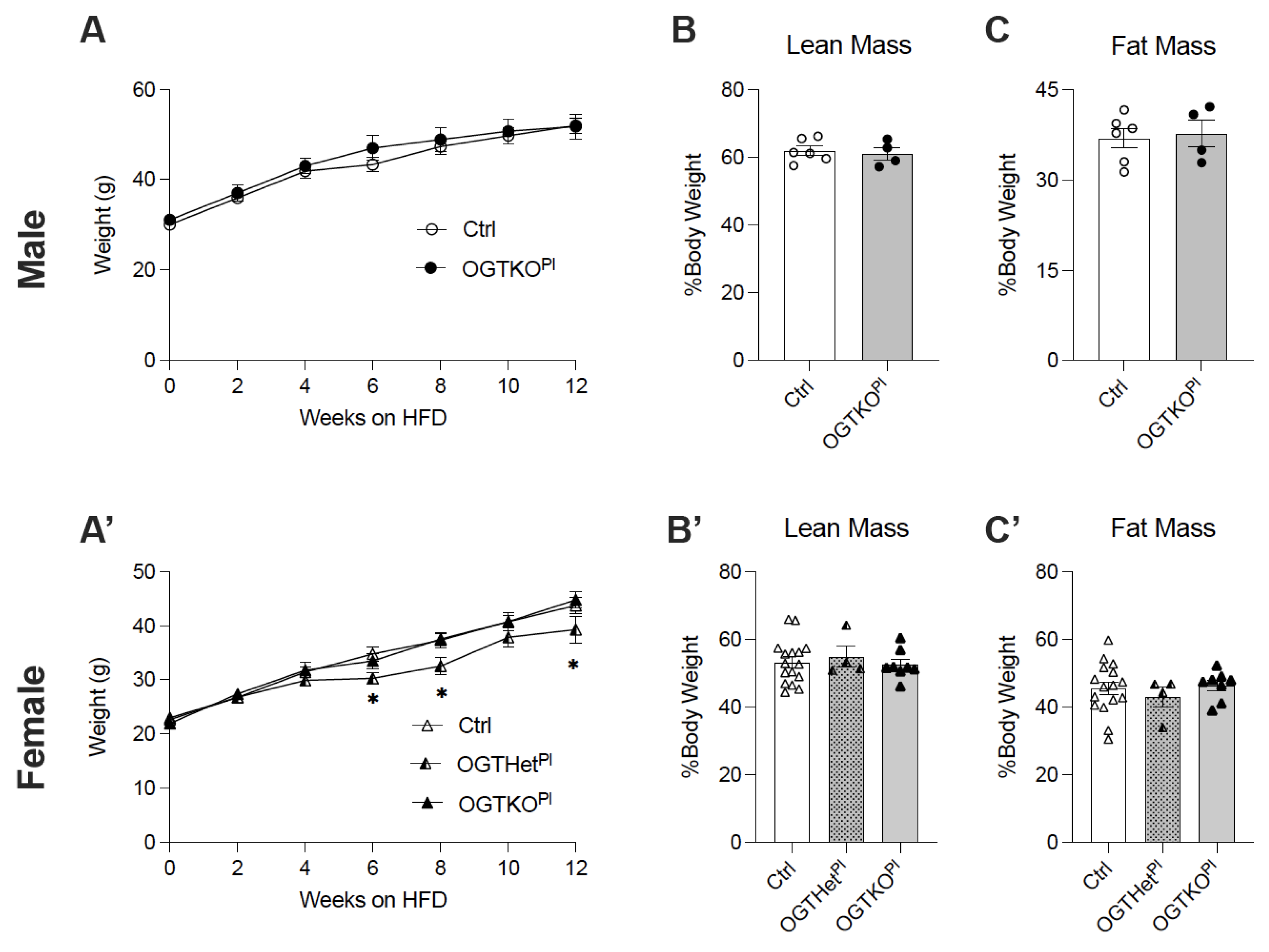
2.2. High-Fat Diet-Induced Weight Gain Was Attenuated in Female Offspring with Reduced Placental OGT
2.3. No Changes in Food Intake or Energy Utilization in Female Mice with Reduced Placental OGT
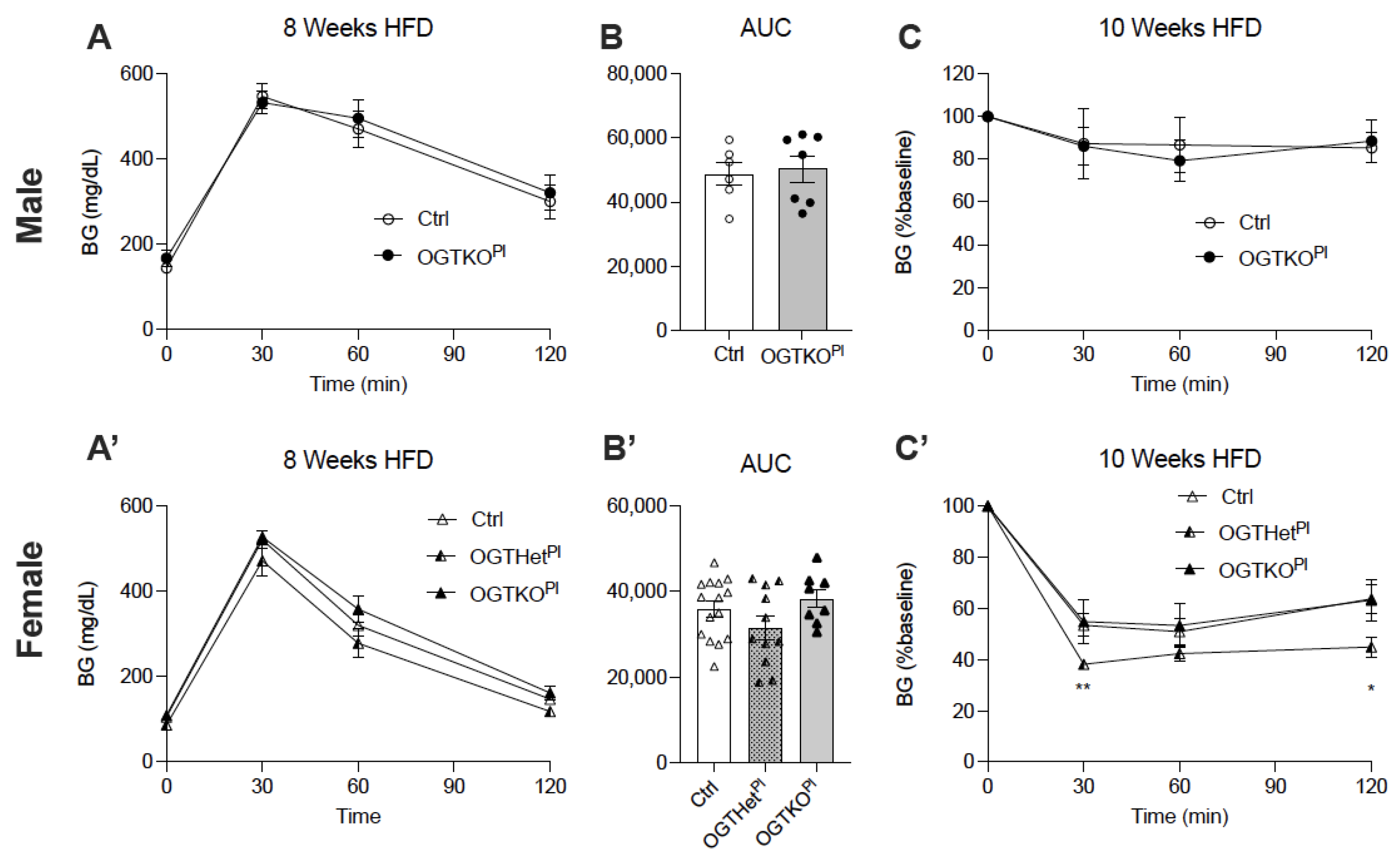
2.4. Heterozygote Placental OGT Female Offspring Demonstrate Increased Insulin Tolerance on HFD
2.5. Female Placental OGT Knockout Offspring on HFD Demonstrated Impaired Insulin Secretion in Response to Glucose and No Change in Beta Cell Mass
2.6. Increased Insulin Sensitivity for OGTHetPl Females Was Associated with Increased Hepatic Insulin Signaling
3. Discussion
4. Materials and Methods
4.1. Generation of Mice
4.2. In Vivo Mouse Studies
4.3. Histological Analysis
4.4. Acute Insulin Treatment In Vivo
4.5. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lima, V.V.; Dela Justina, V.; Dos Passos, R.R., Jr.; Volpato, G.T.; Souto, P.C.S.; San Martin, S.; Giachini, F.R. O-GlcNAc Modification During Pregnancy: Focus on Placental Environment. Front. Physiol. 2018, 9, 1263. [Google Scholar] [CrossRef]
- Barker, D.J. The origins of the developmental origins theory. J. Intern. Med. 2007, 261, 412–417. [Google Scholar] [CrossRef]
- Nugent, B.M.; Bale, T.L. The omniscient placenta: Metabolic and epigenetic regulation of fetal programming. Front. Neuroendocrinol. 2015, 39, 28–37. [Google Scholar] [CrossRef] [Green Version]
- Gheorghe, C.P.; Goyal, R.; Mittal, A.; Longo, L.D. Gene expression in the placenta: Maternal stress and epigenetic responses. Int. J. Dev. Biol. 2010, 54, 507–523. [Google Scholar] [CrossRef] [Green Version]
- Wadhwa, P.D.; Buss, C.; Entringer, S.; Swanson, J.M. Developmental origins of health and disease: Brief history of the approach and current focus on epigenetic mechanisms. Semin. Reprod. Med. 2009, 27, 358–368. [Google Scholar] [CrossRef] [Green Version]
- Vaag, A.; Brøns, C.; Gillberg, L.; Hansen, N.S.; Hjort, L.; Arora, G.P.; Thomas, N.; Broholm, C.; Ribel-Madsen, R.; Grunnet, L.G. Genetic, nongenetic and epigenetic risk determinants in developmental programming of type 2 diabetes. Acta Obstet. Gynecol. Scand. 2014, 93, 1099–1108. [Google Scholar] [CrossRef]
- Baschat, A.A.; Hecher, K. Fetal growth restriction due to placental disease. Semin. Perinatol. 2004, 28, 67–80. [Google Scholar] [CrossRef]
- Alejandro, E.U.; Gregg, B.; Wallen, T.; Kumusoglu, D.; Meister, D.; Chen, A.; Merrins, M.J.; Satin, L.S.; Liu, M.; Arvan, P.; et al. Maternal diet-induced microRNAs and mTOR underlie beta cell dysfunction in offspring. J. Clin. Investig. 2014, 124, 4395–4410. [Google Scholar] [CrossRef] [Green Version]
- Sandovici, I.; Hammerle, C.M.; Ozanne, S.E.; Constância, M. Developmental and environmental epigenetic programming of the endocrine pancreas: Consequences for type 2 diabetes. Cell. Mol. Life Sci. 2013, 70, 1575–1595. [Google Scholar] [CrossRef]
- Jansson, T.; Powell, T.L. IFPA 2005 Award in Placentology Lecture. Human placental transport in altered fetal growth: Does the placenta function as a nutrient sensor?—A review. Placenta 2006, 27 (Suppl. A), S91–S97. [Google Scholar] [CrossRef]
- Lopez-Tello, J.; Perez-Garcia, V.; Khaira, J.; Kusinski, L.C.; Cooper, W.N.; Andreani, A.; Grant, I.; Fernandez de Liger, E.; Lam, B.Y.; Hemberger, M.; et al. Fetal and trophoblast PI3K p110alpha have distinct roles in regulating resource supply to the growing fetus in mice. eLife 2019, 8. [Google Scholar] [CrossRef]
- Diaz, P.; Powell, T.L.; Jansson, T. The role of placental nutrient sensing in maternal-fetal resource allocation. Biol. Reprod. 2014, 91, 82. [Google Scholar] [CrossRef] [Green Version]
- Robertson, S.A.; Moldenhauer, L.M. Immunological determinants of implantation success. Int. J. Dev. Biol. 2014, 58, 205–217. [Google Scholar] [CrossRef] [Green Version]
- Lager, S.; Powell, T.L. Regulation of nutrient transport across the placenta. J. Pregnancy 2012, 2012, 179827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillemain, G.; Filhoulaud, G.; Da Silva-Xavier, G.; Rutter, G.A.; Scharfmann, R. Glucose is necessary for embryonic pancreatic endocrine cell differentiation. J. Biol. Chem. 2007, 282, 15228–15237. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heinis, M.; Simon, M.T.; Ilc, K.; Mazure, N.M.; Pouyssegur, J.; Scharfmann, R.; Duvillie, B. Oxygen tension regulates pancreatic beta-cell differentiation through hypoxia-inducible factor 1alpha. Diabetes 2010, 59, 662–669. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohan, R.; Baumann, D.; Alejandro, E.U. Fetal undernutrition, placental insufficiency, and pancreatic beta-cell development programming in utero. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2018, 315, R867–R878. [Google Scholar] [CrossRef] [Green Version]
- Newsome, C.A.; Shiell, A.W.; Fall, C.H.; Phillips, D.I.; Shier, R.; Law, C.M. Is birth weight related to later glucose and insulin metabolism?—A systematic review. Diabet. Med. 2003, 20, 339–348. [Google Scholar] [CrossRef]
- Fernandez-Twinn, D.S.; Ozanne, S.E. Early life nutrition and metabolic programming. Ann. N. Y. Acad. Sci. 2010, 1212, 78–96. [Google Scholar] [CrossRef]
- Dahri, S.; Snoeck, A.; Reusens-Billen, B.; Remacle, C.; Hoet, J.J. Islet function in offspring of mothers on low-protein diet during gestation. Diabetes 1991, 40 (Suppl. 2), 115–120. [Google Scholar] [CrossRef]
- Alejandro, E.U.; Gregg, B.; Blandino-Rosano, M.; Cras-Meneur, C.; Bernal-Mizrachi, E. Natural history of beta-cell adaptation and failure in type 2 diabetes. Mol. Asp. Med. 2015, 42, 19–41. [Google Scholar] [CrossRef] [Green Version]
- Dumortier, O.; Blondeau, B.; Duvillie, B.; Reusens, B.; Breant, B.; Remacle, C. Different mechanisms operating during different critical time-windows reduce rat fetal beta cell mass due to a maternal low-protein or low-energy diet. Diabetologia 2007, 50, 2495–2503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heltemes, A.; Gingery, A.; Soldner, E.L.; Bozadjieva, N.; Jahr, K.N.; Johnson, B.K.; Gilbert, J.S. Chronic placental ischemia alters amniotic fluid milieu and results in impaired glucose tolerance, insulin resistance and hyperleptinemia in young rats. Exp. Biol. Med. 2010, 235, 892–899. [Google Scholar] [CrossRef]
- Hart, B.; Morgan, E.; Alejandro, E.U. Nutrient sensor signaling pathways and cellular stress in fetal growth restriction. J. Mol. Endocrinol. 2019, 62, R155–R165. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Y.R.; Jang, H.J.; Lee, Y.H.; Kim, I.S.; Lee, H.; Ryu, S.H.; Suh, P.G. O-GlcNAc cycling enzymes control vascular development of the placenta by modulating the levels of HIF-1alpha. Placenta 2015, 36, 1063–1068. [Google Scholar] [CrossRef] [PubMed]
- Howerton, C.L.; Bale, T.L. Targeted placental deletion of OGT recapitulates the prenatal stress phenotype including hypothalamic mitochondrial dysfunction. Proc. Natl. Acad. Sci. USA 2014, 111, 9639–9644. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roos, S.; Jansson, N.; Palmberg, I.; Saljo, K.; Powell, T.L.; Jansson, T. Mammalian target of rapamycin in the human placenta regulates leucine transport and is down-regulated in restricted fetal growth. J. Physiol. 2007, 582, 449–459. [Google Scholar] [CrossRef]
- Rosario, F.J.; Jansson, N.; Kanai, Y.; Prasad, P.D.; Powell, T.L.; Jansson, T. Maternal protein restriction in the rat inhibits placental insulin, mTOR, and STAT3 signaling and down-regulates placental amino acid transporters. Endocrinology 2011, 152, 1119–1129. [Google Scholar] [CrossRef] [Green Version]
- Wullschleger, S.; Loewith, R.; Hall, M.N. TOR signaling in growth and metabolism. Cell 2006, 124, 471–484. [Google Scholar] [CrossRef] [Green Version]
- Hanover, J.A.; Krause, M.W.; Love, D.C. The hexosamine signaling pathway: O-GlcNAc cycling in feast or famine. Biochim. Biophys. Acta 2010, 1800, 80–95. [Google Scholar] [CrossRef] [Green Version]
- Jansson, T.; Aye, I.L.; Goberdhan, D.C. The emerging role of mTORC1 signaling in placental nutrient-sensing. Placenta 2012, 33 (Suppl. 2), e23–e29. [Google Scholar] [CrossRef] [Green Version]
- Wu, D.; Cai, Y.; Jin, J. Potential coordination role between O-GlcNAcylation and epigenetics. Protein Cell 2017, 8, 713–723. [Google Scholar] [CrossRef] [Green Version]
- Lima, V.V.; Giachini, F.R.; Hardy, D.M.; Webb, R.C.; Tostes, R.C. O-GlcNAcylation: A novel pathway contributing to the effects of endothelin in the vasculature. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2011, 300, R236–R250. [Google Scholar] [CrossRef] [Green Version]
- Levine, Z.G.; Walker, S. The Biochemistry of O-GlcNAc Transferase: Which Functions Make It Essential in Mammalian Cells? Annu. Rev. Biochem. 2016, 85, 631–657. [Google Scholar] [CrossRef]
- Jang, H.; Kim, T.W.; Yoon, S.; Choi, S.Y.; Kang, T.W.; Kim, S.Y.; Kwon, Y.W.; Cho, E.J.; Youn, H.D. O-GlcNAc regulates pluripotency and reprogramming by directly acting on core components of the pluripotency network. Cell Stem Cell 2012, 11, 62–74. [Google Scholar] [CrossRef] [Green Version]
- Muter, J.; Alam, M.T.; Vrljicak, P.; Barros, F.S.V.; Ruane, P.T.; Ewington, L.J.; Aplin, J.D.; Westwood, M.; Brosens, J.J. The Glycosyltransferase EOGT Regulates Adropin Expression in Decidualizing Human Endometrium. Endocrinology 2018, 159, 994–1004. [Google Scholar] [CrossRef] [Green Version]
- Pantaleon, M.; Tan, H.Y.; Kafer, G.R.; Kaye, P.L. Toxic effects of hyperglycemia are mediated by the hexosamine signaling pathway and o-linked glycosylation in early mouse embryos. Biol. Reprod. 2010, 82, 751–758. [Google Scholar] [CrossRef] [Green Version]
- Shi, F.T.; Kim, H.; Lu, W.; He, Q.; Liu, D.; Goodell, M.A.; Wan, M.; Songyang, Z. Ten-eleven translocation 1 (Tet1) is regulated by O-linked N-acetylglucosamine transferase (Ogt) for target gene repression in mouse embryonic stem cells. J. Biol. Chem. 2013, 288, 20776–20784. [Google Scholar] [CrossRef] [Green Version]
- Speakman, C.M.; Domke, T.C.; Wongpaiboonwattana, W.; Sanders, K.; Mudaliar, M.; van Aalten, D.M.; Barton, G.J.; Stavridis, M.P. Elevated O-GlcNAc levels activate epigenetically repressed genes and delay mouse ESC differentiation without affecting naive to primed cell transition. Stem Cells 2014, 32, 2605–2615. [Google Scholar] [CrossRef] [Green Version]
- Kim, G.; Cao, L.; Reece, E.A.; Zhao, Z. Impact of protein O-GlcNAcylation on neural tube malformation in diabetic embryopathy. Sci. Rep. 2017, 7, 11107. [Google Scholar] [CrossRef]
- Keembiyehetty, C.; Love, D.C.; Harwood, K.R.; Gavrilova, O.; Comly, M.E.; Hanover, J.A. Conditional knock-out reveals a requirement for O-linked N-Acetylglucosaminase (O-GlcNAcase) in metabolic homeostasis. J. Biol. Chem. 2015, 290, 7097–7113. [Google Scholar] [CrossRef] [Green Version]
- Kafer, G.R.; Carlton, P.M.; Lehnert, S.A. The histone variant H2A.Z is dynamically expressed in the developing mouse placenta and in differentiating trophoblast stem cells. Placenta 2015, 36, 1325–1328. [Google Scholar] [CrossRef]
- Gao, Y.; Wells, L.; Comer, F.I.; Parker, G.J.; Hart, G.W. Dynamic O-glycosylation of nuclear and cytosolic proteins: Cloning and characterization of a neutral, cytosolic beta-N-acetylglucosaminidase from human brain. J. Biol. Chem. 2001, 276, 9838–9845. [Google Scholar] [CrossRef] [Green Version]
- Lim, K.; Chang, H.I. O-GlcNAc inhibits interaction between Sp1 and Elf-1 transcription factors. Biochem. Biophys. Res. Commun. 2009, 380, 569–574. [Google Scholar] [CrossRef]
- Hirosawa, M.; Hayakawa, K.; Yoneda, C.; Arai, D.; Shiota, H.; Suzuki, T.; Tanaka, S.; Dohmae, N.; Shiota, K. Novel O-GlcNAcylation on Ser(40) of canonical H2A isoforms specific to viviparity. Sci. Rep. 2016, 6, 31785. [Google Scholar] [CrossRef] [Green Version]
- Pantaleon, M.; Steane, S.E.; McMahon, K.; Cuffe, J.S.M.; Moritz, K.M. Placental O-GlcNAc-transferase expression and interactions with the glucocorticoid receptor are sex specific and regulated by maternal corticosterone exposure in mice. Sci. Rep. 2017, 7, 2017. [Google Scholar] [CrossRef] [Green Version]
- Ozcan, S.; Andrali, S.S.; Cantrell, J.E. Modulation of transcription factor function by O-GlcNAc modification. Biochim. Biophys. Acta 2010, 1799, 353–364. [Google Scholar] [CrossRef] [Green Version]
- Martinez, M.R.; Dias, T.B.; Natov, P.S.; Zachara, N.E. Stress-induced O-GlcNAcylation: An adaptive process of injured cells. Biochem. Soc. Trans. 2017, 45, 237–249. [Google Scholar] [CrossRef]
- Groves, J.A.; Lee, A.; Yildirir, G.; Zachara, N.E. Dynamic O-GlcNAcylation and its roles in the cellular stress response and homeostasis. Cell Stress Chaperones 2013, 18, 535–558. [Google Scholar] [CrossRef] [Green Version]
- Breier, B.H.; Vickers, M.H.; Ikenasio, B.A.; Chan, K.Y.; Wong, W.P. Fetal programming of appetite and obesity. Mol. Cell. Endocrinol. 2001, 185, 73–79. [Google Scholar] [CrossRef]
- Desai, M.; Hales, C.N. Role of fetal and infant growth in programming metabolism in later life. Biol. Rev. Camb. Philos. Soc. 1997, 72, 329–348. [Google Scholar] [CrossRef] [PubMed]
- Hofman, P.L.; Cutfield, W.S.; Robinson, E.M.; Bergman, R.N.; Menon, R.K.; Sperling, M.A.; Gluckman, P.D. Insulin resistance in short children with intrauterine growth retardation. J. Clin. Endocrinol. Metab. 1997, 82, 402–406. [Google Scholar] [CrossRef]
- Ravelli, A.C.; van Der Meulen, J.H.; Osmond, C.; Barker, D.J.; Bleker, O.P. Obesity at the age of 50 y in men and women exposed to famine prenatally. Am. J. Clin. Nutr. 1999, 70, 811–816. [Google Scholar] [CrossRef]
- Ozanne, S.E.; Nave, B.T.; Wang, C.L.; Shepherd, P.R.; Prins, J.; Smith, G.D. Poor fetal nutrition causes long-term changes in expression of insulin signaling components in adipocytes. Am. J. Physiol. 1997, 273, E46–E51. [Google Scholar] [CrossRef]
- Howerton, C.L.; Morgan, C.P.; Fischer, D.B.; Bale, T.L. O-GlcNAc transferase (OGT) as a placental biomarker of maternal stress and reprogramming of CNS gene transcription in development. Proc. Natl. Acad. Sci. USA 2013, 110, 5169–5174. [Google Scholar] [CrossRef] [Green Version]
- Ozanne, S.E.; Wang, C.L.; Coleman, N.; Smith, G.D. Altered muscle insulin sensitivity in the male offspring of protein-malnourished rats. Am. J. Physiol. 1996, 271, E1128–E1134. [Google Scholar] [CrossRef]
- Yaribeygi, H.; Farrokhi, F.R.; Butler, A.E.; Sahebkar, A. Insulin resistance: Review of the underlying molecular mechanisms. J. Cell. Physiol. 2019, 234, 8152–8161. [Google Scholar] [CrossRef]
- Carvalho, D.S.; Diniz, M.M.; Haidar, A.A.; Cavanal, M.F.; da Silva Alves, E.; Carpinelli, A.R.; Gil, F.Z.; Hirata, A.E. L-Arginine supplementation improves insulin sensitivity and beta cell function in the offspring of diabetic rats through AKT and PDX-1 activation. Eur. J. Pharmacol. 2016, 791, 780–787. [Google Scholar] [CrossRef]
- Cuthbert, C.E.; Foster, J.E.; Ramdath, D.D. A maternal high-fat, high-sucrose diet alters insulin sensitivity and expression of insulin signalling and lipid metabolism genes and proteins in male rat offspring: Effect of folic acid supplementation. Br. J. Nutr 2017, 118, 580–588. [Google Scholar] [CrossRef] [Green Version]
- Alejandro, E.U.; Jo, S.; Akhaphong, B.; Llacer, P.R.; Gianchandani, M.; Gregg, B.; Parlee, S.D.; MacDougald, O.A.; Bernal-Mizrachi, E. Maternal low-protein diet on the last week of pregnancy contributes to insulin resistance and beta-cell dysfunction in the mouse offspring. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2020, 319, R485–R496. [Google Scholar] [CrossRef]
- Migeon, B.R.; Axelman, J.; Jeppesen, P. Differential X reactivation in human placental cells: Implications for reversal of X inactivation. Am. J. Hum. Genet. 2005, 77, 355–364. [Google Scholar] [CrossRef] [Green Version]
- Wenzel, P.L.; Leone, G. Expression of Cre recombinase in early diploid trophoblast cells of the mouse placenta. Genesis 2007, 45, 129–134. [Google Scholar] [CrossRef]
- Lockridge, A.; Jo, S.; Gustafson, E.; Damberg, N.; Mohan, R.; Olson, M.; Abrahante, J.E.; Alejandro, E.U. Islet O-GlcNAcylation Is Required for Lipid Potentiation of Insulin Secretion through SERCA2. Cell Rep. 2020, 31, 107609. [Google Scholar] [CrossRef]
- Peppler, W.T.; Townsend, L.K.; Meers, G.M.; Panasevich, M.R.; MacPherson, R.E.K.; Rector, R.S.; Wright, D.C. Acute administration of IL-6 improves indices of hepatic glucose and insulin homeostasis in lean and obese mice. Am. J. Physiol. Gastrointest. Liver Physiol. 2019, 316, G166–G178. [Google Scholar] [CrossRef]
- Tom, R.Z.; Sjogren, R.J.; Vieira, E.; Glund, S.; Iglesias-Gutierrez, E.; Garcia-Roves, P.M.; Myers, M.G., Jr.; Bjornholm, M. Increased hepatic insulin sensitivity in mice lacking inhibitory leptin receptor signals. Endocrinology 2011, 152, 2237–2246. [Google Scholar] [CrossRef] [Green Version]
- Jo, S.; Lockridge, A.; Mohan, R.; Esch, N.; Schlichting, R.; Panigrahy, N.; Essawy, A.; Gustafson, E.; Alejandro, E.U. Translational Factor eIF4G1 Regulates Glucose Homeostasis and Pancreatic beta-Cell Function. Diabetes 2021, 70, 155–170. [Google Scholar] [CrossRef]



Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Moore, M.; Avula, N.; Jo, S.; Beetch, M.; Alejandro, E.U. Disruption of O-Linked N-Acetylglucosamine Signaling in Placenta Induces Insulin Sensitivity in Female Offspring. Int. J. Mol. Sci. 2021, 22, 6918. https://doi.org/10.3390/ijms22136918
Moore M, Avula N, Jo S, Beetch M, Alejandro EU. Disruption of O-Linked N-Acetylglucosamine Signaling in Placenta Induces Insulin Sensitivity in Female Offspring. International Journal of Molecular Sciences. 2021; 22(13):6918. https://doi.org/10.3390/ijms22136918
Chicago/Turabian StyleMoore, Mackenzie, Nandini Avula, Seokwon Jo, Megan Beetch, and Emilyn U. Alejandro. 2021. "Disruption of O-Linked N-Acetylglucosamine Signaling in Placenta Induces Insulin Sensitivity in Female Offspring" International Journal of Molecular Sciences 22, no. 13: 6918. https://doi.org/10.3390/ijms22136918
APA StyleMoore, M., Avula, N., Jo, S., Beetch, M., & Alejandro, E. U. (2021). Disruption of O-Linked N-Acetylglucosamine Signaling in Placenta Induces Insulin Sensitivity in Female Offspring. International Journal of Molecular Sciences, 22(13), 6918. https://doi.org/10.3390/ijms22136918