
The V-ATPase a3 Subunit: Structure, Function and Therapeutic Potential of an Essential Biomolecule in Osteoclastic Bone Resorption


Abstract
:1. Bone
2. V-ATPase Functions
3. V-ATPase Structure
4. The V-ATPase a Subunit
5. a3-d2-B2
6. Human Diseases Linked to V-ATPase Mutations
7. Membrane Signaling Lipids as Regulators of V-ATPase Localization and/or Activity
8. V-ATPase as a Signalsome
9. The Potential of a3 as a Therapeutic Target for Osteolytic Diseases
10. The Potential of the Correction of a3 Splice Site and Missense Mutations to Treat Osteopetrosis
11. Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lin, X.; Patil, S.; Gao, Y.-G.; Qian, A. The Bone Extracellular Matrix in Bone Formation and Regeneration. Front. Pharmacol. 2020, 11, 757. [Google Scholar] [CrossRef] [PubMed]
- Lee, D.; Kim, D.W.; Cho, J.-Y. Correction to: Role of growth factors in hematopoietic stem cell niche. Cell Biol. Toxicol. 2020, 36, 1. [Google Scholar] [CrossRef]
- Kitaura, H.; Marahleh, A.; Ohori, F.; Noguchi, T.; Shen, W.-R.; Qi, J.; Nara, Y.; Pramusita, A.; Kinjo, R.; Mizoguchi, I. Osteocyte-Related Cytokines Regulate Osteoclast Formation and Bone Resorption. Int. J. Mol. Sci. 2020, 21, 5169. [Google Scholar] [CrossRef] [PubMed]
- Roeder, E.; Matthews, B.G.; Kalajzic, I. Visual reporters for study of the osteoblast lineage. Bone 2016, 92, 189–195. [Google Scholar] [CrossRef] [Green Version]
- Prideaux, M.; Findlay, D.M.; Atkins, G.J. Osteocytes: The master cells in bone remodelling. Curr. Opin. Pharmacol. 2016, 28, 24–30. [Google Scholar] [CrossRef]
- Creecy, A.; Damrath, J.G.; Wallace, J.M. Control of Bone Matrix Properties by Osteocytes. Front. Endocrinol. 2021, 11, 578477. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Wang, Z.; Duan, N.; Zhu, G.; Schwarz, E.M.; Xie, C. Osteoblast-osteoclast interactions. Connect. Tissue Res. 2018, 59, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Jacome-Galarza, C.E.; Percin, G.I.; Muller, J.T.; Mass, E.; Lazarov, T.; Eitler, J.; Rauner, M.; Yadav, V.K.; Crozet, L.; Bohm, M.; et al. Developmental origin, functional maintenance and genetic rescue of osteoclasts. Nat. Cell Biol. 2019, 568, 541–545. [Google Scholar] [CrossRef]
- Yahara, Y.; Barrientos, T.; Tang, Y.J.; Puviindran, V.; Nadesan, P.; Zhang, H.; Gibson, J.R.; Gregory, S.G.; Diao, Y.; Xiang, Y.; et al. Erythromyeloid progenitors give rise to a population of osteoclasts that contribute to bone homeostasis and repair. Nat. Cell Biol. 2020, 22, 49–59. [Google Scholar] [CrossRef]
- Søe, K.; Delaisse, J.-M.; Borggaard, X.G. Osteoclast formation at the bone marrow/bone surface interface: Importance of structural elements, matrix, and intercellular communication. Semin. Cell Dev. Biol. 2020, 112, 8–15. [Google Scholar] [CrossRef]
- Sims, N.A.; Martin, T.J. Osteoclasts Provide Coupling Signals to Osteoblast Lineage Cells Through Multiple Mechanisms. Annu. Rev. Physiol. 2020, 82, 507–529. [Google Scholar] [CrossRef] [PubMed]
- Takito, J.; Nakamura, M. Heterogeneity and Actin Cytoskeleton in Osteoclast and Macrophage Multinucleation. Int. J. Mol. Sci. 2020, 21, 6629. [Google Scholar] [CrossRef] [PubMed]
- Gambari, L.; Grassi, F.; Roseti, L.; Grigolo, B.; Desando, G. Learning from Monocyte-Macrophage Fusion and Multinucleation: Potential Therapeutic Targets for Osteoporosis and Rheumatoid Arthritis. Int. J. Mol. Sci. 2020, 21, 6001. [Google Scholar] [CrossRef] [PubMed]
- Takito, J.; Inoue, S.; Nakamura, M. The Sealing Zone in Osteoclasts: A Self-Organized Structure on the Bone. Int. J. Mol. Sci. 2018, 19, 984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulari, M.T.K.; Zhao, H.; Lakkakorpi, P.T.; Väänänen, H.K. Osteoclast Ruffled Border Has Distinct Subdomains for Secretion and Degraded Matrix Uptake. Traffic 2003, 4, 113–125. [Google Scholar] [CrossRef] [PubMed]
- Toyomura, T.; Murata, Y.; Yamamoto, A.; Oka, T.; Sun-Wada, G.H.; Wada, Y.; Futai, M. From lysosomes to the plasma membrane: Localization of vacuolar-type H+ -ATPase with the a3 isoform during osteoclast differentiation. J. Biol. Chem. 2003, 278, 22023–22030. [Google Scholar] [CrossRef] [Green Version]
- Ng, P.Y.; Ribet, A.B.P.; Pavlos, N.J. Membrane trafficking in osteoclasts and implications for osteoporosis. Biochem. Soc. Trans. 2019, 47, 639–650. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Kim, N. Bone Cell Communication Factors Provide a New Therapeutic Strategy for Osteoporosis. Chonnam. Med. J. 2020, 56, 94–98. [Google Scholar] [CrossRef]
- Wang, H.; Yang, G.; Xiao, Y.; Luo, G.; Li, G.; Li, Z. Friend or Foe? Essential Roles of Osteoclast in Maintaining Skeletal Health. BioMed Res. Int. 2020, 2020, 1–10. [Google Scholar] [CrossRef]
- Corbacho, I.; Teixidó, F.; Olivero, I.; Hernández, L.M. Dependence of Saccharomyces cerevisiae Golgi functions on V-ATPase activity. FEMS Yeast Res. 2012, 12, 341–350. [Google Scholar] [CrossRef] [Green Version]
- Nelson, N. Structure and function of V-ATPases in endocytic and secretory organelles. J. Exp. Biol. 1992, 172, 149–153. [Google Scholar] [CrossRef]
- Perzov, N.; Padler-Karavani, V.; Nelson, H.; Nelson, N. Characterization of yeast V-ATPase mutants lacking Vph1p or Stv1p and the effect on endocytosis. J. Exp. Biol. 2002, 205, 1209–1219. [Google Scholar] [CrossRef] [PubMed]
- Márquez-Sterling, N.; Herman, I.M.; Pesacreta, T.; Arai, H.; Terres, G.; Forgac, M. Immunolocalization of the vacuolar-type (H+)-ATPase from clathrin-coated vesicles. Eur. J. Cell Biol. 1991, 56, 19–33. [Google Scholar]
- Kissing, S.; Hermsen, C.; Repnik, U.; Nesset, C.K.; von Bargen, K.; Griffiths, G.; Ichihara, A.; Lee, B.S.; Schwake, M.; De Brabander, J.; et al. Vacuolar ATPase in Phagosome-Lysosome Fusion. J. Biol. Chem. 2015, 290, 14166–14180. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun-Wada, G.-H.; Wada, Y. Role of vacuolar-type proton ATPase in signal transduction. Biochim. et Biophys. Acta (BBA) Bioenerg. 2015, 1847, 1166–1172. [Google Scholar] [CrossRef] [Green Version]
- Stransky, L.; Cotter, K.; Forgac, M. The Function of V-ATPases in Cancer. Physiol. Rev. 2016, 96, 1071–1091. [Google Scholar] [CrossRef] [PubMed]
- Kibak, H.; Taiz, L.; Starke, T.; Bernasconi, P.; Gogarten, J.P. Evolution of structure and function of V-ATPases. J. Bioenerg. Biomembr. 1992, 24, 415–424. [Google Scholar] [CrossRef]
- Sun-Wada, G.-H.; Wada, Y.; Futai, M. Vacuolar H+ pumping ATPases in luminal acidic organelles and extracellular compartments: Common rotational mechanism and diverse physiological roles. J. Bioenerg. Biomembr. 2003, 35, 347–358. [Google Scholar] [CrossRef]
- Futai, M.; Oka, T.; Sun-Wada, G.; Moriyama, Y.; Kanazawa, H.; Wada, Y. Luminal acidification of diverse organelles by V-ATPase in animal cells. J. Exp. Biol. 2000, 203, 107–116. [Google Scholar] [CrossRef]
- Esmail, S.; Kartner, N.; Yao, Y.; Kim, J.W.; Reithmeier, R.A.; Manolson, M.F. Molecular mechanisms of cutis laxa– and distal renal tubular acidosis–causing mutations in V-ATPase a subunits, ATP6V0A2 and ATP6V0A4. J. Biol. Chem. 2018, 293, 2787–2800. [Google Scholar] [CrossRef] [Green Version]
- Mindell, J.A. Lysosomal Acidification Mechanisms. Annu. Rev. Physiol. 2012, 74, 69–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Futai, M.; Sun-Wada, G.H.; Wada, Y.; Matsumoto, N.; Nakanishi-Matsui, M. Vacuolar-type ATPase: A proton pump to lysosomal trafficking. Proc. Jpn. Acad. Ser. B Phys. Biol. Sci. 2019, 95, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, X.; Meima, M.E.; Nelson, J.K.; Sorrentino, V.; Loregger, A.; Scheij, S.; Dekkers, D.H.; Mulder, M.T.; Demmers, J.A.; M-Dallinga-Thie, G.; et al. Identification of the (Pro)renin Receptor as a Novel Regulator of Low-Density Lipoprotein MetabolismNovelty and Significance. Circ. Res. 2016, 118, 222–229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeganeh, B.; Ghavami, S.; Kroeker, A.L.; Mahood, T.H.; Stelmack, G.L.; Klonisch, T.; Coombs, K.; Halayko, A.J. Suppression of influenza A virus replication in human lung epithelial cells by noncytotoxic concentrations bafilomycin A1. Am. J. Physiol. Cell. Mol. Physiol. 2015, 308, L270–L286. [Google Scholar] [CrossRef] [PubMed]
- Müller, K.H.; E Kainov, D.; El Bakkouri, K.; Saelens, X.; De Brabander, J.K.; Kittel, C.; Samm, E.; Muller, C.P. The proton translocation domain of cellular vacuolar ATPase provides a target for the treatment of influenza A virus infections. Br. J. Pharmacol. 2011, 164, 344–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gary-Bobo, M.; Nirdé, P.; Jeanjean, A.; Morère, A.; Garcia, M. Mannose 6-Phosphate Receptor Targeting and its Applications in Human Diseases. Curr. Med. Chem. 2007, 14, 2945–2953. [Google Scholar] [CrossRef] [Green Version]
- Abu-Remaileh, M.; Wyant, G.A.; Kim, C.; Laqtom, N.N.; Abbasi, M.; Chan, S.H.; Freinkman, E.; Sabatini, D.M. Lysosomal metabolomics reveals V-ATPase- and mTOR-dependent regulation of amino acid efflux from lysosomes. Science 2017, 358, 807–813. [Google Scholar] [CrossRef] [Green Version]
- Efeyan, A.; Zoncu, R.; Sabatini, D.M. Amino acids and mTORC1: From lysosomes to disease. Trends Mol. Med. 2012, 18, 524–533. [Google Scholar] [CrossRef]
- Zoncu, R.; Bar-Peled, L.; Efeyan, A.; Wang, S.; Sancak, Y.; Sabatini, D.M. mTORC1 Senses Lysosomal Amino Acids Through an Inside-Out Mechanism That Requires the Vacuolar H+-ATPase. Science 2011, 334, 678–683. [Google Scholar] [CrossRef] [Green Version]
- Stransky, L.A.; Forgac, M. Amino Acid Availability Modulates Vacuolar H+-ATPase Assembly. J. Biol. Chem. 2015, 290, 27360–27369. [Google Scholar] [CrossRef] [Green Version]
- Rama, S.; Boumedine-Guignon, N.; Sangiardi, M.; Youssouf, F.; Maulet, Y.; Lévêque, C.; Belghazi, M.; Seagar, M.; Debanne, D.; El Far, O. Chromophore-Assisted Light Inactivation of the V-ATPase V0c Subunit Inhibits Neurotransmitter Release Downstream of Synaptic Vesicle Acidification. Mol. Neurobiol. 2018, 56, 3591–3602. [Google Scholar] [CrossRef]
- Morel, N.; Poëa-Guyon, S. The membrane domain of vacuolar H+ATPase: A crucial player in neurotransmitter exocytotic release. Cell. Mol. Life Sci. 2015, 72, 2561–2573. [Google Scholar] [CrossRef] [PubMed]
- Morel, N. Neurotransmitter release: The dark side of the vacuolar-H+ATPase. Biol. Cell 2003, 95, 453–457. [Google Scholar] [CrossRef] [Green Version]
- Marshansky, V.R.; Grüber, G. Eukaryotic V-ATPase: Novel structural findings and functional insights. Biochim. Biophys. Acta 2014, 1837, 23. [Google Scholar] [CrossRef] [Green Version]
- Sun-Wada, G.H.; Toyomura, T.; Murata, Y.; Yamamoto, A.; Futai, M.; Wada, Y. The a3 isoform of V-ATPase regulates insulin secretion from pancreatic beta-cells. J. Cell. Sci. 2006, 119, 4531–4540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun-Wada, G.-H.; Tabata, H.; Kawamura, N. Selective Assembly of V-ATPase Subunit Isoforms in Mouse Kidney. J. Bioenerg. Biomembr. 2005, 37, 415–418. [Google Scholar] [CrossRef] [PubMed]
- Brown, D.; Sabolic, I.; Gluck, S. Polarized targeting of V-ATPase in kidney epithelial cells. J. Exp. Biol. 1992, 172, 231–243. [Google Scholar] [CrossRef]
- Hermo, L.; I Adamali, H.; Andonian, S. Immunolocalization of CA II and H+ V-ATPase in epithelial cells of the mouse and rat epididymis. J. Androl. 2000, 21, 376–391. [Google Scholar]
- Pietrement, C.; Sun-Wada, G.-H.; Da Silva, N.; McKee, M.; Marshansky, V.; Brown, D.; Futai, M.; Breton, S. Distinct Expression Patterns of Different Subunit Isoforms of the V-ATPase in the Rat Epididymis1. Biol. Reprod. 2006, 74, 185–194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qin, A.; Cheng, T.; Pavlos, N.; Lin, Z.; Dai, K.; Zheng, M. V-ATPases in osteoclasts: Structure, function and potential inhibitors of bone resorption. Int. J. Biochem. Cell Biol. 2012, 44, 1422–1435. [Google Scholar] [CrossRef]
- Kartner, N.; Manolson, M. V-ATPase subunit interactions: The long road to therapeutic targeting. Curr. Protein Pept. Sci. 2012, 13, 164–179. [Google Scholar] [CrossRef]
- Hinton, A.; Bond, S.; Forgac, M. V-ATPase functions in normal and disease processes. Pflügers Arch. Eur. J. Physiol. 2009, 457, 589–598. [Google Scholar] [CrossRef]
- Frattini, A.; Orchard, P.J.; Sobacchi, C.; Giliani, S.; Abinun, M.; Mattsson, J.P.; Keeling, D.J.; Andersson, A.-K.; Wallbrandt, P.; Zecca, L.; et al. Defects in TCIRG1 subunit of the vacuolar proton pump are responsible for a subset of human autosomal recessive osteopetrosis. Nat. Genet. 2000, 25, 343–346. [Google Scholar] [CrossRef]
- Nakhoul, N.L.; Hamm, L.L. Vacuolar H(+)-ATPase in the kidney. J. Nephrol. 2002, 15, 22–31. [Google Scholar]
- Kartner, N.; Yao, Y.; Li, K.; Crasto, G.J.; Datti, A.; Manolson, M.F. Inhibition of Osteoclast Bone Resorption by Disrupting Vacuolar H+-ATPase a3-B2 Subunit Interaction. J. Biol. Chem. 2010, 285, 37476–37490. [Google Scholar] [CrossRef] [Green Version]
- Blair, H.C.; Teitelbaum, S.L.; Ghiselli, R.; Gluck, S. Osteoclastic bone resorption by a polarized vacuolar proton pump. Science 1989, 245, 855–857. [Google Scholar] [CrossRef] [PubMed]
- Collins, M.P.; Forgac, M. Regulation of V-ATPase Assembly in Nutrient Sensing and Function of V-ATPases in Breast Cancer Metastasis. Front. Physiol. 2018, 9, 902. [Google Scholar] [CrossRef] [PubMed]
- Michel, V.; Munoz, Y.L.; Trujillo, K.; Bisoffi, M.; Parra, K.J. Inhibitors of vacuolar ATPase proton pumps inhibit human prostate cancer cell invasion and prostate-specific antigen expression and secretion. Int. J. Cancer 2013, 132, E1–E10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, Y.S.; Jun, S.; Kim, M.J.; Lee, S.H.; Suh, H.N.; Lien, E.M.; Jung, H.Y.; Lee, S.; Zhang, J.; Yang, J.I.; et al. TMEM9 promotes intestinal tumorigenesis through vacuolar-ATPase-activated Wnt/beta-catenin signalling. Nat. Cell Biol. 2018, 20, 1421–1433. [Google Scholar] [CrossRef]
- Wang, D.; Epstein, D.; Khalaf, O.; Srinivasan, S.; Williamson, W.R.; Fayyazuddin, A.; Quiocho, F.A.; Hiesinger, P.R. Ca2+-Calmodulin regulates SNARE assembly and spontaneous neurotransmitter release via v-ATPase subunit V0a1. J. Cell Biol. 2014, 205, 21–31. [Google Scholar] [CrossRef] [Green Version]
- Portela, M.; Yang, L.; Paul, S.; Li, X.; Veraksa, A.; Parsons, L.M.; Richardson, H.E. Lgl reduces endosomal vesicle acidification and Notch signaling by promoting the interaction between Vap33 and the V-ATPase complex. Sci. Signal. 2018, 11, eaar1976. [Google Scholar] [CrossRef] [Green Version]
- Abbas, Y.M.; Wu, D.; Bueler, S.A.; Robinson, C.V.; Rubinstein, J.L. Structure of V-ATPase from the mammalian brain. Science 2020, 367, 1240–1246. [Google Scholar] [CrossRef]
- Jansen, E.J.; Scheenen, W.J.; Hafmans, T.G.; Martens, G.J. Accessory subunit Ac45 controls the V-ATPase in the regulated secretory pathway. Biochim. Biophys. Acta (BBA) Bioenerg. 2008, 1783, 2301–2310. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazhab-Jafari, M.T.; Rohou, A.; Schmidt, C.; Bueler, S.A.; Benlekbir, S.; Robinson, C.; Rubinstein, J.L. Atomic model for the membrane-embedded VO motor of a eukaryotic V-ATPase. Nat. Cell Biol. 2016, 539, 118–122. [Google Scholar] [CrossRef]
- Kawasaki-Nishi, S.; Nishi, T.; Forgac, M. Proton translocation driven by ATP hydrolysis in V-ATPases. FEBS Lett. 2003, 545, 76–85. [Google Scholar] [CrossRef] [Green Version]
- Flannery, A.R.; Graham, L.A.; Stevens, T.H. Topological Characterization of the c, c′, and c″ Subunits of the Vacuolar ATPase from the Yeast Saccharomyces cerevisiae. J. Biol. Chem. 2004, 279, 39856–39862. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Cipriano, D.J.; Forgac, M. Arrangement of Subunits in the Proteolipid Ring of the V-ATPase. J. Biol. Chem. 2007, 282, 34058–34065. [Google Scholar] [CrossRef] [Green Version]
- Kawasaki-Nishi, S.; Nishi, T.; Forgac, M. Arg-735 of the 100-kDa subunit a of the yeast V-ATPase is essential for proton translocation. Proc. Natl. Acad. Sci. USA 2001, 98, 12397–12402. [Google Scholar] [CrossRef] [Green Version]
- Oot, R.A.; Kane, P.M.; Berry, E.A.; Wilkens, S. Crystal structure of yeast V1-ATPase in the autoinhibited state. EMBO J. 2016, 35, 1694–1706. [Google Scholar] [CrossRef]
- Smardon, A.M.; Tarsio, M.; Kane, P.M. The RAVE Complex Is Essential for Stable Assembly of the Yeast V-ATPase. J. Biol. Chem. 2002, 277, 13831–13839. [Google Scholar] [CrossRef] [Green Version]
- Kane, P.M. Targeting Reversible Disassembly as a Mechanism of Controlling V-ATPase Activity. Curr. Protein Pept. Sci. 2012, 13, 117–123. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Oot, R.A.; Wilkens, S. MgATP hydrolysis destabilizes the interaction between subunit H and yeast V1-ATPase, highlighting H’s role in V-ATPase regulation by reversible disassembly. J. Biol. Chem. 2018, 293, 10718–10730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, M.; Holliday, L.S.; Zhang, L.; Dunn, W.A., Jr.; Gluck, S.L. Interaction between aldolase and vacuolar H+-ATPase: Evidence for direct coupling of glycolysis to the ATP-hydrolyzing proton pump. J. Biol. Chem. 2001, 276, 30407–30413. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, M.; Sautin, Y.; Holliday, L.S.; Gluck, S.L. The Glycolytic Enzyme Aldolase Mediates Assembly, Expression, and Activity of Vacuolar H+-ATPase. J. Biol. Chem. 2004, 279, 8732–8739. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Zhang, C.-S.; Zong, Y.; Feng, J.-W.; Ma, T.; Hu, M.; Lin, Z.; Li, X.; Xie, C.; Wu, Y.; et al. Transient Receptor Potential V Channels Are Essential for Glucose Sensing by Aldolase and AMPK. Cell Metab. 2019, 30, 508–524. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Banerjee, S.; Kane, P.M. Regulation of V-ATPase Activity and Organelle pH by Phosphatidylinositol Phosphate Lipids. Front. Cell Dev. Biol. 2020, 8, 510. [Google Scholar] [CrossRef]
- Fogarty, F.M.; O’Keeffe, J.; Zhadanov, A.; Papkovsky, D.; Ayllón, V.; O’Connor, R. HRG-1 enhances cancer cell invasive potential and couples glucose metabolism to cytosolic/extracellular pH gradient regulation by the vacuolar-H+ ATPase. Oncogene 2013, 33, 4653–4663. [Google Scholar] [CrossRef] [Green Version]
- Marshansky, V.; Futai, M. The V-type H+-ATPase in vesicular trafficking: Targeting, regulation and function. Curr. Opin. Cell Biol. 2008, 20, 415–426. [Google Scholar] [CrossRef] [PubMed]
- Qi, J.; Wang, Y.; Forgac, M. The vacuolar (H+)-ATPase: subunit arrangement and in vivo regulation. J. Bioenerg. Biomembr. 2007, 39, 423–426. [Google Scholar] [CrossRef]
- Rahman, S.; Yamato, I.; Murata, T. Function and Regulation of Mammalian V-ATPase Isoforms. In Regulation of Ca2+-ATPases, V-ATPases and F-ATPases; Springer Science and Business Media LLC: Cham, Switzerland, 2016; Volume 14, pp. 283–299. [Google Scholar]
- Lafourcade, C.; Sobo, K.; Kieffer-Jaquinod, S.; Garin, J.; Van Der Goot, F.G. Regulation of the V-ATPase along the Endocytic Pathway Occurs through Reversible Subunit Association and Membrane Localization. PLoS ONE 2008, 3, e2758. [Google Scholar] [CrossRef] [Green Version]
- Esmail, S.; Kartner, N.; Yao, Y.; Kim, J.W.; Reithmeier, R.A.F.; Manolson, M.F. N-linked glycosylation of a subunit isoforms is critical for vertebrate vacuolar H(+) -ATPase (V-ATPase) biosynthesis. J. Cell Biochem. 2018, 119, 861–875. [Google Scholar] [CrossRef]
- Manolson, M.F.; Wu, B.; Proteau, D.; Taillon, B.E.; Roberts, B.T.; Hoyt, M.A.; Jones, E.W. STV1 gene encodes functional homologue of 95-kDa yeast vacuolar H(+)-ATPase subunit Vph1p. J. Biol. Chem. 1994, 269, 14064–14074. [Google Scholar] [CrossRef]
- Srinivasan, S.; Vyas, N.K.; Baker, M.L.; Quiocho, F.A. Erratum to “Crystal Structure of the Cytoplasmic N-Terminal Domain of Subunit I, a Homolog of Subunit a, of V-ATPase” [J. Mol. Biol. 412/1 (2011) 14–21]. J. Mol. Biol. 2011, 413, 523. [Google Scholar] [CrossRef]
- Kawasaki-Nishi, S.; Nishi, T.; Forgac, M.; Garofano, A.; Zwicker, K.; Kerscher, S.; Okun, P.; Brandt, U. Interacting Helical Surfaces of the Transmembrane Segments of Subunits a and c′ of the Yeast V-ATPase Defined by Disulfide-mediated Cross-linking. J. Biol. Chem. 2003, 278, 41908–41913. [Google Scholar] [CrossRef] [Green Version]
- Ochotny, N.; Flenniken, A.M.; Owen, C.; Voronov, I.; A Zirngibl, R.; Osborne, L.R.; E Henderson, J.; Adamson, S.L.; Rossant, J.; Manolson, M.; et al. The V-ATPase a3 subunit mutation R740S is dominant negative and results in osteopetrosis in mice. J. Bone Miner. Res. 2011, 26, 1484–1493. [Google Scholar] [CrossRef]
- Kawasaki-Nishi, S.; Bowers, K.; Nishi, T.; Forgac, M.; Stevens, T.H. The Amino-terminal Domain of the Vacuolar Proton-translocating ATPase a Subunit Controls Targeting and in Vivo Dissociation, and the Carboxyl-terminal Domain Affects Coupling of Proton Transport and ATP Hydrolysis. J. Biol. Chem. 2001, 276, 47411–47420. [Google Scholar] [CrossRef] [Green Version]
- Finnigan, G.C.; Cronan, G.E.; Park, H.J.; Srinivasan, S.; Quiocho, F.A.; Stevens, T.H. Sorting of the yeast vacuolar-type, proton-translocating ATPase enzyme complex (V-ATPase): Identification of a necessary and sufficient Golgi/endosomal retention signal in Stv1p. J. Biol. Chem. 2012, 287, 19487–194500. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.; Wang, D.; Volk, E.; Bellen, H.J.; Hiesinger, P.R.; Quiocho, F.A. V-ATPase V0 sector subunit a1 in neurons is a target of calmodulin. J. Biol. Chem. 2008, 283, 294–300. [Google Scholar] [CrossRef] [Green Version]
- Saw, N.M.N.; Kang, S.-Y.A.; Parsaud, L.; Han, G.A.; Jiang, T.; Grzegorczyk, K.; Surkont, M.; Sun-Wada, G.-H.; Wada, Y.; Li, L.; et al. Vacuolar H+-ATPase subunits Voa1 and Voa2 cooperatively regulate secretory vesicle acidification, transmitter uptake, and storage. Mol. Biol. Cell 2011, 22, 3394–3409. [Google Scholar] [CrossRef]
- Matsumoto, N.; Sekiya, M.; Tohyama, K.; Ishiyama-Matsuura, E.; Sun-Wada, G.-H.; Wada, Y.; Futai, M.; Nakanishi-Matsui, M. Essential Role of the a3 Isoform of V-ATPase in Secretory Lysosome Trafficking via Rab7 Recruitment. Sci. Rep. 2018, 8, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, N.; Daido, S.; Sun-Wada, G.-H.; Wada, Y.; Futai, M.; Nakanishi-Matsui, M. Diversity of proton pumps in osteoclasts: V-ATPase with a3 and d2 isoforms is a major form in osteoclasts. Biochim. et Biophys. Acta (BBA) Bioenerg. 2014, 1837, 744–749. [Google Scholar] [CrossRef] [Green Version]
- Oka, T.; Murata, Y.; Namba, M.; Yoshimizu, T.; Toyomura, T.; Yamamoto, A.; Sun-Wada, G.-H.; Hamasaki, N.; Wada, Y.; Futai, M. a4, a Unique Kidney-specific Isoform of Mouse Vacuolar H+-ATPase Subunit a. J. Biol. Chem. 2001, 276, 40050–40054. [Google Scholar] [CrossRef] [Green Version]
- McGuire, C.M.; Collins, M.P.; Sun-Wada, G.; Wada, Y.; Forgac, M. Isoform-specific gene disruptions reveal a role for the V-ATPase subunit a4 isoform in the invasiveness of 4T1-12B breast cancer cells. J. Biol. Chem. 2019, 294, 11248–11258. [Google Scholar] [CrossRef] [Green Version]
- Toyomura, T.; Oka, T.; Yamaguchi, C.; Wada, Y.; Futai, M. Three subunit a isoforms of mouse vacuolar H(+)-ATPase. Preferential expression of the a3 isoform during osteoclast differentiation. J. Biol. Chem. 2000, 275, 8760–8765. [Google Scholar] [CrossRef] [Green Version]
- Kornak, U.; Schulz, A.; Friedrich, W.; Uhlhaas, S.; Kremens, B.; Voit, T.; Hasan, C.; Bode, U.; Jentsch, T.J.; Kubisch, C. Mutations in the a3 subunit of the vacuolar H(+)-ATPase cause infantile malignant osteopetrosis. Hum. Mol. Genet. 2000, 9, 2059–2063. [Google Scholar] [CrossRef] [Green Version]
- Zirngibl, R.A.; Wang, A.; Yao, Y.; Manolson, M.; Krueger, J.; Dupuis, L.; Mendoza-Londono, R.; Voronov, I. Novel c.G630A TCIRG1 mutation causes aberrant splicing resulting in an unusually mild form of autosomal recessive osteopetrosis. J. Cell. Biochem. 2019, 120, 17180–17193. [Google Scholar] [CrossRef]
- Duan, X.; Yang, S.; Zhang, L.; Yang, T. V-ATPases and osteoclasts: Ambiguous future of V-ATPases inhibitors in osteoporosis. Theranostics 2018, 8, 5379–5399. [Google Scholar] [CrossRef]
- Kartner, N.; Manolson, M.F. The Vacuolar Proton ATPase (V-ATPase): Regulation and Therapeutic Targeting. In Regulation of Ca2+-ATPases, V-ATPases and F-ATPases, 1st ed.; Chakraborti, S., Dhalla, N.S., Eds.; Springer International Publishing: Cham, Switzerland, 2016; Volume 14, pp. 407–437. [Google Scholar]
- Nishi, T.; Kawasaki-Nishi, S.; Forgac, M. Expression and Function of the Mouse V-ATPase d Subunit Isoforms. J. Biol. Chem. 2003, 278, 46396–46402. [Google Scholar] [CrossRef] [Green Version]
- Nishi, T.; Forgac, M. Molecular Cloning and Expression of Three Isoforms of the 100-kDa a Subunit of the Mouse Vacuolar Proton-translocating ATPase. J. Biol. Chem. 2000, 275, 6824–6830. [Google Scholar] [CrossRef] [Green Version]
- Crasto, G.J.; Kartner, N.; Yao, Y.; Li, K.; Bullock, L.; Datti, A.; Manolson, M.F. Luteolin inhibition of V-ATPase a3-d2 interaction decreases osteoclast resorptive activity. J. Cell Biochem. 2013, 114, 929–941. [Google Scholar] [CrossRef]
- Lee, B.S.; Holliday, L.S.; Ojikutu, B.; Krits, I.; Gluck, S.L. Osteoclasts express the B2 isoform of vacuolar H(+)-ATPase intracellularly and on their plasma membranes. Am. J. Physiol. Physiol. 1996, 270, C382–C388. [Google Scholar] [CrossRef]
- Smith, A.N.; Jouret, F.; Bord, S.; Borthwick, K.J.; Al-Lamki, R.S.; Wagner, C.A.; Ireland, D.C.; Cormier-Daire, V.; Frattini, A.; Villa, A.; et al. Vacuolar H+-ATPase d2 Subunit: Molecular Characterization, Developmental Regulation, and Localization to Specialized Proton Pumps in Kidney and Bone. J. Am. Soc. Nephrol. 2005, 16, 1245–1256. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.-H.; Rho, J.; Jeong, D.; Sul, J.-Y.; Kim, T.; Kim, N.; Kang, J.-S.; Miyamoto, T.; Suda, T.; Lee, S.-K.; et al. v-ATPase V0 subunit d2–deficient mice exhibit impaired osteoclast fusion and increased bone formation. Nat. Med. 2006, 12, 1403–1409. [Google Scholar] [CrossRef]
- Jobst-Schwan, T.; Klämbt, V.; Tarsio, M.; Heneghan, J.F.; Majmundar, A.J.; Shril, S.; Buerger, F.; Ottlewski, I.; Shmukler, B.E.; Topaloglu, R.; et al. Whole exome sequencing identified ATP6V1C2 as a novel candidate gene for recessive distal renal tubular acidosis. Kidney Int. 2020, 97, 567–579. [Google Scholar] [CrossRef]
- Aoto, K.; Kato, M.; Akita, T.; Nakashima, M.; Mutoh, H.; Akasaka, N.; Tohyama, J.; Nomura, Y.; Hoshino, K.; Ago, Y.; et al. ATP6V0A1 encoding the a1-subunit of the V0 domain of vacuolar H+-ATPases is essential for brain development in humans and mice. Nat. Commun. 2021, 12, 1–17. [Google Scholar] [CrossRef]
- Howaldt, A.; Nampoothiri, S.; Quell, L.-M.; Ozden, A.; Fischer-Zirnsak, B.; Collet, C.; de Vernejoul, M.-C.; Doneray, H.; Kayserili, H.; Kornak, U. Sclerosing bone dysplasias with hallmarks of dysosteosclerosis in four patients carrying mutations in SLC29A3 and TCIRG1. Bone 2019, 120, 495–503. [Google Scholar] [CrossRef]
- Cao, W.-H.; Wei, W.-B.; Yu, G.; Li, L.; Wu, Q. Complex Heterozygous Mutation in the T-cell Immune Regulator 1 Gene Associated with Severe Ocular Characteristics of Osteopetrosis in an Infant. Chin. Med J. 2018, 131, 354–356. [Google Scholar] [CrossRef]
- Palagano, E.; Blair, H.C.; Pangrazio, A.; Tourkova, I.; Strina, D.; Angius, A.; Cuccuru, G.; Oppo, M.; Uva, P.; Van Hul, W.; et al. Buried in the Middle but Guilty: Intronic Mutations in the TCIRG1 Gene Cause Human Autosomal Recessive Osteopetrosis. J. Bone Miner. Res. 2015, 30, 1814–1821. [Google Scholar] [CrossRef]
- Sobacchi, C.; Pangrazio, A.; Lopez, A.G.; Gomez, D.P.; Caldana, M.E.; Susani, L.; Vezzoni, P.; Villa, A. As Little as Needed: The Extraordinary Case of a Mild Recessive Osteopetrosis Owing to a Novel Splicing Hypomorphic Mutation in the TCIRG1 Gene. J. Bone Miner. Res. 2014, 29, 1646–1650. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.-Y.; He, J.-W.; Fu, W.-Z.; Wang, C.; Zhang, Z.-L. Novel mutations of TCIRG1 cause a malignant and mild phenotype of autosomal recessive osteopetrosis (ARO) in four Chinese families. Acta Pharmacol. Sin. 2017, 38, 1456–1465. [Google Scholar] [CrossRef]
- Voronov, I.; Ochotny, N.; Jaumouillé, V.; Owen, C.; Manolson, M.F.; Aubin, J.E. The R740S mutation in the V-ATPase a3 subunit increases lysosomal pH, impairs NFATc1 translocation, and decreases in vitro osteoclastogenesis. J. Bone Miner. Res. 2012, 28, 108–118. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, A.; Voronov, I.; Wang, Y.; Glogauer, M.; Kartner, N.; Manolson, M.F. Osteopetrosis Mutation R444L Causes Endoplasmic Reticulum Retention and Misprocessing of Vacuolar H+-ATPase a3 Subunit. J. Biol. Chem. 2012, 287, 26829–26839. [Google Scholar] [CrossRef] [Green Version]
- Ochotny, N.; Van Vliet, A.; Chan, N.; Yao, Y.; Morel, M.; Kartner, N.; von Schroeder, H.P.; Heersche, J.N.M.; Manolson, M.F. Effects of Human a3 and a4 Mutations That Result in Osteopetrosis and Distal Renal Tubular Acidosis on Yeast V-ATPase Expression and Activity. J. Biol. Chem. 2006, 281, 26102–26111. [Google Scholar] [CrossRef] [Green Version]
- Matsumoto, N.; Matsukawa, R.; Takahashi, S.; Kudo, K.; Sun-Wada, G.-H.; Wada, Y.; Nakanishi-Matsui, M. V-ATPase a3 isoform mutations identified in osteopetrosis patients abolish its expression and disrupt osteoclast function. Exp. Cell Res. 2020, 389, 111901. [Google Scholar] [CrossRef]
- Matsumoto, N.; Sekiya, M.; Fujimoto, Y.; Haga, S.; Sun-Wada, G.-H.; Wada, Y.; Nakanishi-Matsui, M. Functional complementation of V-ATPase a subunit isoforms in osteoclasts. J. Biochem. 2021, 169, 459–466. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Clapp, K.; Tarsio, M.; Kane, P.M. Interaction of the late endo-lysosomal lipid PI(3,5)P2 with the Vph1 isoform of yeast V-ATPase increases its activity and cellular stress tolerance. J. Biol. Chem. 2019, 294, 9161–9171. [Google Scholar] [CrossRef] [PubMed]
- Banerjee, S.; Kane, P.M. Direct interaction of the Golgi V-ATPase a-subunit isoform with PI(4)P drives localization of Golgi V-ATPases in yeast. Mol. Biol. Cell 2017, 28, 2518–2530. [Google Scholar] [CrossRef]
- Vasanthakumar, T.; Bueler, S.A.; Wu, D.; Beilsten-Edmands, V.; Robinson, C.V.; Rubinstein, J.L. Structural comparison of the vacuolar and Golgi V-ATPases from Saccharomyces cerevisiae. Proc. Natl. Acad. Sci. USA 2019, 116, 7272–7277. [Google Scholar] [CrossRef] [Green Version]
- Al-Bataineh, M.M.; Gong, F.; Marciszyn, A.L.; Myerburg, M.M.; Pastor-Soler, N.M. Regulation of proximal tubule vacuolar H(+)-ATPase by PKA and AMP-activated protein kinase. Am. J. Physiol. Physiol. 2014, 306, F981–F995. [Google Scholar] [CrossRef] [Green Version]
- Alzamora, R.; Al-Bataineh, M.M.; Liu, W.; Gong, F.; Li, H.; Thali, R.F.; Joho-Auchli, Y.; Brunisholz, R.A.; Satlin, L.M.; Neumann, D.; et al. AMP-activated protein kinase regulates the vacuolar H+-ATPase via direct phosphorylation of the A subunit (ATP6V1A) in the kidney. Am. J. Physiol. Physiol. 2013, 305, F943–F956. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soliman, M.; Seo, J.-Y.; Kim, D.-S.; Kim, J.-Y.; Park, J.-G.; Alfajaro, M.M.; Baek, Y.-B.; Cho, E.-H.; Kwon, J.; Choi, J.-S.; et al. Activation of PI3K, Akt, and ERK during early rotavirus infection leads to V-ATPase-dependent endosomal acidification required for uncoating. PLoS Pathog. 2018, 14, e1006820. [Google Scholar] [CrossRef] [Green Version]
- Soliman, M.; Kim, D.-S.; Park, J.-G.; Kim, J.-Y.; Alfajaro, M.M.; Baek, Y.-B.; Cho, E.-H.; Park, C.-H.; Kang, M.-I.; Park, S.-I.; et al. Phosphatidylinositol 3-Kinase/Akt and MEK/ERK Signaling Pathways Facilitate Sapovirus Trafficking and Late Endosomal Acidification for Viral Uncoating in LLC-PK Cells. J. Virol. 2018, 92, 24. [Google Scholar] [CrossRef] [Green Version]
- Marjuki, H.; Gornitzky, A.; Marathe, B.M.; Ilyushina, N.A.; Aldridge, J.R.; Desai, G.; Webby, R.J.; Webster, R.G. Influenza A virus-induced early activation of ERK and PI3K mediates V-ATPase-dependent intracellular pH change required for fusion. Cell. Microbiol. 2010, 13, 587–601. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roy, M.; Roux, S. Rab GTPases in Osteoclastic Bone Resorption and Autophagy. Int. J. Mol. Sci. 2020, 21, 7655. [Google Scholar] [CrossRef] [PubMed]
- Stenmark, H. Rab GTPases as coordinators of vesicle traffic. Nat. Rev. Mol. Cell Biol. 2009, 10, 513–525. [Google Scholar] [CrossRef] [PubMed]
- Hosokawa, H.; Dip, P.V.; Merkulova, M.; Bakulina, A.; Zhuang, Z.; Khatri, A.; Jian, X.; Keating, S.M.; Bueler, S.A.; Rubinstein, J.L.; et al. The N termini of a-subunit isoforms are involved in signaling between vacuolar H+-ATPase (V-ATPase) and cytohesin-2. J. Biol. Chem. 2013, 288, 5896–5913. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bar-Peled, L.; Schweitzer, L.D.; Zoncu, R.; Sabatini, D.M. Ragulator Is a GEF for the Rag GTPases that Signal Amino Acid Levels to mTORC1. Cell 2012, 150, 1196–1208. [Google Scholar] [CrossRef] [Green Version]
- Shen, K.; Sabatini, D.M. Ragulator and SLC38A9 activate the Rag GTPases through noncanonical GEF mechanisms. Proc. Natl. Acad. Sci. USA 2018, 115, 9545–9550. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.-P.; Chen, W.; Liang, Y.; Li, E.; Stashenko, P. Atp6i-deficient mice exhibit severe osteopetrosis due to loss of osteoclast-mediated extracellular acidification. Nat. Genet. 1999, 23, 447–451. [Google Scholar] [CrossRef]
- Bollerslev, J.; Marks, S.C., Jr.; Pockwinse, S.; Kassem, M.; Brixen, K.; Steiniche, T.; Mosekilde, L. Ultrastructural investigations of bone resorptive cells in two types of autosomal dominant osteopetrosis. Bone 1993, 14, 865–869. [Google Scholar] [CrossRef]
- Karsdal, M.A.; Martin, T.J.; Bollerslev, J.; Christiansen, C.; Henriksen, K. Are Nonresorbing Osteoclasts Sources of Bone Anabolic Activity? J. Bone Miner. Res. 2007, 22, 487–494. [Google Scholar] [CrossRef] [PubMed]
- Black, D.M.; Greenspan, S.L.; Ensrud, K.E.; Palermo, L.; McGowan, J.A.; Lang, T.F.; Garnero, P.; Bouxsein, M.L.; Bilezikian, J.P.; Rosen, C.J.; et al. The effects of parathyroid hormone and alendronate alone or in combination in postmenopausal osteoporosis. N. Engl. J. Med. 2003, 349, 1207–1215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finkelstein, J.S.; Hayes, A.; Hunzelman, J.L.; Wyland, J.J.; Lee, H.; Neer, R.M. The Effects of Parathyroid Hormone, Alendronate, or Both in Men with Osteoporosis. N. Engl. J. Med. 2003, 349, 1216–1226. [Google Scholar] [CrossRef] [PubMed]
- Santos-Pereira, C.; Rodrigues, L.R.; Côrte-Real, M. Emerging insights on the role of V-ATPase in human diseases: Therapeutic challenges and opportunities. Med. Res. Rev. 2021, 41, 1927–1964. [Google Scholar] [CrossRef]
- Toro, E.J.; Zuo, J.; Ostrov, D.A.; Catalfamo, D.; Bradaschia-Correa, V.; Arana-Chavez, V.; Caridad, A.R.; Neubert, J.K.; Wronski, T.J.; Wallet, S.M.; et al. Enoxacin Directly Inhibits Osteoclastogenesis without Inducing Apoptosis. J. Biol. Chem. 2012, 287, 17894–17904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ostrov, D.A.; Magis, A.T.; Wronski, T.J.; Chan, E.K.L.; Toro, E.J.; Donatelli, R.E.; Sajek, K.; Haroun, I.N.; Nagib, M.I.; Piedrahita, A.; et al. Identification of Enoxacin as an Inhibitor of Osteoclast Formation and Bone Resorption by Structure-Based Virtual Screening. J. Med. Chem. 2009, 52, 5144–5151. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Xu, X.; Liu, X.; Mao, C.; Qin, A.; Lu, E. Bis-enoxacin blocks alveolar bone resorption in rats with ovariectomy-induced osteoporosis. Mol. Med. Rep. 2017, 17, 3232–3238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Toro, E.; Zuo, J.; Guiterrez, A.; La Rosa, R.; Gawron, A.; Bradaschia-Correa, V.; Arana-Chavez, V.; Dolce, C.; Rivera, M.; Kesavalu, L.; et al. Bis-enoxacin Inhibits Bone Resorption and Orthodontic Tooth Movement. J. Dent. Res. 2013, 92, 925–931. [Google Scholar] [CrossRef] [Green Version]
- Kartner, N.; Yao, Y.; Bhargava, A.; Manolson, M.F. Topology, glycosylation and conformational changes in the membrane domain of the vacuolar H+-ATPase a subunit. J. Cell Biochem. 2013, 114, 1474–1487. [Google Scholar] [CrossRef] [PubMed]
- Kulshrestha, A.; Katara, G.K.; Ibrahim, S.A.; Riehl, V.E.; Schneiderman, S.; Bilal, M.; Young, A.N.; Levine, S.; Fleetwood, S.; Dolan, J.; et al. In vivo anti-V-ATPase antibody treatment delays ovarian tumor growth by increasing anti-tumor immune responses. Mol. Oncol. 2020, 14, 2436–2454. [Google Scholar] [CrossRef]
- Kulshrestha, A.; Katara, G.K.; Ibrahim, S.; Pamarthy, S.; Jaiswal, M.K.; Sachs, A.G.; Beaman, K.D. Vacuolar ATPase ‘a2’ isoform exhibits distinct cell surface accumulation and modulates matrix metalloproteinase activity in ovarian cancer. Oncotarget 2015, 6, 3797–3810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, Y.; Nyman, J.; Muhonen, P.; Väänanen, H.K.; Laitala-Leinonen, T. Inhibition of the osteoclast V-ATPase by small interfering RNAs. FEBS Lett. 2005, 579, 4937–4942. [Google Scholar] [CrossRef] [Green Version]
- Jiang, H.; Chen, W.; Zhu, G.; Zhang, L.; Tucker, B.; Hao, L.; Feng, S.; Ci, H.; Ma, J.; Wang, L.; et al. RNAi-Mediated Silencing of Atp6i and Atp6i Haploinsufficiency Prevents Both Bone Loss and Inflammation in a Mouse Model of Periodontal Disease. PLOS ONE 2013, 8, e58599. [Google Scholar] [CrossRef] [Green Version]
- Capecci, J.; Forgac, M. The Function of Vacuolar ATPase (V-ATPase) a Subunit Isoforms in Invasiveness of MCF10a and MCF10CA1a Human Breast Cancer Cells. J. Biol. Chem. 2013, 288, 32731–32741. [Google Scholar] [CrossRef] [Green Version]
- Nishisho, T.; Hata, K.; Nakanishi, M.; Morita, Y.; Sun-Wada, G.-H.; Wada, Y.; Yasui, N.; Yoneda, T. The a3 Isoform Vacuolar Type H+-ATPase Promotes Distant Metastasis in the Mouse B16 Melanoma Cells. Mol. Cancer Res. 2011, 9, 845–855. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinton, A.; Sennoune, S.R.; Bond, S.; Fang, M.; Reuveni, M.; Sahagian, G.G.; Jay, D.; Martinez-Zaguilan, R.; Forgac, M. Function of a Subunit Isoforms of the V-ATPase in pH Homeostasis and in Vitro Invasion of MDA-MB231 Human Breast Cancer Cells. J. Biol. Chem. 2009, 284, 16400–16408. [Google Scholar] [CrossRef] [Green Version]
- Flinck, M.; Hagelund, S.; Gorbatenko, A.; Severin, M.; Pedraz-Cuesta, E.; Novak, I.; Stock, C.; Pedersen, S.F. The Vacuolar H+ ATPase α3 Subunit Negatively Regulates Migration and Invasion of Human Pancreatic Ductal Adenocarcinoma Cells. Cells 2020, 9, 465. [Google Scholar] [CrossRef] [Green Version]
- Maurizi, A.; Capulli, M.; Patel, R.; Curle, A.; Rucci, N.; Teti, A. RNA interference therapy for autosomal dominant osteopetrosis type 2. Towards the preclinical development. Bone 2018, 110, 343–354. [Google Scholar] [CrossRef]
- Havens, M.A.; Hastings, M.L. Splice-switching antisense oligonucleotides as therapeutic drugs. Nucleic Acids Res. 2016, 44, 6549–6563. [Google Scholar] [CrossRef]
- Couto-Vieira, J.; Nicolau-Neto, P.; Costa, E.P.; Figueira, F.F.; Simão, T.D.A.; Okorokova-Façanha, A.L.; Pinto, L.F.R.; Façanha, A.R. Multi-cancer V-ATPase molecular signatures: A distinctive balance of subunit C isoforms in esophageal carcinoma. EBioMedicine 2020, 51, 102581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pangrazio, A.; Caldana, M.E.; Lo Iacono, N.; Mantero, S.; Vezzoni, P.; Villa, A.; Sobacchi, C. Autosomal recessive osteopetrosis: Report of 41 novel mutations in the TCIRG1 gene and diagnostic implications. Osteoporos. Int. 2012, 23, 2713–2718. [Google Scholar] [CrossRef]
- Cao, W.; Wei, W.; Wu, Q. Ophthalmic phenotype of TCIRG1 gene mutations in Chinese infantile malignant osteopetrosis. BMJ Open Ophthalmol. 2018, 3, e000180. [Google Scholar] [CrossRef] [Green Version]
- Barvencik, F.; Kurth, I.; Koehne, T.; Stauber, T.; Zustin, J.; Tsiakas, K.; Ludwig, C.F.; Beil, F.T.; Pestka, J.M.; Hahn, M.; et al. CLCN7andTCIRG1Mutations Differentially Affect Bone Matrix Mineralization in Osteopetrotic Individuals. J. Bone Miner. Res. 2013, 29, 982–991. [Google Scholar] [CrossRef]
- Susani, L.; Pangrazio, A.; Sobacchi, C.; Taranta, A.; Mortier, G.; Savarirayan, R.; Villa, A.; Orchard, P.; Vezzoni, P.; Albertini, A.; et al. TCIRG1-dependent recessive osteopetrosis: Mutation analysis, functional identification of the splicing defects, andin vitro rescue by U1 snRNA. Hum. Mutat. 2004, 24, 225–235. [Google Scholar] [CrossRef]
- Sobacchi, C.; Frattini, A.; Orchard, P.; Porras, O.; Tezcan, I.; Andolina, M.; Babul-Hirji, R.; Baric, I.; Canham, N.; Chitayat, D.; et al. The mutational spectrum of human malignant autosomal recessive osteopetrosis. Hum. Mol. Genet. 2001, 10, 1767–1773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazzolari, E.; Forino, C.; Razza, A.; Porta, F.; Villa, A.; Notarangelo, L.D. A single-center experience in 20 patients with infantile malignant osteopetrosis. Am. J. Hematol. 2009, 84, 473–479. [Google Scholar] [CrossRef]
- Bahr, T.; Lund, T.; Sando, N.M.; Orchard, P.J.; Miller, W.P. Haploidentical transplantation with post-transplant cyclophosphamide following reduced-intensity conditioning for osteopetrosis: Outcomes in three children. Bone Marrow Transpl. 2016, 51, 1546–1548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, P.; Yue, Z.; Sun, L.; Huang, W.; Hu, B.; Yang, Z.; Hu, Y.; Xiao, H.; Shi, H.; Zhou, Q.; et al. Novel mutation of TCIRG1 and clinical pictures of two infantile malignant osteopetrosis patients. J. Bone Miner. Metab. 2010, 29, 251–256. [Google Scholar] [CrossRef]
- Hernandez-Martinez, C.; Guzman-Martinez, M.N.; Scheffler-Mendoza, S.; Espinosa-Padilla, S.E.; Sobacchi, C.; Blancas-Galicia, L. [Identification of new mutations in TCIRG1 as a cause of infantile malignant osteopetrosis in two Mexican patients]. Rev. Alerg. Mex. 2018, 65, 108–116. [Google Scholar]
- Scimeca, J.-C.; Quincey, D.; Parrinello, H.; Romatet, D.; Grosgeorge, J.; Gaudray, P.; Philip, N.; Fischer, A.; Carle, G.F. Novel mutations in theTCIRG1gene encoding the a3 subunit of the vacuolar proton pump in patients affected by infantile malignant osteopetrosis. Hum. Mutat. 2003, 21, 151–157. [Google Scholar] [CrossRef]
- Chavez-Guitron, L.E.; Ceron-Torres, T.; Sobacchi, C.; Ochoa-Ruiz, E.; Villegas-Huesca, S. Autosomal recessive osteopetrosis type I: Description of pathogenic variant of TCIRG1 gene. Bol. Med. Hosp. Infant. Mex. 2018, 75, 255–259. [Google Scholar] [PubMed]
- Shamriz, O.; Shaag, A.; Yaacov, B.; Eddin, A.N.; Weintraub, M.; Elpeleg, O.; Stepensky, P. The Use of Whole Exome Sequencing for the Diagnosis of Autosomal Recessive Malignant Infantile Osteopetrosis. Clin. Genet. 2016, 92, 80–85. [Google Scholar] [CrossRef]
- Ajmal, M.; Mir, A.; Wahid, S.; Khor, C.C.; Foo, J.N.; Siddiqi, S.; Kauser, M.; Malik, S.A.; Nasir, M. Identification and in silico characterization of a novel p.P208PfsX1 mutation in V-ATPase a3 subunit associated with autosomal recessive osteopetrosis in a Pakistani family. BMC Med Genet. 2017, 18, 148. [Google Scholar] [CrossRef] [Green Version]
- Al-Seraihy, A.; Al-Saedi, H.; Al-Ahmari, A.; Ghemlas, I.; Ayas, M. T-cell replete haploidentical transplantation with reduced post-transplant cyclophosphamide in six children with infantile osteopetrosis. Bone Marrow Transpl. 2021, 1–4. [Google Scholar] [CrossRef]
- Souraty, N.; Noun, P.; Djambas-Khayat, C.; Chouery, E.; Pangrazio, A.; Villa, A.; Lefranc, G.; Frattini, A.; Mégarbané, A. Molecular study of six families originating from the Middle-East and presenting with autosomal recessive osteopetrosis. Eur. J. Med. Genet. 2007, 50, 188–199. [Google Scholar] [CrossRef]
- Capo, V.; Penna, S.; Merelli, I.; Barcella, M.; Scala, S.; Basso-Ricci, L.; Draghici, E.; Palagano, E.; Zonari, E.; DeSantis, G.; et al. Expanded circulating hematopoietic stem/progenitor cells as novel cell source for the treatment of TCIRG1 osteopetrosis. Haematology 2020, 106, 74–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taranta, A.; Migliaccio, S.; Recchia, I.; Caniglia, M.; Luciani, M.; De Rossi, G.; Dionisi-Vici, C.; Pinto, R.M.; Francalanci, P.; Boldrini, R.; et al. Genotype-Phenotype Relationship in Human ATP6i-Dependent Autosomal Recessive Osteopetrosis. Am. J. Pathol. 2003, 162, 57–68. [Google Scholar] [CrossRef] [Green Version]
- Wada, K.; Harada, D.; Michigami, T.; Tachikawa, K.; Nakano, Y.; Kashiwagi, H.; Yamashita, S.; Sano, T.; Seino, Y. A case of autosomal dominant osteopetrosis type II with a novel TCIRG1 gene mutation. J. Pediatr. Endocrinol. Metab. 2013, 26, 575–577. [Google Scholar] [CrossRef]
- Vomero, A.; Tapie, A.; Arroyo, C.; Raggio, V.; Peluffo, G.; Dufort, G. Malignant Infantile osteopetrosis. Rev. Chil. Pediatr. 2019, 90, 443–447. [Google Scholar] [CrossRef]
- Bliznetz, E.A.; Tverskaya, S.M.; A Zinchenko, R.; Abrukova, A.V.; Savaskina, E.N.; Nikulin, M.V.; Kirillov, A.G.; Ginter, E.K.; Polyakov, A.V. Genetic analysis of autosomal recessive osteopetrosis in Chuvashiya: The unique splice site mutation in TCIRG1 gene spread by the founder effect. Eur. J. Hum. Genet. 2009, 17, 664–672. [Google Scholar] [CrossRef]
- Michigami, T.; Kageyama, T.; Satomura, K.; Shima, M.; Yamaoka, K.; Nakayama, M.; Ozono, K. Novel mutations in the a3 subunit of vacuolar H+-adenosine triphosphatase in a Japanese patient with infantile malignant osteopetrosis. Bone 2002, 30, 436–439. [Google Scholar] [CrossRef]
- Yu, T.; Yu, Y.; Wang, J.; Yin, L.; Zhou, Y.; Ying, D.; Huang, R.; Chen, H.; Wu, S.; Shen, Y.; et al. Identification of TCIRG1 and CLCN7 gene mutations in a patient with autosomal recessive osteopetrosis. Mol. Med. Rep. 2014, 9, 1191–1196. [Google Scholar] [CrossRef]
- Siddaiahgari, S.R.; Makadia, D.; Shah, N.; Devi, R.R.; Lingappa, L. Identification of Novel Mutation in Autosomal Recessive Infantile Malignant Osteopetrosis. Indian J. Pediatr. 2013, 81, 969–970. [Google Scholar] [CrossRef]
- Palagano, E.; Susani, L.; Menale, C.; Ramenghi, U.; Berger, M.; Uva, P.; Oppo, M.; Vezzoni, P.; Villa, A.; Sobacchi, C. Synonymous Mutations Add a Layer of Complexity in the Diagnosis of Human Osteopetrosis. J. Bone Miner. Res. 2016, 32, 99–105. [Google Scholar] [CrossRef]
- Del Fattore, A.; Peruzzi, B.; Rucci, N.; Recchia, I.; Cappariello, A.; Longo, M.; Fortunati, D.; Ballanti, P.; Iacobini, M.; Luciani, M.; et al. Clinical, genetic, and cellular analysis of 49 osteopetrotic patients: Implications for diagnosis and treatment. J. Med Genet. 2005, 43, 315–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gheorghe, G.; Galambos, C.; Jain, S.; Krishnamurti, L.; Jaffe, R. A Novel TCIRG1 Gene Mutation Leads to Severe Osteopetrosis with Altered Content of Monocytes/Macrophages in Several Organs. Pediatr. Dev. Pathol. 2012, 15, 156–159. [Google Scholar] [CrossRef]
- Engiz, O.; Kara, S.; Bagrul, D.; Lahr, G.; Alioglu, B.; Bilge, Y.D.; Arikan, I. Infantile malignant osteopetrosis: A rare cause of neonatal hypocalcemia. J. Pediatr. Endocrinol. Metab. 2012, 25, 1205–1207. [Google Scholar] [CrossRef]
- Lanzi, G.; Ferraro, R.M.; Masneri, S.; Piovani, G.; Barisani, C.; Sobacchi, C.; Villa, A.; Vezzoni, P.; Giliani, S. Generation of 3 clones of induced pluripotent stem cells (iPSCs) from a patient affected by Autosomal Recessive Osteopetrosis due to mutations in TCIRG1 gene. Stem Cell Res. 2020, 42, 101660. [Google Scholar] [CrossRef]
- Chen, W.; Twaroski, K.; Eide, C.; Riddle, M.J.; Orchard, P.J.; Tolar, J. TCIRG1 Transgenic Rescue of Osteoclast Function Using Induced Pluripotent Stem Cells Derived from Patients with Infantile Malignant Autosomal Recessive Osteopetrosis. J. Bone Jt. Surg. Am. Vol. 2019, 101, 1939–1947. [Google Scholar] [CrossRef] [PubMed]
- Phadke, S.R.; Fischer, B.; Gupta, N.; Ranganath, P.; Kabra, M.; Kornak, U. Novel mutations in Indian patients with autosomal recessive infantile malignant osteopetrosis. Indian J. Med Res. 2010, 131, 508–514. [Google Scholar]
- Demir, K.; Nalbantoğlu, Ö.; Karaer, K.; Korkmaz, H.A.; Yıldız, M.; Tunç, S.; Özkan, B. Genetic Diagnosis Using Whole Exome Analysis in Two Cases with Malignant Osteopetrosis of Infancy. J. Clin. Res. Pediatr. Endocrinol. 2015, 7, 356–357. [Google Scholar] [CrossRef] [PubMed]


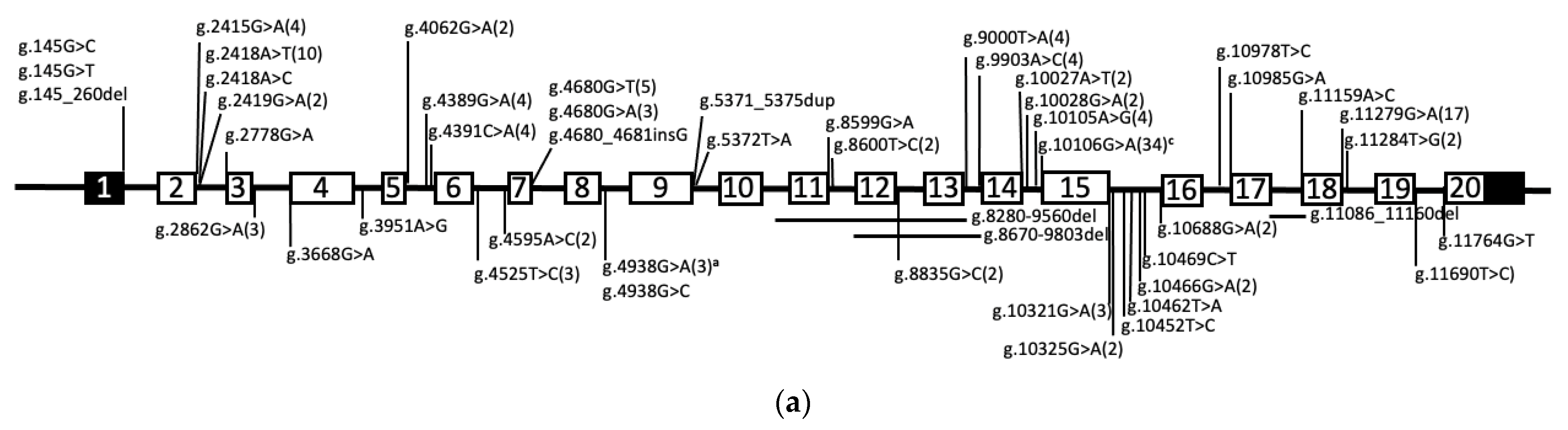
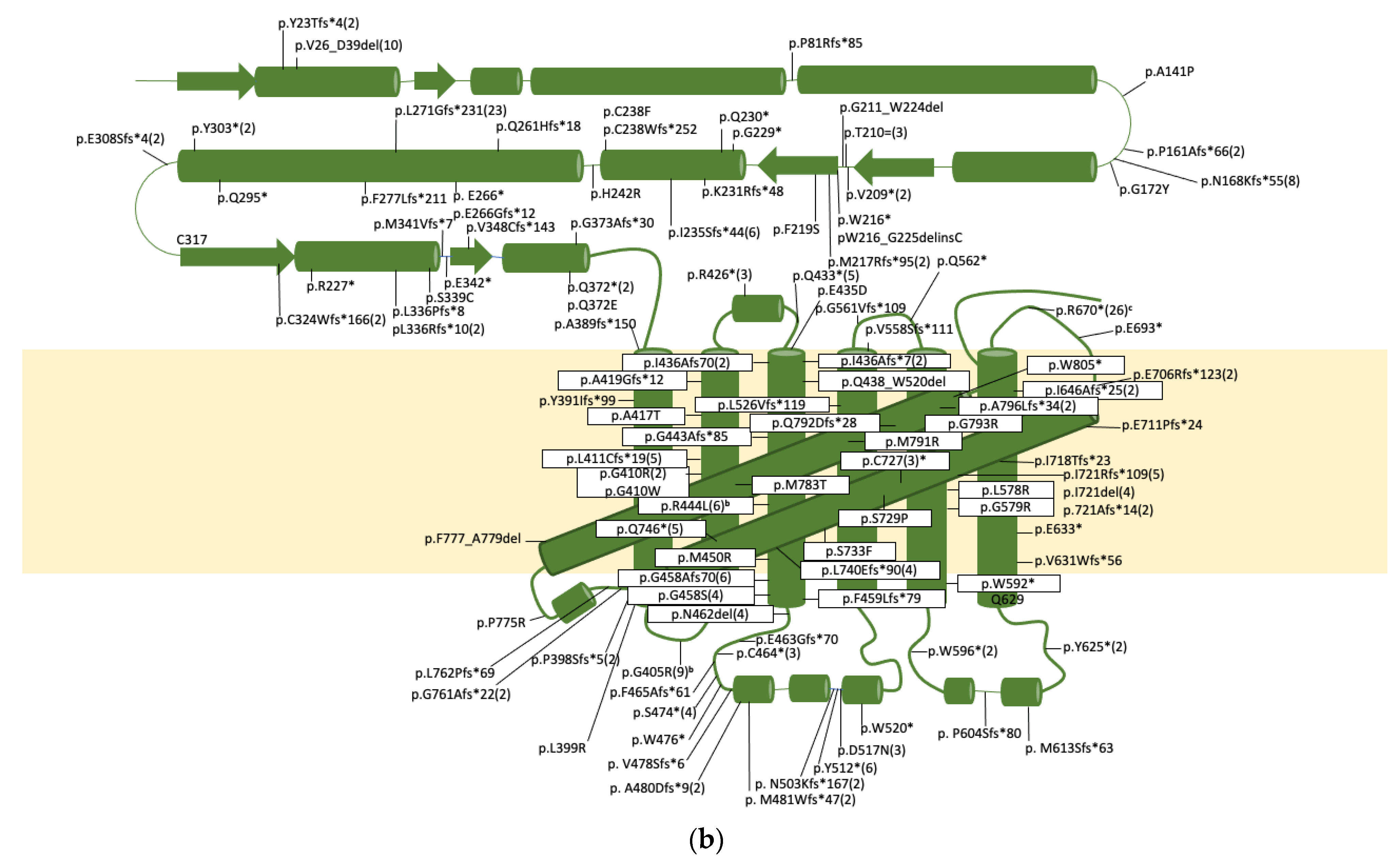

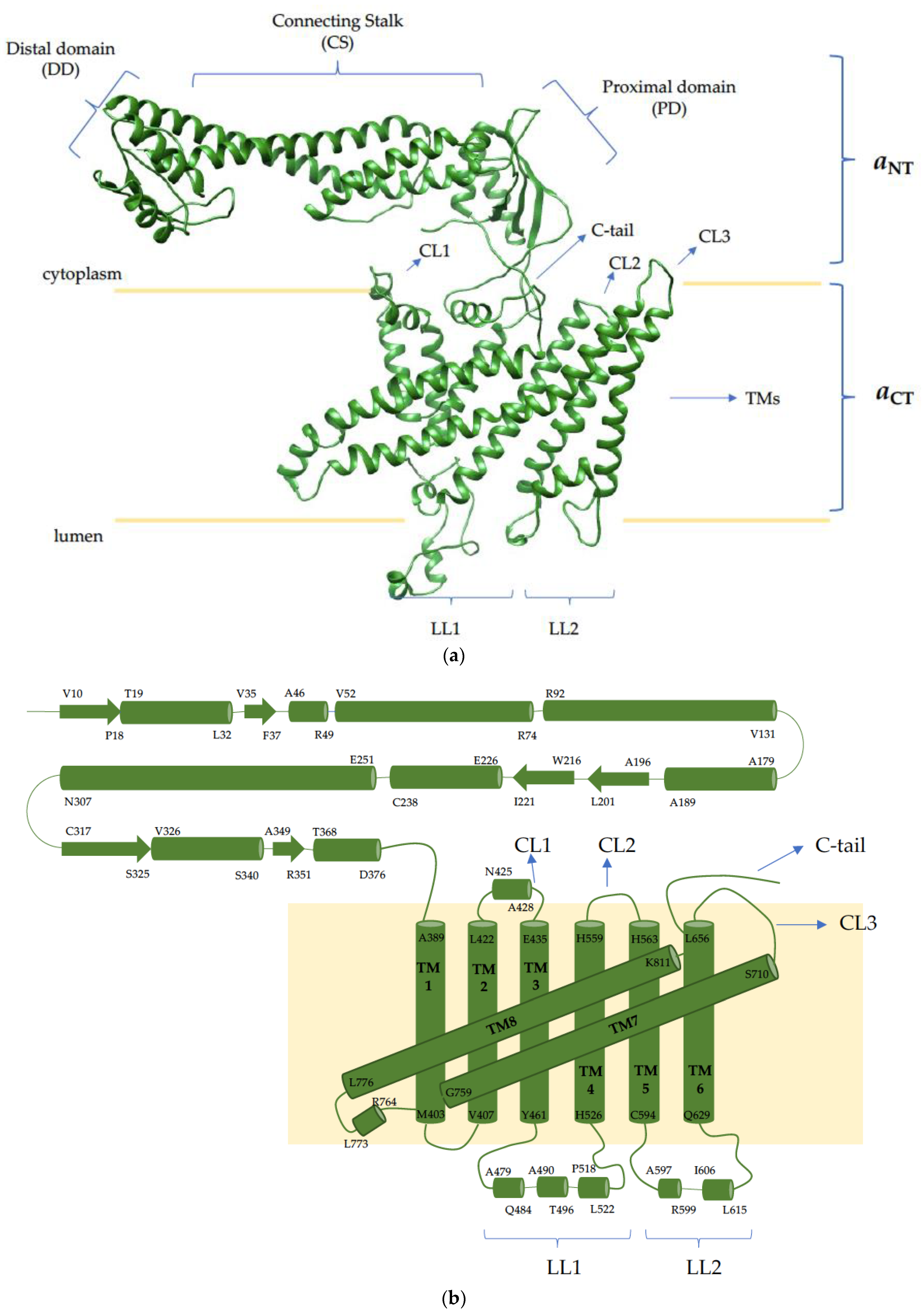
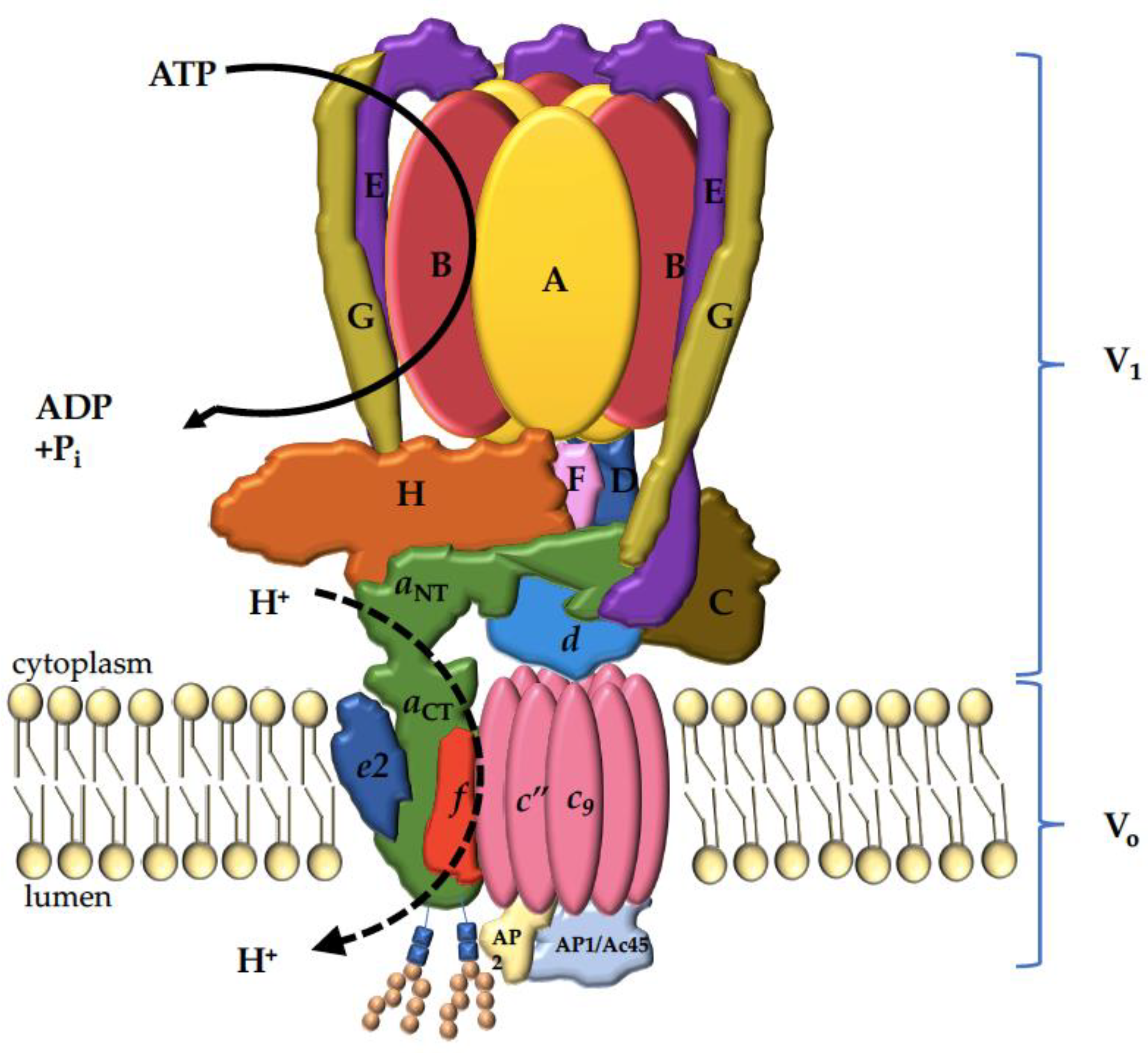{kind=link}
{kind=link}
{kind=link}
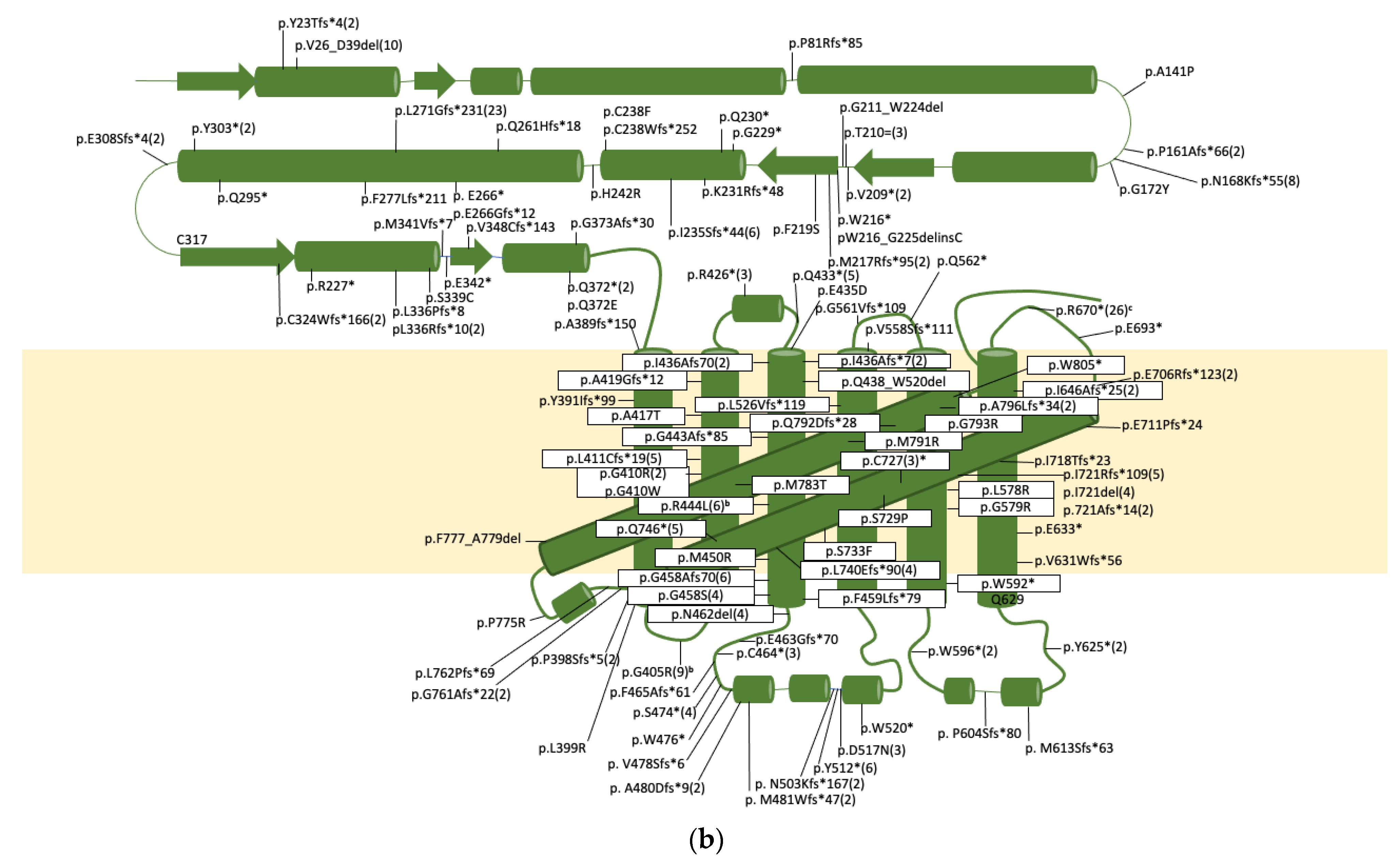{kind=link}
| Subunit Designation a | Yeast Gene b | Human Gene b | Function | Human Disease (OMIM#) c |
|---|---|---|---|---|
| V1 Subunits | ||||
| A | VMA1 | ATP6V1A | ATP binding/hydrolysis | #617403-Cutis laxa, type IID (AR) #618012 Developmental and epileptic encephalopathy 93 (AD) |
| B | VMA2 | ATP6V1B1 | ATP binding/hydrolysis | # 267300-Renal tubular acidosis, distal, with progressive nerve deafness (AR) |
| ATP6V1B2 | # 124480-Deafness, congenital, with onychodystrophy; DDOD (AD) # 616455-Zimmermann-Laband syndrome 2; ZLS2 (AD) | |||
| C | VMA5 | ATP6V1C1 | ||
| ATP6V1C2 | Recessive renal tubular acidosis [106] | |||
| ATP6V1C3 | ||||
| D | VMA6 | ATP6V1D | Torque transmission | |
| E | VMA4 | ATP6V1E1 | Stator function | # 617402-Cutis laxa, type IIC; ARCL2C (AR) |
| ATP6V1E2 | ||||
| F | VMA7 | ATP6V1F | Torque transmission | |
| G | VMA10 | ATP6V1G1 | Stator function | |
| ATP6V1G2 | ||||
| ATP6V1G3 | ||||
| H | VMA13 | ATP6V1H | ||
| VO Subunit | ||||
| a | VPH1/STV1 | ATP6V0A1 | Stator function, proton transport | Developmental and epileptic encephalopathy [107] |
| ATP6V0A2 | # 219200-Cutis laxa, type IIA; ARCL2A(AR) # 278250-Wrinkly skin syndrome; WSS (AR) | |||
| TCIRG1 | # 259700-Osteopetrosis, type B1; OPTB1 (AR) | |||
| ATP6V0A4 | # 602722-Renal tubular acidosis, distal; RTADR (AR) | |||
| c | VMA3 | ATP6V0C | Rotation, proton transport | |
| C′ | VMA11 | |||
| c″ | VMA16 | ATP6V0B | ||
| d | VMA6 | ATP6V0D1 | Transmission of torque, coupling ratio | |
| ATP6V0D2 | ||||
| e | VMA9 | ATP6V0E1 | ||
| ATP6V0E2 | ||||
| f | YPR170W-B | RNASEK | ||
| Ac45 | VOA1 | ATP6AP1 | VO assembly | # 300972-Immunodeficiency 47; IMD47 (XLR) |
| M8-9 | ATP6AP2 | VO assembly | # 301045-Congenital disorder of glycosylation, type IIr; CDG2R (XLR) # 300423-Mental retardation, X-linked, syndromic, hedera type; MRXSH (XLR) # 300911-Parkinsonism with spasticity, X-linked; XPDS (XLR) | |
| Chaperone | ||||
| VMA12 | VPH2 | TMEM199 | Stabilizes Vph1p, binds Vma22p | # 616829-Congenital disorder of glycosylation, type IIp; CDG2P (AR) |
| VMA21 | VMA21 | Assembles VO, exports VO from ER to Golgi | # 310440-myopathy, X-linked, with excessive autophagy; MEAX (XLR) | |
| VMA22 | CCDC115 | Stabilizes Vph1p, binds Vma12p | # 616828-Congenital disorder of glycosylation, type IIo; CDG2O (AR) | |
| Genomic a | cDNA b | Protein c | Allele Freq. d | Comment. [Ref] | ||
|---|---|---|---|---|---|---|
| Domain | Position | Position | Domain | Position | ||
| Int1 | g.145G>C | 1 | [153] | |||
| Int1 | g.145G>T | 1 | [153] | |||
| Int1 | g.145_260del | 1 | [153] | |||
| E2 | g.2363del | c.66del | PD | p.Y23Tfs*4 | 2 | Listed as c.65(E2):delC; p.22A>Afs5 [112,154] |
| Int2 | g.2415G>A | c.117+1G>A | 4 | [155,156,157] | ||
| g.2418A>T | c.117+4A>T | PD | p.V26_D39del | 10 | Activates cryptic splice site in E2 resulting in a deletion. [96,156,157,158,159] | |
| Int2 | g.2418A>C | c.117+4A>C | 1 | [108] | ||
| Int2 | g.2419G>A | c.117+5G>A | 2 | [154,156] | ||
| Int2 | g.2778G>A | c118-1G>a | 1 | [153] | ||
| Int3 | g.2862G>A | c.196+5G>A | 3 | [154] | ||
| Int3 | g.3668G>A | c.197-1G>A | 1 | [153] | ||
| E4 | g.3714del | c.242del | CS | p.P81Rfs*85 | 1 | [160] |
| Int4 | g.3951A>G | c.418-21A>G | 1 | [154] | ||
| E5 | g.3975G>C | c.421G>C | DD | p.A141P | 1 | [156] |
| E5 | DD | p.G159Rfs*68 | 1 | Listed with no details [161] | ||
| E5 | g.4034dup | c.480dup | DD | p.P161Afs*66 | 2 | Listed as c.475dupC [153] |
| Int5 | g.4062G>A | c.503+5G>A | 2 | [162] | ||
| Int5 | g.4389G>A | c.504-8G>A | DD | p.N168Kfs*55 and/or p.N168Kfs*8 | 4 | Activates cryptic splice in E6 resulting in a 11bp deletion of E6 and/or skipping of E6 [156] |
| Int5 | g.4391C>A | c.504-6C>A | DD | p.N168Kfs*55 and/or p.N168Kfs*8 | 4 | Activates cryptic splice in E6 resulting in a 11bp deletion of E6 and/or skipping of E6 [53,158,163] |
| E6 | g.4406_4407delinsTA | c.514_515delinsTA | DD | p.G172Y | 1 | [164] |
| E6 | g.4517del | c.624del | DD | p.V209* | 2 | Described as p.P208Pfs*1 [165] |
| E6 | g.4523G>A | c.630G>A | DD | p.T210 | 3 | We showed exon skipping and activation of cryptic splice site [162] |
| Int6 | g.4525T>C | c.630+2T>C | 3 | [156,157,158] | ||
| Int6 | g.4595A>C | c.631-2A>C | 2 | [154] | ||
| E7 | g.4613G>A | c.647G>A | DD | p.W216* | 1 | [155] |
| E7 | g.4614_4640del | c.648_674del | DD | p.W216_G225delinsC | 1 | [153] |
| E7 | g.4615_8590del | c.649_1297del | DD | p.M217Rfs*95 | 2 | Deletes most of E7 into E11 [154] |
| E7 | g.4622T>C | c.656T>C | DD | p.F219S | 1 | [166] |
| E7 | g.4637G>A | c.671G>A | DD | p.G211_W224del | 1 | Activates splice acceptor in E7 resulting in deletion [53] |
| E7 | g.4651G>T | c.685G>T | DD | p.G229* | 1 | [154] |
| E7 | g.4654C>T | c.688C>T | DD | p.Q330* | 2 | [157] |
| E7 | g.4658del | c.692del | DD | p.K231Rfs*48 | 1 | [112] |
| E7 | g.4668del | c.702del | DD | p.I235Sfs*44 | 6 | [156,167] |
| E7 | g.4679G>T | c.713G>T | DD | p.C238F | 1 | Assumes no alt. splicing as this is the last nt of E7 [53] |
| Int7 | g.4680G>T | c.713+1G>T | 5 | [157,159] | ||
| Int7 | g.4680G>A | c.713+1G>A | 3 | [156,168,169] | ||
| Int7 | g.4680_4681insG | c.713+1_713+2insG | DD | p.C238Wfs*252 | 1 | Assumes no alt splicing [153] |
| E8 | g.4851A>G | c.725A>G | DD | p.H242R | 1 | De novo mutation, not other mutation found, potential dominant [170] |
| E8 | g.4909del | c.783del | CS | p.Q261Hfs*18 | 1 | [153] |
| E8 | g.4922G>T | c.796G>T | CS | p.E266* | 1 | [154] |
| E8 | g.4923del | c.797del | CS | p.E266Gfs*12 | 1 | [171] |
| Int8 | g.4938G>A | c.807+5G>A | CS | p.L271Gfs*231 | 23 | Founder mutation in Chuvashiya population. Resulted in activation of cryptic splice donor 37nt downstream. [164,172] |
| Int8 | g.4938G>C | c.807+5G>T | 1 | [153] | ||
| E9 | g.5181_5186delinsA | c.831_836delinsA | CS | p.F277Lfs*211 | 1 | [173] |
| E9 | g.5233C>T | c.883C>T | CS | p.Q295* | 1 | [153] |
| E9 | g.5259C>A | c.909C>A | CS | pY303* | 2 | [112,174] |
| E9 | g.5272del | c.922del | CS | p.E308Sfs*4 | 2 | [53,156] |
| E9 | g.5321_5322insG | c.971_972insG | PD | p.C324Wfs*166 | 2 | [157] |
| E9 | g.5329C>T | c.978C>T | PD | pR327* | 1 | [153] |
| E9 | g.5357del | c.1007del | PD | p.L336Rfs*10 | 2 | [155] |
| E9 | g.5357_5363del | c.1007_1013del | PD | p.L336Pfs*8 | 2 | [154] |
| E9 | g.5365A>T | c.1015A>T | PD | p.S339C | 1 | [154] |
| E9 | g.5369_5370insGGTGA | c.1019_1020insGGTGA | PD | p.M341Vfs*7 | 1 | Described as p.340S>Sfs151 [154] |
| Int9 | g.5371_5375dup | c.1020+1_1020+5dup | 1 | [165] | ||
| Int9 | g.5372T>A | c.1020+2T>A | 1 | [153] | ||
| E10 | g.5988G>T | c.1024G>T | PD | p.E342* | 1 | [96] |
| E10 | g.6000_6001dupGTGC | c.1037_1040dup | PD | p.V348Cfs*143 | 1 | Described as c.1036_1037insGTGC [154] |
| E10 | g.6078G>T | c.1114C>T | PD | p.Q372* | 2 | [154,160] |
| E10 | g.6078C>G | 1 | listed as p.Q372* [157], but would be pQ372E if it is g.6078C>G | |||
| E10 | g.6082del | c.1118del | PD | pG373Afs*30 | 1 | [153] |
| g.8280_9560del | c.1166_1554del | TM1 | p.A389Dfs*151 | 4 | Deletion includes E11-13 [156,158,168] | |
| E11 | g.8464_8465insA | c.1171_1174insA | TM1 | p.Y391Ifs*99 | 2 | Listed as g.8464insA [153,157] |
| E11 | g.8484del | c.1191del | TM1 | p.F398Sfs*5 | 2 | Listed as c.1188delC; p.P397Pfs6 [154] |
| E11 | g.8489T>G | c.1196T>G | TM1 | p.L399R | 1 | [153] |
| E11 | g.8506G>A | c.1213G>A | TM1 | p.G405R | 23 | Founder mutation effect in Costa Rica [112,153,154,156,157,162,168] |
| E11 | g.8521G>A | c.1228G>A | TM2 | p.G410R | 1 | [168] |
| E11 | g.8521G>T | c.1228G>T | TM2 | p.G410W | 1 | Listed as p.G410R [154] |
| E11 | g.8521G>C | c.1228G>C | TM2 | p.G410R | 1 | [153] |
| E11 | g.8523del | c.1230del | TM2 | p.L411Cfs*19 | 5 | Listed as Pt1 delG8521; p>G410fsX429 or Pt20 g.8521delG [158,168,169] |
| E11 | g.8542G>A | c.1249G>A | TM2 | p.A417T | 1 | [153] |
| E11 | g.8548_8549insGG | c.1255_1256insGG | TM2 | p.A419Gfs*12 | 1 | [153] |
| E11 | g.8569C>T | c.1276C>t | CL1 | p.R426* | 3 | [153,175] |
| E11 | g.8590C>T | c.1297C>T | CL2 | p.Q433* | 5 | [156,157] |
| E11 | g.8598G>C | c.1305G>T | TM3 | p.E435D | 1 | [153] |
| Int11 | g.8599G>A | c.1305+1G>A | 1 | [153] | ||
| Int11 | g.8600T>C | c.1305+2T>C | 2 | [110,159] | ||
| g.8670_9803del | c.1306_1554del | TM3 | p.Q438_W520del | 1 | Deletion includes E12-13. Checked both DNA and cDNA [153] | |
| E12 | g.8695del | c.1328del | TM3 | p.G443Afs*85 | 1 | [153] |
| E12 | g.8698G>T | c.1331G>T | TM3 | p.R444L | 6 | Founder effect in Cost Rica population [157] |
| E12 | g.8716T>G | c.1349T>G | TM3 | p.M450R | 1 | [153] |
| E12 | g.8738del | c.1371del | TM3 | p.G458Afs*70 | 6 | Listed as c.1370delc; p.T457Tfs71 [154] |
| E12 | g.8738C>A | c.1371C>A | TM3 | p.I436Afs*70 | 2 | Creates cryptic splice acceptor, results in the deletion of 67nt of E12 [176] |
| E12 | g.8739G>A | c.1372G>A | TM3 | p.G458S | 3 | [153,154] |
| E12 | TM3 | p.F459Lfs*79 | 1 | No other details provided [161] | ||
| E12 | g.8749_8751del | c.1382_1384del | LL1 | p.N462del | 4 | [156,157] |
| E12 | g.8755delinsGCTTCATCTACAACG | c.1387delinsGCTTCATCTACAACG | LL1 | p.E463Gfs*70 | 1 | Listed as c.1387insGCTTCATCTACAACG; pGlu463Glyfs [171] |
| E12 | g.8759C>A | c.1392C>A | LL1 | p.C464* | 3 | [53,156,157] |
| E12 | g.876-_8766del | c.1393_1399del | LL1 | p.F465Afs*61 | 1 | [153] |
| E12 | g.8788C>A | c.1421C>A | LL1 | p.S474* | 5 | [156,167] |
| E12 | g.8795G>A | c.1428G>A | LL1 | p.W476* | 1 | [153] |
| E12 | g.8799_8816delinsT | c.1432_1449delinsT | LL1 | p.V478Sfs*6 | 1 | [153] |
| E12 | g.8805_8806del | c.1438_1439del | LL1 | p.A480Dfs*9 | 2 | [96] |
| E12 | g.8807del | c.1430del | LL1 | p.M481Wfs*47 | 2 | [177] |
| Int12 | g.8835G>C | c.1463+5G>C | 2 | [178] | ||
| E13 | g.8952_8553insA | c.1507_1508insA | LL1 | p.N503Kfs*167 | 2 | [179] |
| E13 | g.8980C>A | c.1536C>A | LL1 | p.Y512* | 6 | [156,158,169,180] |
| E113 | g.8993G>A | c.1549G>A | LL1 | p.D517N | 3 | [156,181] |
| Int13 | g.9000T>A | c.1554+2T>A | 4 | [157,182] | ||
| Int13 | g.9903A>C | c.1555-2A>C | 4 | [154,168] | ||
| E14 | g.9909G>A | c.1559G>A | LL1 | p.W520* | 1 | [110] |
| E14 | g.10003_10004insGTGG | c.1653_1654insGTGG | TM4 | p.L552Vfs*119 | 1 | Listed as RT p.V451fsX670 [182] |
| Int14 | g.10027A>T | c.1673+4A>T | TM4 | p.V558Afs111 | 2 | Not obvious how V558 becomes A [164] |
| Int14 | g.10028G>A | c.1673+5G>A | 2 | Retains int14 or skips E14/15 [156,157] | ||
| Int14 | g.10105A>G | c.1674-2A>G | 3 | Appears as if intronic seq are retained [162] | ||
| Int14 | g.10106G>A | c.1674-1G>A | 34 | Founder mutation if Flanders population [53,156,157,159,168,169] | ||
| E15 | g.10115delinsTT | c.1682delinsTT | LL2 | p.G561Vfs*109 | 1 | [168] |
| E15 | g.10117C>T | c.1684C>T | LL2 | p.Q562* | 1 | [182] |
| E15 | g.10166T>G | c.1733T>G | TM5 | p.L578R | 1 | [154] |
| E15 | g.10168 | c.1735G>A | TM5 | pG579R | 1 | [153] |
| E15 | g.10208G>A | c.1775G>A | TM5 | p.W592* | 1 | [154] |
| E15 | g.10220G>A | c.1787G>A | LL2 | p.W596* | 2 | [96] |
| E15 | g.10242_10251del | c.1809_1818del | LL2 | p.P604Sfs*80 | 1 | [163] |
| E15 | g.10270_10273del | c.1837_1840del | LL2 | p.M613Sfs*63 | 1 | [154] |
| E15 | g.10311C>A | c.1878C>A | LL2 | p.Y625* | 2 | [168] |
| Int15 | g.10321G>A | c.1887+1G>A | 3 | [154] | ||
| Int15 | g.10325G>A | c.1887+5G>A | 2 | Listed as c.1941+5G>A. Makes 32% normal transcript [111] | ||
| Int15 | g.10452T>C | c.1887+132T>C | 1 | [110] | ||
| Int15 | g.10462T>A | c.1887+142T>A | 1 | retains 6.7% wt splicing [110] | ||
| Int15 | g.10466G>A | c.1887+146G>A | 2 | retains 5.5% wt splicing [110] | ||
| Int15 | g.10469C>T | c.1887+149C>T | 1 | [110] | ||
| Int15 | g.10688G>A | c.1888-1G>A | 2 | Listed asc.1874-1G>A [168] | ||
| E16 | g.10692del | c.1891del | TM6 | p.V631Wfs*56 | 1 | Listed as c.1878delG [153] |
| E16 | g.10698C>T | c.1897C>T | TM6 | p.E633* | 1 | [154] |
| E16 | g.10735_10736insGGCA | c.1934_1935insGGCA | TM6 | p.I646Afs*25 | 2 | [166] |
| E16 | g. 10809C>T | c.2008C>T | CL3 | p.Arg670* | 26 | Founder variant in Flander population [154,156,157,168,174] |
| Int16 | g.10978T>C | c.2014-8T>C | [157] | |||
| Int16 | g.10985G>A | c.2014-1G>A | [153] | |||
| E17 | g.11049G>T | c.2077G>T | CL3 | p.E693* | 1 | [153] |
| E17 | g.11086_11160del | c.2114_2119-1del | CL3 | p.E706Rfs*123 | 2 | Deletes last 5nt of E17 and all if Int17 [157] |
| Int17 | g.11159A>C | c.2119-2A>C | 1 | [153] | ||
| E18 | g.11172_11190del | c.2130_2148del | TM7 | p.E711Pfs*24 | 1 | [153] |
| E18 | g.11195del | c.2153del | TM7 | p.I718Tfs*23 | 1 | [156] |
| E18 | g.11202_11204del | c.2160_2162del | TM7 | p.I721Rfs*109 | 5 | [182] |
| E18 | g.11203_11205del | c.2161_2163del | TM7 | p.I721del | 4 | [154,156,166] |
| E18 | TM7 | pI721Afs*14 | 2 | No data shown [161] | ||
| E18 | g.11223C>A | c.2181C>A | TM7 | p.C727* | 3 | [112,154] |
| E18 | g.11227T>C | c.2185C>T | TM7 | p.S729P | 1 | [153] |
| E18 | g.11240C>T | c.2198C>T | TM7 | p.S733F | 1 | [153] |
| E18 | g.11260_11261del | c.2218_2219del | TM7 | p.L740Qfs*90 | 4 | [154,168] |
| E18 | g.11278C>T | c.2236C>T | TM7 | p.Q746* | 5 | Assumes splicing to E19 [159,169,181,183]. |
| Int18 | g.11279G>A | c.2236+1G>A | 17 | PCR analysis shows minor wt transcript, and at least 5 alternative splice events [156,157,158,162,168,169] | ||
| E19 | g.11556del | c.2282del | p.G761Afs*22 | 2 | [153] | |
| E19 | g.11558_11559insC | c.2284_2285insC | p.L762Pfs*69 | 1 | [153] | |
| E19 | g.11598C>G | c.2324C>G | TM8 | p.P775R | 6 | [156,157,168] |
| E19 | g.11602_11610del | c.2328_2336del | TM8 | p.F777_A779del | 1 | [157] |
| E19 | g.11622T>C | c.2348T>C | TM8 | pM783T | 2 | [110,158] |
| E19 | g.11646T>G | c.2372T>G | TM8 | p.M791R | 1 | [153] |
| E19 | g.11647_11650del | c.2376_2379del | TM8 | p.Q792Dfs*28 | 2 | [157,158] |
| E19 | g.11651G>C | c.2377G>C | TM8 | p.G793R | 1 | [168] |
| E19 | g.11654_11655del | c.2380_2381del | TM8 | p.A796Lfs*34 | 1 | [108] |
| E19 | g.11657_11658del | c.2383_2384del | TM8 | p.A796Lfs*34 | 1 | [168] |
| Int19 | g.11690T>C | c.2414+2T>C | 1 | [173] | ||
| Int19 | g.11764G>T | c.2115-1G>T | 1 | [155] | ||
| E20 | g.11765G>A | c.2415G>A | TM8 | p.W805* | 1 | Assumes correct splicing. This is 1st nt in E20 [96]. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chu, A.; Zirngibl, R.A.; Manolson, M.F. The V-ATPase a3 Subunit: Structure, Function and Therapeutic Potential of an Essential Biomolecule in Osteoclastic Bone Resorption. Int. J. Mol. Sci. 2021, 22, 6934. https://doi.org/10.3390/ijms22136934
Chu A, Zirngibl RA, Manolson MF. The V-ATPase a3 Subunit: Structure, Function and Therapeutic Potential of an Essential Biomolecule in Osteoclastic Bone Resorption. International Journal of Molecular Sciences. 2021; 22(13):6934. https://doi.org/10.3390/ijms22136934
Chicago/Turabian StyleChu, Anh, Ralph A. Zirngibl, and Morris F. Manolson. 2021. "The V-ATPase a3 Subunit: Structure, Function and Therapeutic Potential of an Essential Biomolecule in Osteoclastic Bone Resorption" International Journal of Molecular Sciences 22, no. 13: 6934. https://doi.org/10.3390/ijms22136934
APA StyleChu, A., Zirngibl, R. A., & Manolson, M. F. (2021). The V-ATPase a3 Subunit: Structure, Function and Therapeutic Potential of an Essential Biomolecule in Osteoclastic Bone Resorption. International Journal of Molecular Sciences, 22(13), 6934. https://doi.org/10.3390/ijms22136934