
Electrophysiological and Structural Remodeling of the Atria in a Mouse Model of Troponin-I Mutation Linked Hypertrophic Cardiomyopathy: Implications for Atrial Fibrillation

 ,
,  ,
,
Abstract
1. Introduction
2. Results
2.1. Animal Characteristics
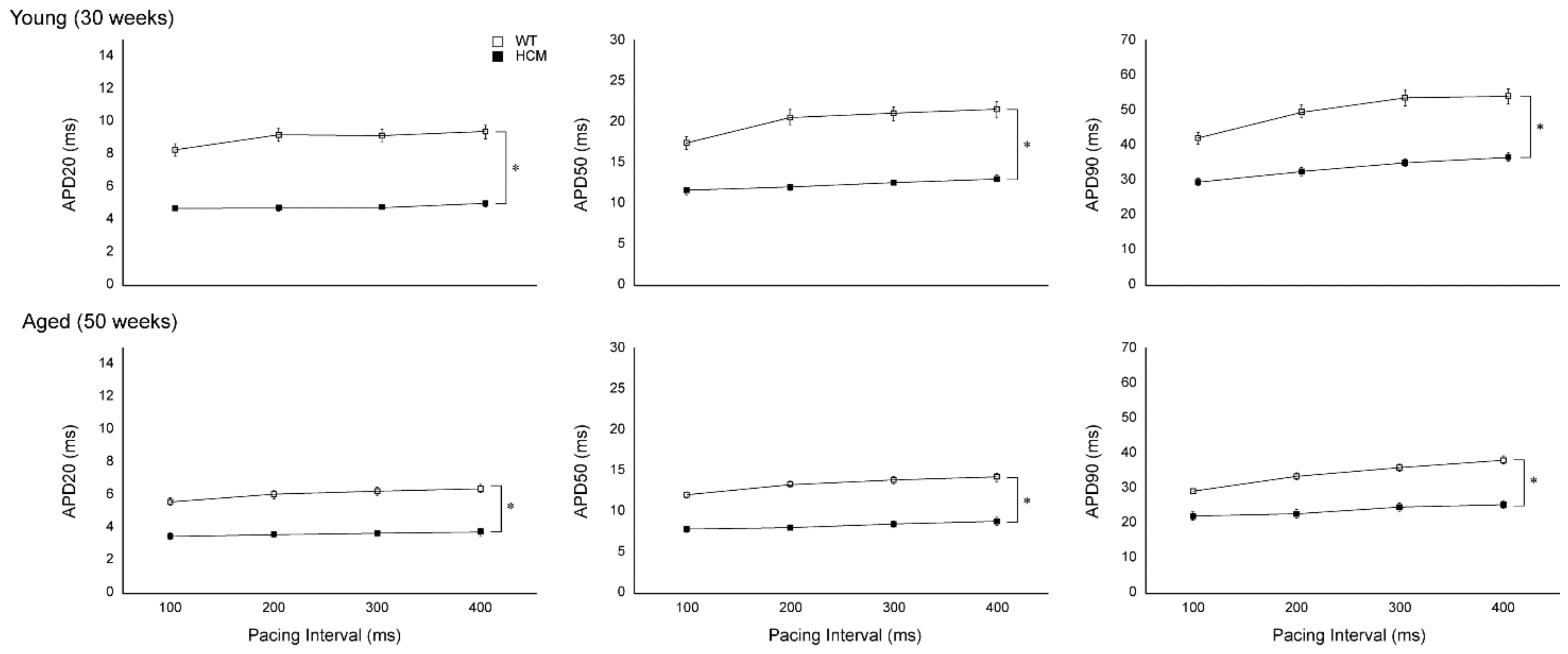
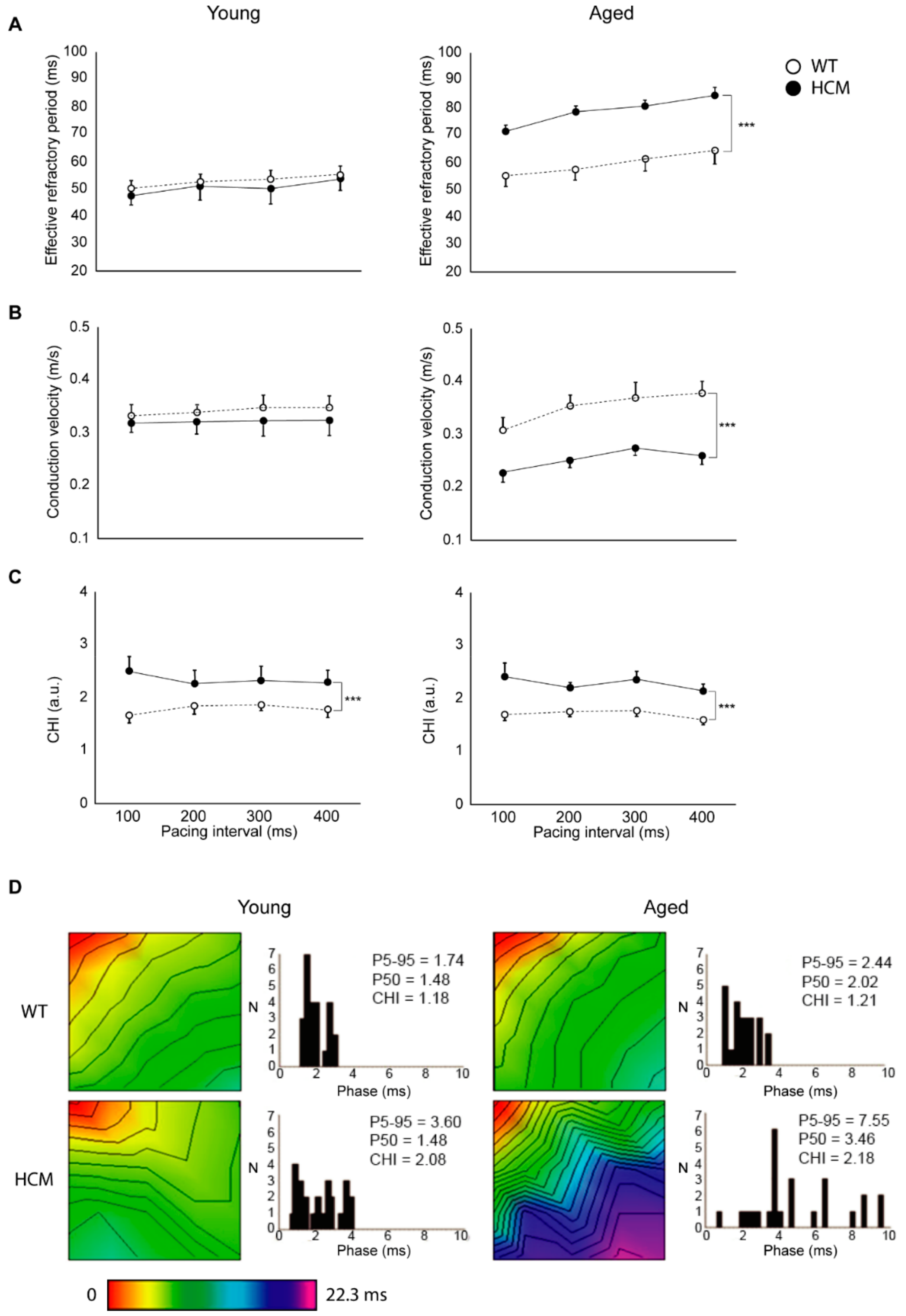
2.2. Atrial Action Potential Duration and Effective Refractory Period
2.3. Atrial Conduction
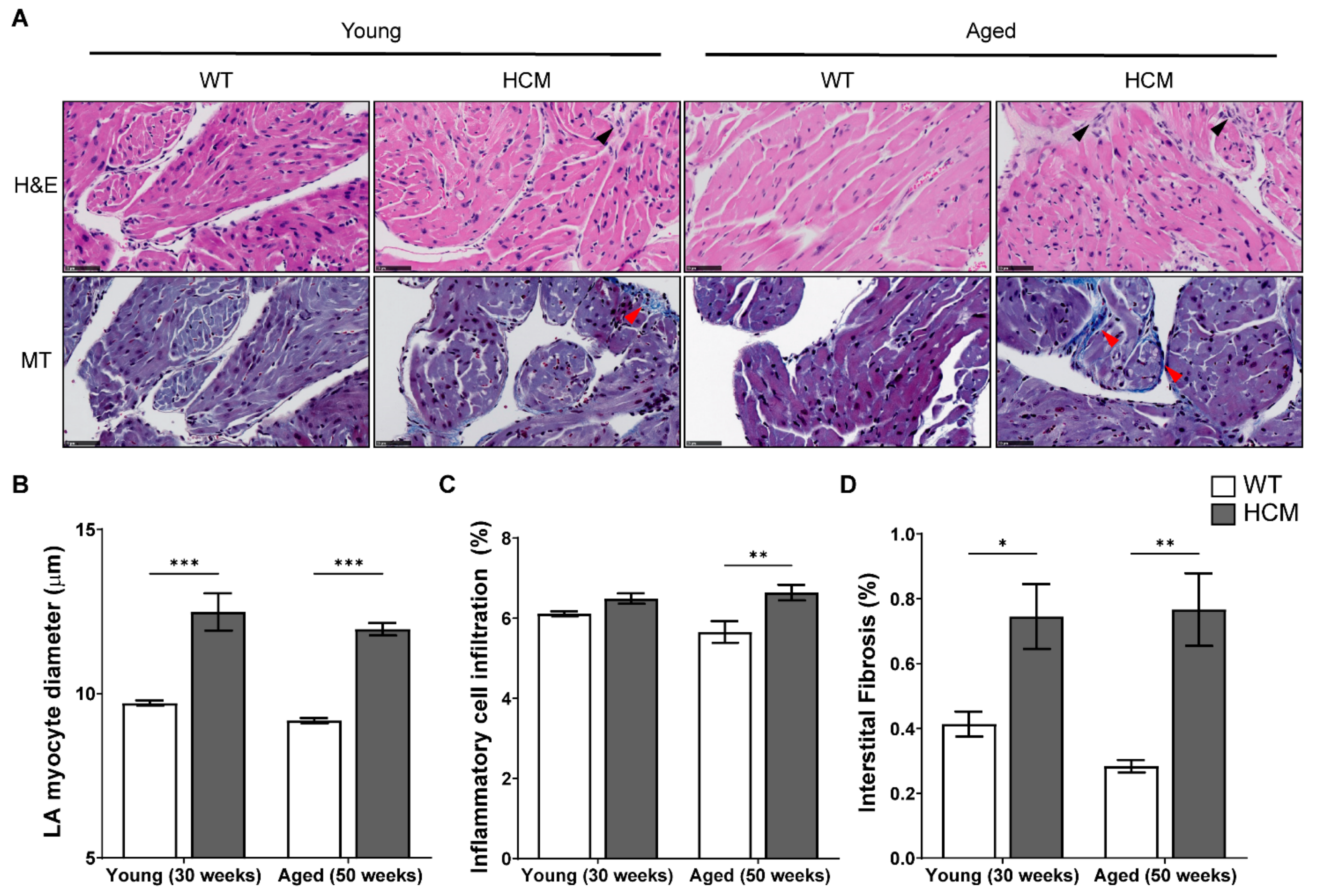
2.4. Atrial Structural Remodeling
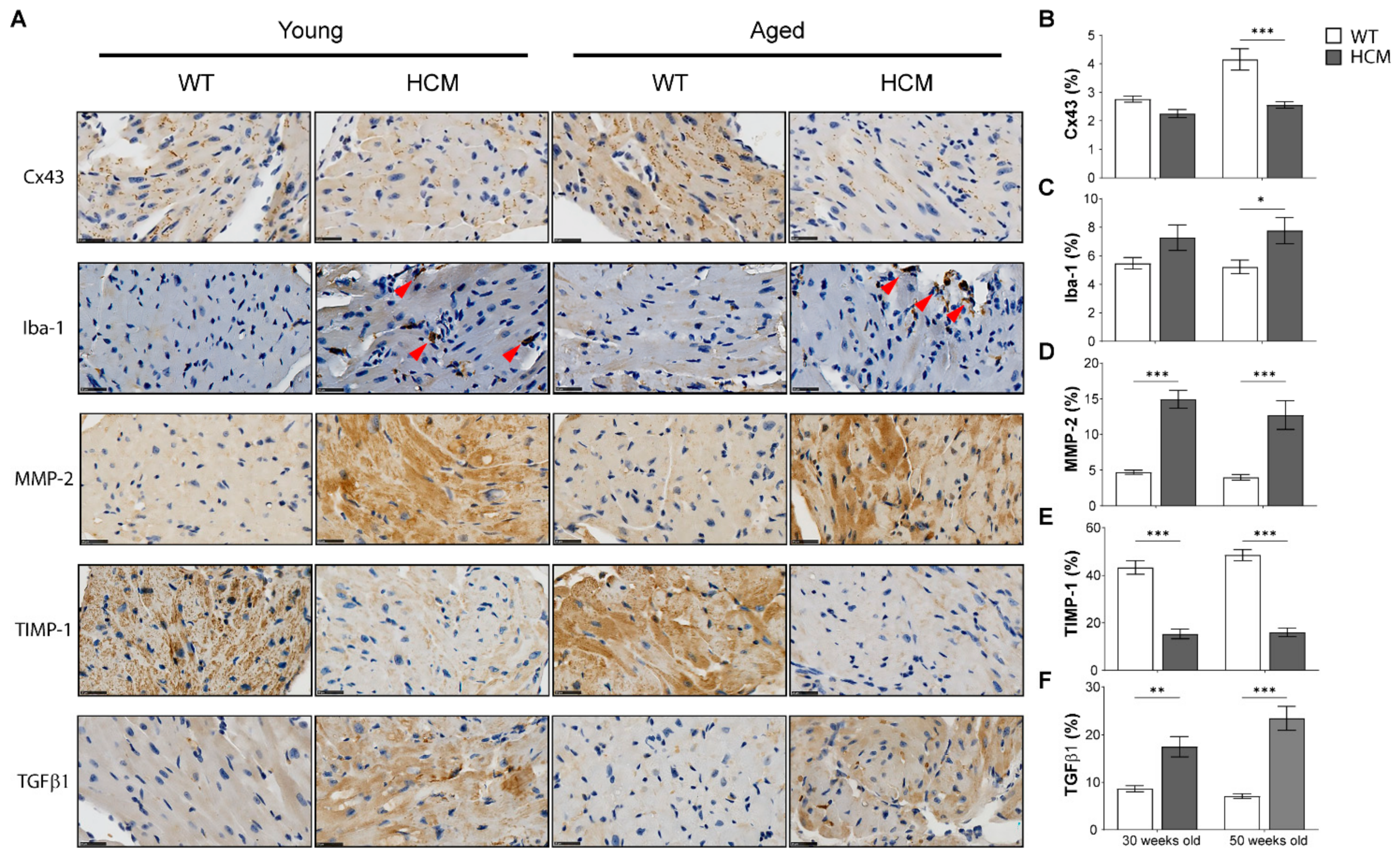
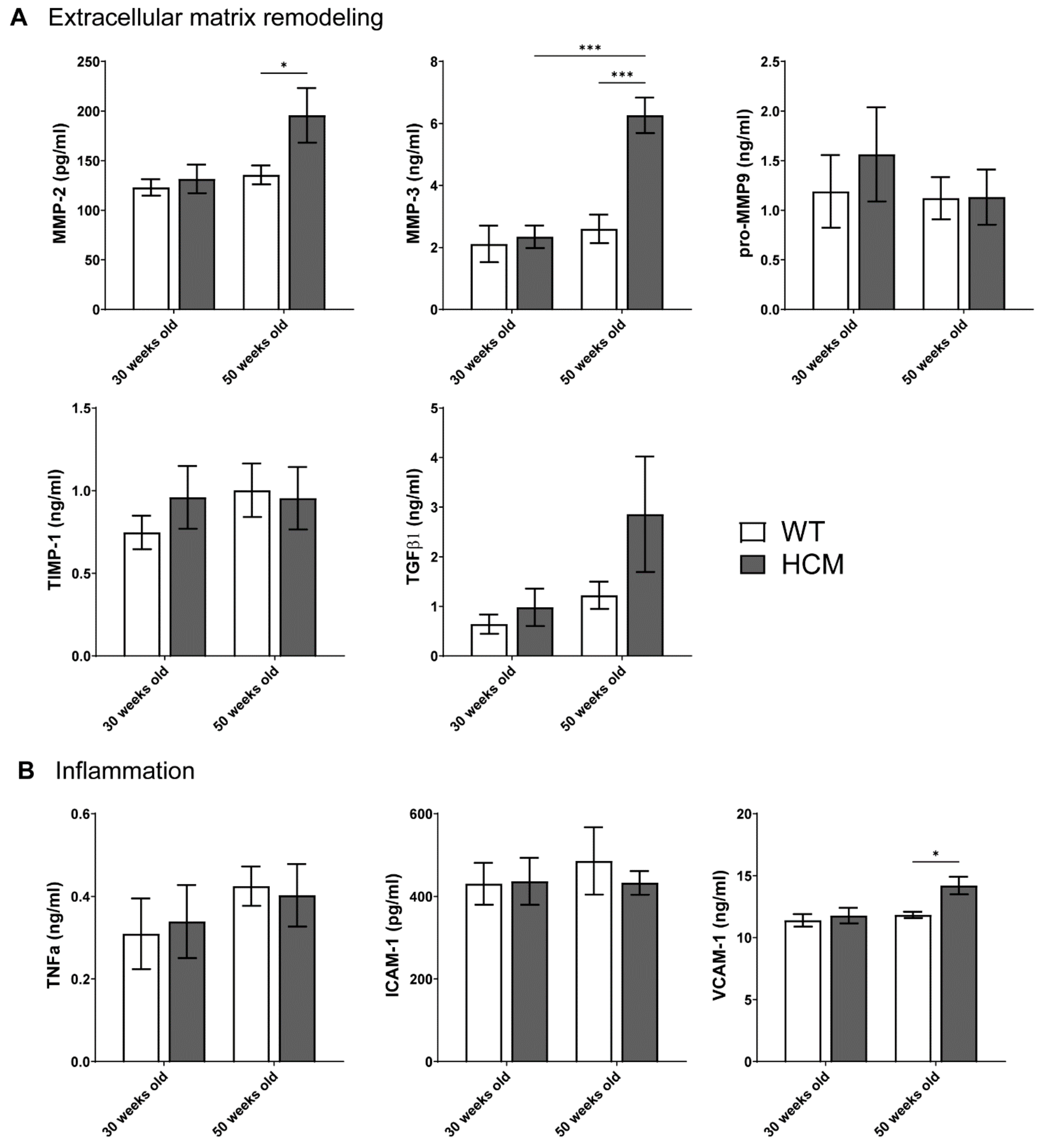
2.5. Biomarkers of Extracellular Matrix Remodeling and Inflammation
3. Discussion
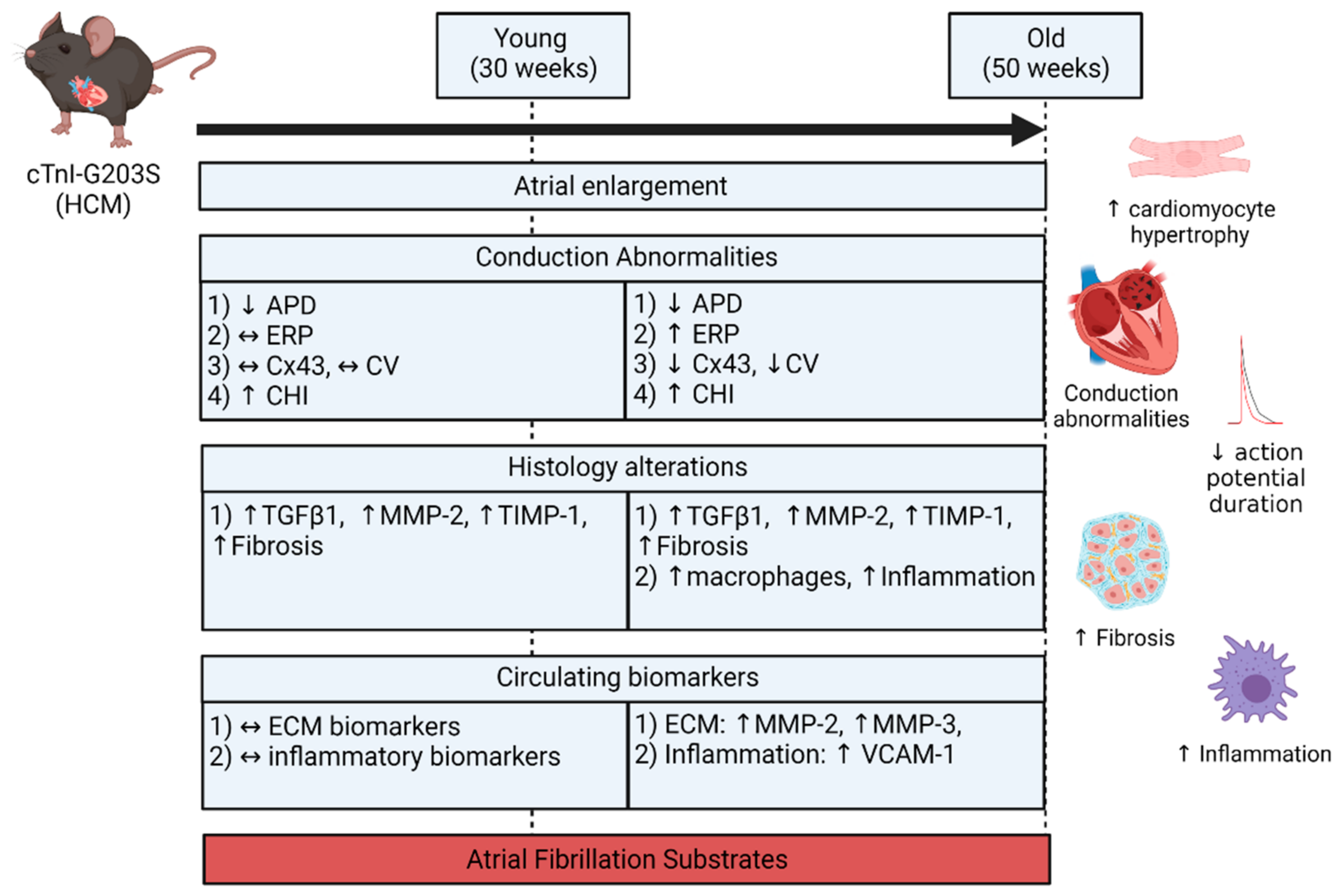
- Both atria were enlarged with an increase in atrial myocardial mass in young and aged HCM mice.
- Marked atrial structural abnormalities with myocyte hypertrophy and fibrosis was observed in young HCM mice and aged HCM mice demonstrated myocyte hypertrophy, inflammatory cell infiltration, and fibrosis.
- Electrophysiological abnormalities within the atria were observed from a young age with abbreviations in the action potential duration and an increase in conduction heterogeneity. However, abnormal refractoriness and conduction velocity were only observed with increasing age.
- Aged HCM mice demonstrated increased circulating levels of extracellular matrix remodeling MMP-2, MMP-3, and inflammation VCAM-1, which were unaltered in the younger cohort.
3.1. Atrial Substrate Predisposing to AF
3.2. Atrial Remodeling in HCM
3.3. Potential Mechanisms of HCM-Induced Atrial Remodeling
3.4. Study Limitations
3.5. Conclusions
4. Materials and Methods
4.1. Animal Model
4.2. Heart Rate and Blood Pressure
4.3. Action Potential Duration and Multi-Electrode Array Electrophysiology Study
4.4. Atrial Refractoriness
4.5. Atrial Conduction Analyses
4.6. Atrial Histology
4.7. Multiplex Enzyme-Linked Immunoassay
4.8. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AF | Atrial fibrillation |
| APD | Action potential duration |
| CV | Conduction velocity |
| CHI | Conduction heterogeneity index |
| CRP | C-reactive protein |
| Cx43 | Connexin 43 |
| DAB | Diaminobenzidine tetrahydrochloride |
| ERP | Effective refractory period |
| GLM ANOVA | General linear model analysis of variance |
| HCM | Hypertrophic cardiomyopathy |
| H&E | Hematoxylin and eosin |
| Iba-1 | Ionized calcium binding adaptor molecule 1 |
| ICAM-1 | Intercellular Adhesion Molecule 1 |
| IL-6 | Interleukin-6 |
| LA | Left atria |
| LAA | Left atrial appendage |
| LAFW | Left atrial free wall |
| LV | Left ventricle |
| MEA | Multi-electrode array |
| MMP | Matrix metalloproteinase |
| MT | Masson’s trichrome |
| MYBPC3 | Myosin-binding protein C3 |
| MYH7 | Myosin heavy chain 7 |
| RA | Right atria |
| SEM | Standard error of the mean |
| TAT | Total activation time |
| TIMP | Tissue inhibitor of metalloproteinase |
| TGFβ1 | Transforming growth factor beta 1 |
| TNFα | Tumor necrosis factor alpha |
| TnI | Troponin I |
| VCAM-1 | Vascular cell adhesion molecule 1 |
| WT | Wild-type |
References
- Maron, B.J. Clinical course and management of hypertrophic cardiomyopathy. N. Engl. J. Med. 2018, 379, 655–668. [Google Scholar] [CrossRef]
- Semsarian, C.; Ingles, J.; Maron, M.S.; Maron, B.J. New perspectives on the prevalence of hypertrophic cardiomyopathy. J. Am. Coll. Cardiol. 2015, 65, 1249–1254. [Google Scholar] [CrossRef] [PubMed]
- Canepa, M.; Fumagalli, C.; Tini, G.; Vincent-Tompkins, J.; Day, S.M.; Ashley, E.A.; Mazzarotto, F.; Ware, J.S.; Michels, M.; Jacoby, D.; et al. Temporal trend of age at diagnosis in hypertrophic cardiomyopathy: An analysis of the International Sarcomeric Human Cardiomyopathy Registry. Circ. Heart Fail. 2020, 13, e007230. [Google Scholar] [CrossRef]
- Bagnall, R.D.; Weintraub, R.G.; Ingles, J.; Duflou, J.; Yeates, L.; Lam, L.; Davis, A.M.; Thompson, T.; Connell, V.; Wallace, J.; et al. A prospective study of sudden cardiac death among children and young adults. N. Engl. J. Med. 2016, 374, 2441–2452. [Google Scholar] [CrossRef] [PubMed]
- Ho, C.Y.; Day, S.M.; Ashley, E.A.; Michels, M.; Pereira, A.C.; Jacoby, D.; Cirino, A.L.; Fox, J.C.; Lakdawala, N.K.; Ware, J.S.; et al. Genotype and lifetime burden of disease in hypertrophic cardiomyopathy: Insights from the sarcomeric human cardiomyopathy registry (SHaRe). Circulation 2018, 138, 1387–1398. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.-E.; Park, J.-K.; Uhm, J.-S.; Kim, J.Y.; Pak, H.-N.; Lee, M.-H.; Joung, B. Impact of atrial fibrillation on the clinical course of apical hypertrophic cardiomyopathy. Heart 2017, 103, 1496–1501. [Google Scholar] [CrossRef]
- Alphonse, P.; Virk, S.; Collins, J.; Campbell, T.; Thomas, S.P.; Semsarian, C.; Kumar, S. Prognostic impact of atrial fibrillation in hypertrophic cardiomyopathy: A systematic review. Clin. Res. Cardiol. 2021, 110, 544–554. [Google Scholar] [CrossRef]
- Guttmann, O.P.; Rahman, M.S.; O’Mahony, C.; Anastasakis, A.; Elliott, P.M. Atrial fibrillation and thromboembolism in patients with hypertrophic cardiomyopathy: Systematic review. Heart 2014, 100, 465–472. [Google Scholar] [CrossRef]
- Hindricks, G.; Potpara, T.; Dagres, N.; Arbelo, E.; Bax, J.J.; Blomström-Lundqvist, C.; Boriani, G.; Castella, M.; Dan, G.-A.; Dilaveris, P.E.; et al. 2020 ESC guidelines for the diagnosis and management of atrial fibrillation developed in collaboration with the European Association for Cardio-Thoracic Surgery (EACTS): The task force for the diagnosis and management of atrial fibrillation of the European Society of Cardiology (ESC) developed with the special contribution of the European Heart Rhythm Association (EHRA) of the ESC. Eur. Heart J. 2021, 42, 373–498. [Google Scholar] [CrossRef]
- Zoni-Berisso, M.; Lercari, F.; Carazza, T.; Domenicucci, S. Epidemiology of atrial fibrillation: European perspective. Clin. Epidemiol. 2014, 6, 213–220. [Google Scholar] [CrossRef]
- Rowin, E.J.; Hausvater, A.; Link, M.S.; Abt, P.; Gionfriddo, W.; Wang, W.; Rastegar, H.; Estes, N.A.M.; Maron, M.S.; Maron, B.J. Clinical profile and consequences of atrial fibrillation in hypertrophic cardiomyopathy. Circulation 2017, 136, 2420–2436. [Google Scholar] [CrossRef]
- Siontis, K.C.; Geske, J.B.; Ong, K.; Nishimura, R.A.; Ommen, S.R.; Gersh, B.J. Atrial fibrillation in hypertrophic cardiomyopathy: Prevalence, clinical correlations, and mortality in a large high-risk population. J. Am. Heart Assoc. 2014, 3, e001002. [Google Scholar] [CrossRef]
- Ball, J.; Carrington, M.J.; McMurray, J.J.V.; Stewart, S. Atrial fibrillation: Profile and burden of an evolving epidemic in the 21st century. Int. J. Cardiol. 2013, 167, 1807–1824. [Google Scholar] [CrossRef]
- Goldberger, J.J.; Arora, R.; Green, D.; Greenland, P.; Lee, D.C.; Lloyd-Jones, D.M.; Markl, M.; Ng, J.; Shah, S.J. Evaluating the atrial myopathy underlying atrial fibrillation: Identifying the arrhythmogenic and thrombogenic substrate. Circulation 2015, 132, 278–291. [Google Scholar] [CrossRef]
- Tsoutsman, T.; Chung, J.; Doolan, A.; Nguyen, L.; Williams, I.A.; Tu, E.; Lam, L.; Bailey, C.G.; Rasko, J.E.J.; Allen, D.G.; et al. Molecular insights from a novel cardiac troponin I mouse model of familial hypertrophic cardiomyopathy. J. Mol. Cell. Cardiol. 2006, 41, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Lim, W.-W.; Baumert, M.; Neo, M.; Kuklik, P.; Ganesan, A.N.; Lau, D.H.; Tsoutsman, T.; Semsarian, C.; Sanders, P.; Saint, D.A. Slowed atrial and atrioventricular conduction and depressed HRV in a murine model of hypertrophic cardiomyopathy. Clin. Exp. Pharmacol. Physiol. 2016, 43, 95–101. [Google Scholar] [CrossRef] [PubMed]
- Bonda, T.A.; Szynaka, B.; Sokołowska, M.; Dziemidowicz, M.; Winnicka, M.M.; Chyczewski, L.; Kamiński, K.A. Remodeling of the intercalated disc related to aging in the mouse heart. J. Cardiol. 2016, 68, 261–268. [Google Scholar] [CrossRef]
- Wijffels, M.C.; Kirchhof, C.J.; Dorland, R.; Allessie, M.A. Atrial fibrillation begets atrial fibrillation. A study in awake chronically instrumented goats. Circulation 1995, 92, 1954–1968. [Google Scholar] [CrossRef] [PubMed]
- Morillo, C.A.; Klein, G.J.; Jones, D.L.; Guiraudon, C.M. Chronic rapid atrial pacing. Structural, functional, and electrophysiological characteristics of a new model of sustained atrial fibrillation. Circulation 1995, 91, 1588–1595. [Google Scholar] [CrossRef]
- Li, D.; Fareh, S.; Leung, T.K.; Nattel, S. Promotion of atrial fibrillation by heart failure in dogs: Atrial remodeling of a different sort. Circulation 1999, 100, 87–95. [Google Scholar] [CrossRef] [PubMed]
- Sanders, P.; Morton, J.B.; Davidson, N.C.; Spence, S.J.; Vohra, J.K.; Sparks, P.B.; Kalman, J.M. Electrical remodeling of the atria in congestive heart failure. Circulation 2003, 108, 1461–1468. [Google Scholar] [CrossRef]
- Shinagawa, K.; Shi, Y.-F.; Tardif, J.-C.; Leung, T.-K.; Nattel, S. Dynamic nature of atrial fibrillation substrate during development and reversal of heart failure in dogs. Circulation 2002, 105, 2672–2678. [Google Scholar] [CrossRef] [PubMed]
- Alasady, M.; Shipp, N.J.; Brooks, A.G.; Lim, H.S.; Lau, D.H.; Barlow, D.; Kuklik, P.; Worthley, M.I.; Roberts-Thomson, K.C.; Saint, D.A.; et al. Myocardial infarction and atrial fibrillation: Importance of atrial ischemia. Circ. Arrhythm. Electrophysiol. 2013, 6, 738–745. [Google Scholar] [CrossRef] [PubMed]
- Mahajan, R.; Lau, D.H.; Brooks, A.G.; Shipp, N.J.; Manavis, J.; Wood, J.P.M.; Finnie, J.W.; Samuel, C.S.; Royce, S.G.; Twomey, D.J.; et al. Electrophysiological, electroanatomical, and structural remodeling of the atria as consequences of sustained obesity. J. Am. Coll. Cardiol. 2015, 66, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Iwasaki, Y.-K.; Kato, T.; Xiong, F.; Shi, Y.-F.; Naud, P.; Maguy, A.; Mizuno, K.; Tardif, J.-C.; Comtois, P.; Nattel, S. Atrial fibrillation promotion with long-term repetitive obstructive sleep apnea in a rat model. J. Am. Coll. Cardiol. 2014, 64, 2013–2023. [Google Scholar] [CrossRef]
- Lau, D.H.; Mackenzie, L.; Kelly, D.J.; Psaltis, P.J.; Brooks, A.G.; Worthington, M.; Rajendram, A.; Kelly, D.R.; Zhang, Y.; Kuklik, P.; et al. Hypertension and atrial fibrillation: Evidence of progressive atrial remodeling with electrostructural correlate in a conscious chronically instrumented ovine model. Heart Rhythm 2010, 7, 1282–1290. [Google Scholar] [CrossRef]
- Lau, D.H.; Psaltis, P.J.; Mackenzie, L.; Kelly, D.J.; Carbone, A.; Worthington, M.; Nelson, A.J.; Zhang, Y.; Kuklik, P.; Wong, C.X.; et al. Atrial remodeling in an ovine model of anthracycline-induced nonischemic cardiomyopathy: Remodeling of the same sort. J. Cardiovas. Electrophysiol. 2011, 175–182. [Google Scholar] [CrossRef]
- Sutanto, H.; Cluitmans, M.J.M.; Dobrev, D.; Volders, P.G.A.; Bébarová, M.; Heijman, J. Acute effects of alcohol on cardiac electrophysiology and arrhythmogenesis: Insights from multiscale in silico analyses. J. Mol. Cell. Cardiol. 2020, 146, 69–83. [Google Scholar] [CrossRef]
- Chen, Q.; Yi, Z.; Cheng, J. Atrial fibrillation in aging population. Aging Med. 2018, 1, 67–74. [Google Scholar] [CrossRef]
- Cherry, E.M.; Fenton, F.H. Effects of boundaries and geometry on the spatial distribution of action potential duration in cardiac tissue. J. Theor. Biol. 2011, 285, 164–176. [Google Scholar] [CrossRef][Green Version]
- Bishop, M.J.; Connolly, A.; Plank, G. Structural heterogeneity modulates effective refractory period: A mechanism of focal arrhythmia initiation. PLoS ONE 2014, 9, e109754. [Google Scholar] [CrossRef] [PubMed]
- Geisterfer-Lowrance, A.A.; Christe, M.; Conner, D.A.; Ingwall, J.S.; Schoen, F.J.; Seidman, C.E.; Seidman, J.G. A mouse model of familial hypertrophic cardiomyopathy. Science 1996, 272, 731–734. [Google Scholar] [CrossRef] [PubMed]
- McConnell, B.K.; Fatkin, D.; Semsarian, C.; Jones, K.A.; Georgakopoulos, D.; Maguire, C.T.; Healey, M.J.; Mudd, J.O.; Moskowitz, I.P.; Conner, D.A.; et al. Comparison of two murine models of familial hypertrophic cardiomyopathy. Circ. Res. 2001, 88, 383–389. [Google Scholar] [CrossRef]
- Prabhakar, R.; Boivin, G.P.; Grupp, I.L.; Hoit, B.; Arteaga, G.; Solaro, R.J.; Wieczorek, D.F. A familial hypertrophic cardiomyopathy alpha-tropomyosin mutation causes severe cardiac hypertrophy and death in mice. J. Mol. Cell. Cardiol. 2001, 33, 1815–1828. [Google Scholar] [CrossRef]
- Vikstrom, K.L.; Factor, S.M.; Leinwand, L.A. Mice expressing mutant myosin heavy chains are a model for familial hypertrophic cardiomyopathy. Mol. Med. 1996, 2, 556–567. [Google Scholar] [CrossRef] [PubMed]
- Dharmaprani, D.; Schopp, M.; Kuklik, P.; Chapman, D.; Lahiri, A.; Dykes, L.; Xiong, F.; Aguilar, M.; Strauss, B.; Mitchell, L.; et al. Renewal theory as a universal quantitative framework to characterize phase singularity regeneration in mammalian cardiac fibrillation. Circ. Arrhythm. Electrophysiol. 2019, 12, e007569. [Google Scholar] [CrossRef]
- Dharmaprani, D.; Jenkins, E.; Aguilar, M.; Quah, J.X.; Lahiri, A.; Tiver, K.; Mitchell, L.; Kuklik, P.; Meyer, C.; Willems, S.; et al. M/M/Infinity birth-death processes–A quantitative representational framework to summarize and explain phase singularity and wavelet dynamics in atrial fibrillation. Front. Physiol. 2021, 11, 616866. [Google Scholar] [CrossRef]
- Van Hunnik, A.; Zeemering, S.; Podziemski, P.; Kuklik, P.; Kuiper, M.; Verheule, S.; Schotten, U. Bi-atrial high-density mapping reveals inhibition of wavefront turning and reduction of complex propagation patterns as main antiarrhythmic mechanisms of vernakalant. Europace 2021, euab026. [Google Scholar] [CrossRef]
- Kistler, P.M.; Sanders, P.; Fynn, S.P.; Stevenson, I.H.; Spence, S.J.; Vohra, J.K.; Sparks, P.B.; Kalman, J.M. Electrophysiologic and electroanatomic changes in the human atrium associated with age. J. Am. Coll. Cardiol. 2004, 44, 109–116. [Google Scholar] [CrossRef]
- Osadchii, O.E. Role of abnormal repolarization in the mechanism of cardiac arrhythmia. Acta Physiol. 2017, 220, 1–71. [Google Scholar] [CrossRef]
- Spach, M.S.; Boineau, J.P. Microfibrosis produces electrical load variations due to loss of side-to-side cell connections; A major mechanism of structural heart disease arrhythmias. Pacing Clin. Electrophysiol. 1997, 20, 397–413. [Google Scholar] [CrossRef]
- Roldán, V.; Marín, F.; Gimeno, J.R.; Ruiz-Espejo, F.; González, J.; Feliu, E.; García-Honrubia, A.; Saura, D.; de la Morena, G.; Valdés, M.; et al. Matrix metalloproteinases and tissue remodeling in hypertrophic cardiomyopathy. Am. Heart J. 2008, 156, 85–91. [Google Scholar] [CrossRef] [PubMed]
- Bi, X.; Yang, C.; Song, Y.; Yuan, J.; Cui, J.; Hu, F.; Qiao, S. Matrix metalloproteinases increase because of hypoperfusion in obstructive hypertrophic cardiomyopathy. Ann. Thorac. Surg. 2021, 111, 915–922. [Google Scholar] [CrossRef] [PubMed]
- Meschiari, C.A.; Ero, O.K.; Pan, H.; Finkel, T.; Lindsey, M.L. The impact of aging on cardiac extracellular matrix. Geroscience 2017, 39, 7–18. [Google Scholar] [CrossRef]
- Xu, J.; Cui, G.; Esmailian, F.; Plunkett, M.; Marelli, D.; Ardehali, A.; Odim, J.; Laks, H.; Sen, L. Atrial extracellular matrix remodeling and the maintenance of atrial fibrillation. Circulation 2004, 109, 363–368. [Google Scholar] [CrossRef]
- Mukherjee, R.; Akar, J.G.; Wharton, J.M.; Adams, D.K.; McClure, C.D.; Stroud, R.E.; Rice, A.D.; DeSantis, S.M.; Spinale, F.G.; Gold, M.R. Plasma profiles of matrix metalloproteinases and tissue inhibitors of the metalloproteinases predict recurrence of atrial fibrillation following cardioversion. J. Cardiovasc. Transl. Res. 2013, 6, 528–535. [Google Scholar] [CrossRef] [PubMed]
- Tsoutsman, T.; Wang, X.; Garchow, K.; Riser, B.; Twigg, S.; Semsarian, C. CCN2 Plays a key role in extracellular matrix gene expression in severe hypertrophic cardiomyopathy and heart failure. J. Mol. Cell. Cardiol. 2013, 62, 164–178. [Google Scholar] [CrossRef]
- Teekakirikul, P.; Eminaga, S.; Toka, O.; Alcalai, R.; Wang, L.; Wakimoto, H.; Nayor, M.; Konno, T.; Gorham, J.M.; Wolf, C.M.; et al. Cardiac fibrosis in mice with hypertrophic cardiomyopathy is mediated by non-myocyte proliferation and requires Tgf-β. J. Clin. Investig. 2010, 120, 3520–3529. [Google Scholar] [CrossRef]
- Harada, M.; Van Wagoner, D.R.; Nattel, S. Role of inflammation in atrial fibrillation pathophysiology and management. Circ. J. 2015, 79, 495–502. [Google Scholar] [CrossRef]
- Heijman, J.; Muna, A.P.; Veleva, T.; Molina, C.E.; Sutanto, H.; Tekook, M.; Wang, Q.; Abu-Taha, I.H.; Gorka, M.; Künzel, S.; et al. Atrial myocyte NLRP3/CaMKII nexus forms a substrate for postoperative atrial fibrillation. Circ. Res. 2020, 127, 1036–1055. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Alcaide, P.; Liu, L.; Sun, J.; He, A.; Luscinskas, F.W.; Shi, G.-P. Regulation of endothelial cell adhesion molecule expression by mast cells, macrophages, and neutrophils. PLoS ONE 2011, 6, e14525. [Google Scholar] [CrossRef]
- Schultz, C.D.; Rangneker, G.; Lim, H.S.; Fraudeau, A.; Young, G.; Roberts-Thomson, K.; John, B.; Worthley, M.; Sanders, P.; Willoughby, S.R. Characterization of thrombogenic, endothelial and inflammatory markers in supraventricular tachycardia: A study in patients with structurally normal hearts. Clin. Exp. Pharmacol. Physiol. 2014, 41, 551–557. [Google Scholar] [CrossRef] [PubMed]
- Kitz, S.; Fonfara, S.; Hahn, S.; Hetzel, U.; Kipar, A. Feline hypertrophic cardiomyopathy: The consequence of cardiomyocyte-initiated and macrophage-driven remodeling processes? Vet. Pathol. 2019, 56, 565–575. [Google Scholar] [CrossRef] [PubMed]
- Willeit, K.; Pechlaner, R.; Willeit, P.; Skroblin, P.; Paulweber, B.; Schernthaner, C.; Toell, T.; Egger, G.; Weger, S.; Oberhollenzer, M.; et al. Association between vascular cell adhesion molecule 1 and atrial fibrillation. JAMA Cardiol. 2017, 2, 516–523. [Google Scholar] [CrossRef]
- Vaidya, K.; Semsarian, C.; Chan, K.H. Atrial fibrillation in hypertrophic cardiomyopathy. Heart Lung Circ. 2017, 26, 975–982. [Google Scholar] [CrossRef] [PubMed]
- Pujadas, S.; Vidal-Perez, R.; Hidalgo, A.; Leta, R.; Carreras, F.; Barros, A.; Bayes-Genis, A.; Subirana, M.T.; Pons-Llado, G. Correlation between myocardial fibrosis and the occurrence of atrial fibrillation in hypertrophic cardiomyopathy: A cardiac magnetic resonance imaging study. Eur. J. Radiol. 2010, 75, e88–e91. [Google Scholar] [CrossRef]
- Gruver, E.J.; James Gruver, E.; Fatkin, D.; Alfred Dodds, G.; Kisslo, J.; Maron, B.J.; Seidman, J.G.; Seidman, C.E. Familial hypertrophic cardiomyopathy and atrial fibrillation caused by Arg663His beta–cardiac myosin heavy chain mutation. Am. J. Cardiol. 1999, 83, 13–18. [Google Scholar] [CrossRef]
- Thanigaimani, S.; Brooks, A.G.; Kuklik, P.; Twomey, D.J.; Franklin, S.; Noschka, E.; Chapman, D.; Pathak, R.K.; Mahajan, R.; Sanders, P.; et al. Spatiotemporal characteristics of atrial fibrillation electrograms: A novel marker for arrhythmia stability and termination. J. Arrhythm. 2017, 33, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Dinshaw, L.; Münkler, P.; Schäffer, B.; Klatt, N.; Jungen, C.; Dickow, J.; Tamenang, A.; Schleberger, R.; Pecha, S.; Pinnschmidt, H.; et al. Ablation of atrial fibrillation in patients with hypertrophic cardiomyopathy: Treatment strategy, characteristics of consecutive atrial tachycardia and long-term outcome. J. Am. Heart Assoc. 2021, 10, e017451. [Google Scholar] [CrossRef] [PubMed]
- Neo, M.; Morris, D.G.; Kuklik, P.; Lau, D.H.; Dimitri, H.; Lim, W.-W.; Sanders, P.; Saint, D.A. Simultaneous conduction mapping and intracellular membrane potential recording in isolated atria. Can. J. Physiol. Pharmacol. 2016, 94, 563–569. [Google Scholar] [CrossRef]
- Dahab, G.M.; Kheriza, M.M.; El-Beltagi, H.M.; Fouda, A.-M.M.; El-Din, O.A.S. Digital quantification of fibrosis in liver biopsy sections: Description of a new method by photoshop software. J. Gastroenterol. Hepatol. 2004, 19, 78–85. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| 30 Weeks Old | 50 Weeks Old | |||
|---|---|---|---|---|
| WT | HCM | WT | HCM | |
| Body weight (g) | 31.3 ± 1.2 | 30.3 ± 1.0 | 30.3 ± 0.3 | 31.3 ± 0.5 |
| Heart rate (bpm) | 295.5 ± 14.9 | 254.9 ± 20.4 | 268.7 ± 7.3 | 273.8 ± 12.4 |
| Systolic blood pressure (mmHg) | 94.5 ± 2.8 | 95.3 ± 1.5 | 86.4 ± 1.6 ǂ | 85.2 ± 1.7 ǂ |
| LA mass (mg) | 3.9 ± 0.4 | 6.8 ± 0.9 * | 3.7 ± 0.1 | 8.0 ± 2.0 * |
| RA mass (mg) | 4.1 ± 0.4 | 5.3 ± 0.6 * | 3.6 ± 0.3 | 4.4 ± 0.5 * |
| LV wall thickness (mm) | 1.2 ± 0.1 | 1.5 ± 0.1 * | 1.2 ± 0.1 | 1.7 ± 0.1 * |
| Septal wall thickness (mm) | 1.0 ± 0.1 | 1.3 ± 0.1 * | 0.9 ± 0.1 | 1.4 ± 0.1 * |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lim, W.-W.; Neo, M.; Thanigaimani, S.; Kuklik, P.; Ganesan, A.N.; Lau, D.H.; Tsoutsman, T.; Kalman, J.M.; Semsarian, C.; Saint, D.A.; et al. Electrophysiological and Structural Remodeling of the Atria in a Mouse Model of Troponin-I Mutation Linked Hypertrophic Cardiomyopathy: Implications for Atrial Fibrillation. Int. J. Mol. Sci. 2021, 22, 6941. https://doi.org/10.3390/ijms22136941
Lim W-W, Neo M, Thanigaimani S, Kuklik P, Ganesan AN, Lau DH, Tsoutsman T, Kalman JM, Semsarian C, Saint DA, et al. Electrophysiological and Structural Remodeling of the Atria in a Mouse Model of Troponin-I Mutation Linked Hypertrophic Cardiomyopathy: Implications for Atrial Fibrillation. International Journal of Molecular Sciences. 2021; 22(13):6941. https://doi.org/10.3390/ijms22136941
Chicago/Turabian StyleLim, Wei-Wen, Melissa Neo, Shivshankar Thanigaimani, Pawel Kuklik, Anand N. Ganesan, Dennis H. Lau, Tatiana Tsoutsman, Jonathan M. Kalman, Christopher Semsarian, David A. Saint, and et al. 2021. "Electrophysiological and Structural Remodeling of the Atria in a Mouse Model of Troponin-I Mutation Linked Hypertrophic Cardiomyopathy: Implications for Atrial Fibrillation" International Journal of Molecular Sciences 22, no. 13: 6941. https://doi.org/10.3390/ijms22136941
APA StyleLim, W.-W., Neo, M., Thanigaimani, S., Kuklik, P., Ganesan, A. N., Lau, D. H., Tsoutsman, T., Kalman, J. M., Semsarian, C., Saint, D. A., & Sanders, P. (2021). Electrophysiological and Structural Remodeling of the Atria in a Mouse Model of Troponin-I Mutation Linked Hypertrophic Cardiomyopathy: Implications for Atrial Fibrillation. International Journal of Molecular Sciences, 22(13), 6941. https://doi.org/10.3390/ijms22136941