
TENT4A Non-Canonical Poly(A) Polymerase Regulates DNA-Damage Tolerance via Multiple Pathways That Are Mutated in Endometrial Cancer

 , , , ,
, , , ,
Abstract
1. Introduction
2. Results
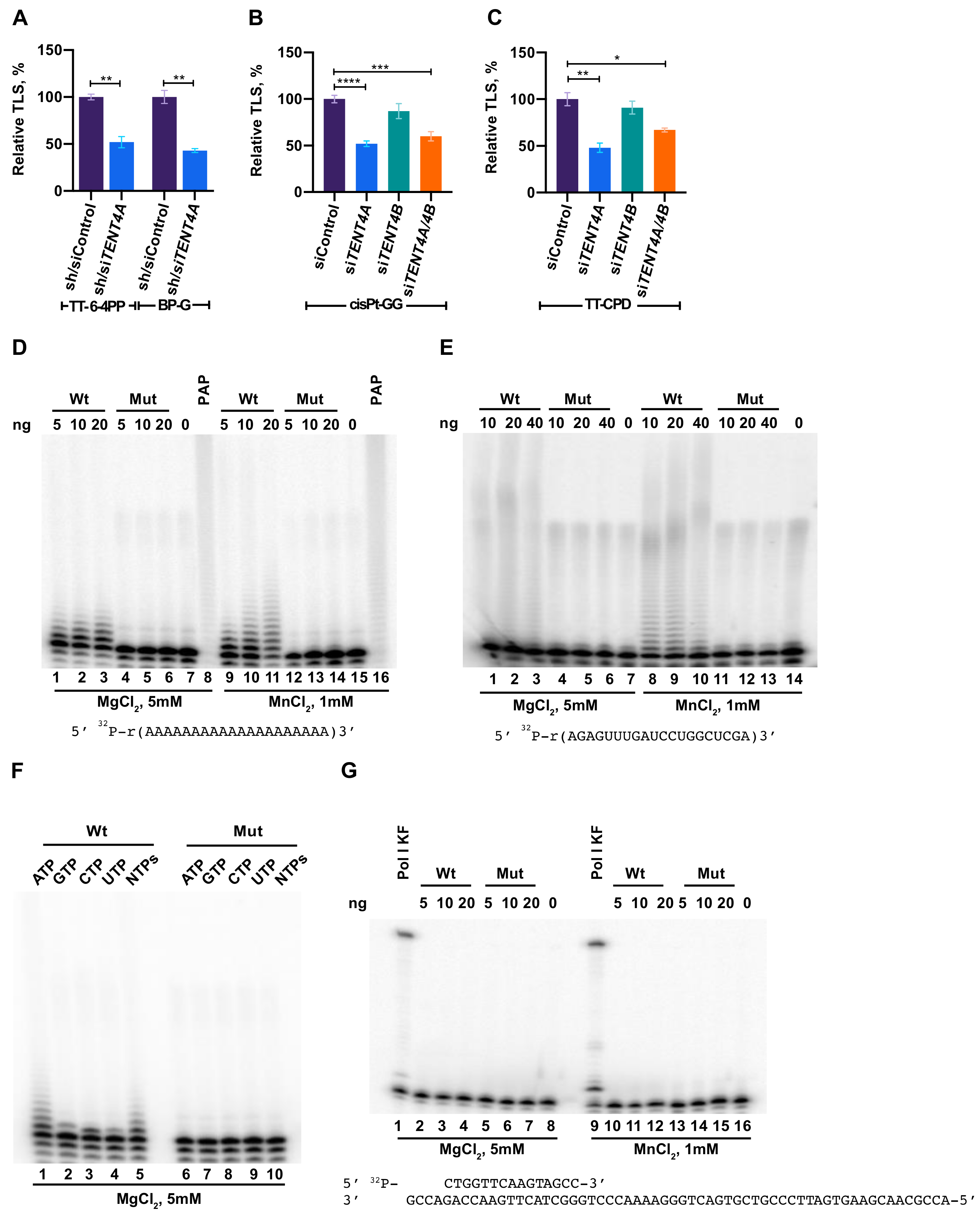
2.1. TENT4A Is Needed for Effective TLS
2.2. Purified TENT4A Is a Non-Canonical Poly(A) RNA Polymerase, Which Also Incorporates Other Ribonucleotides
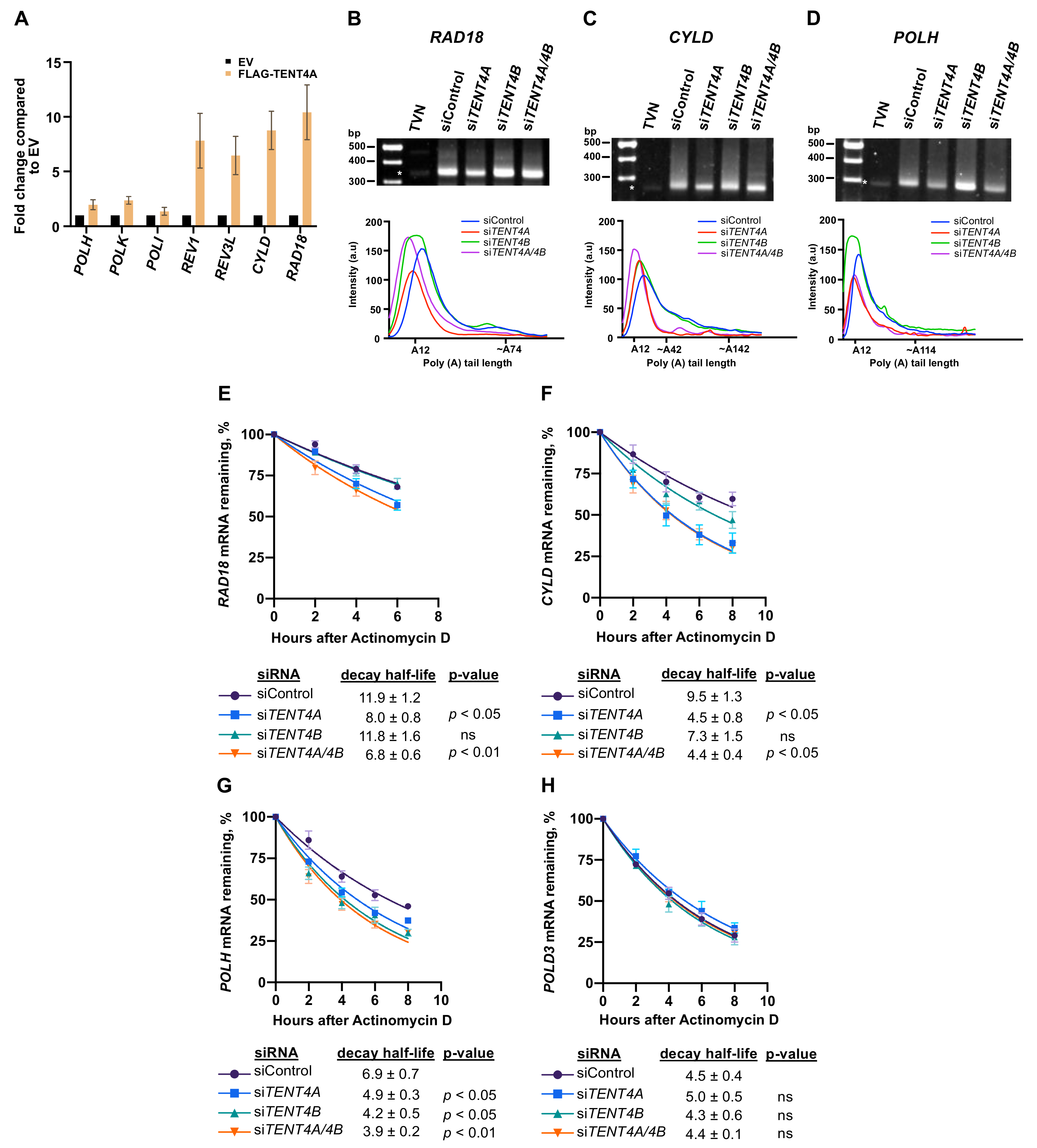
2.3. Effect of TENT4A on mRNA of Genes Involved in TLS
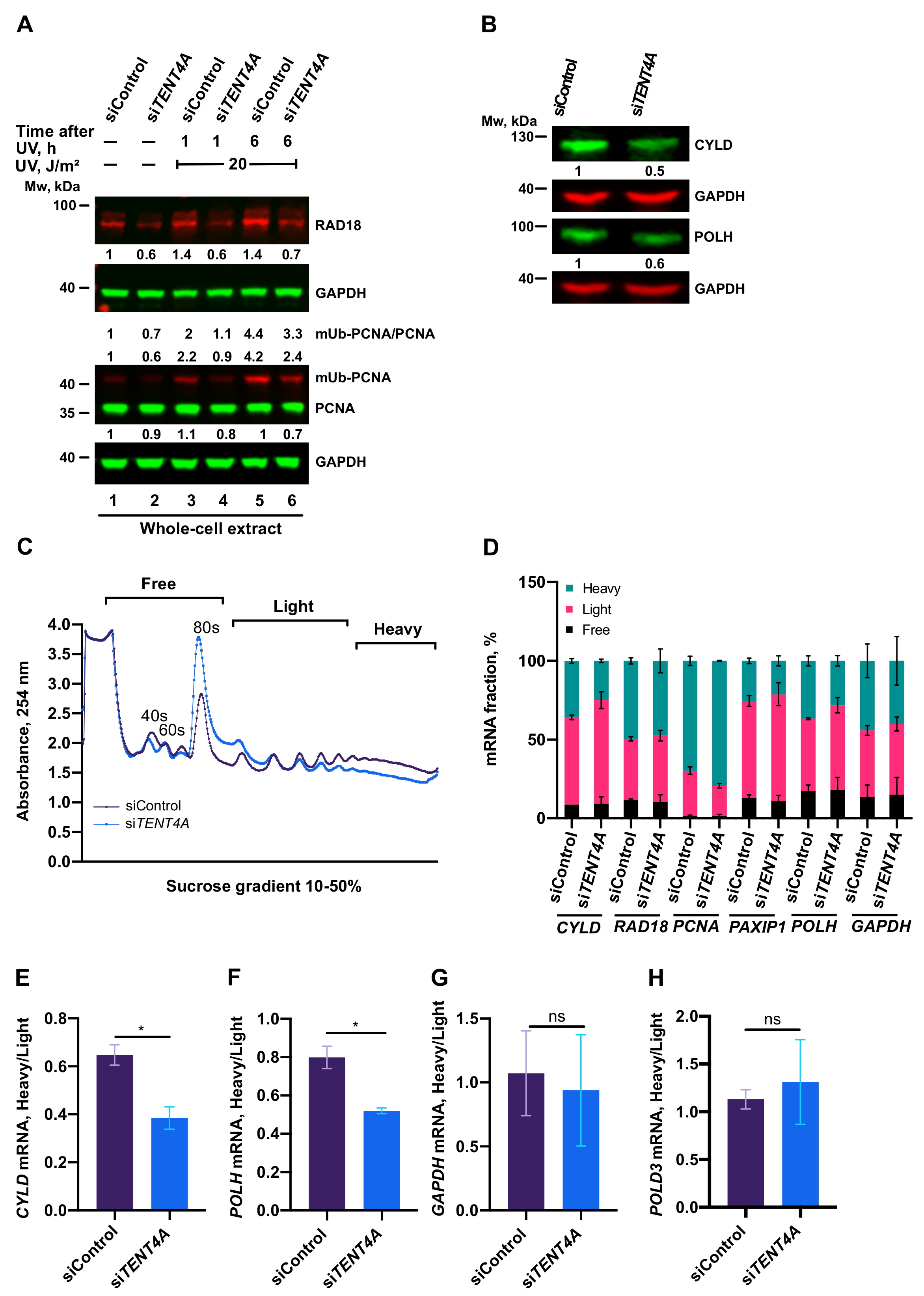
2.4. TENT4A Affects the Amounts of RAD18, CYLD and POLH Proteins via Different Mechanisms
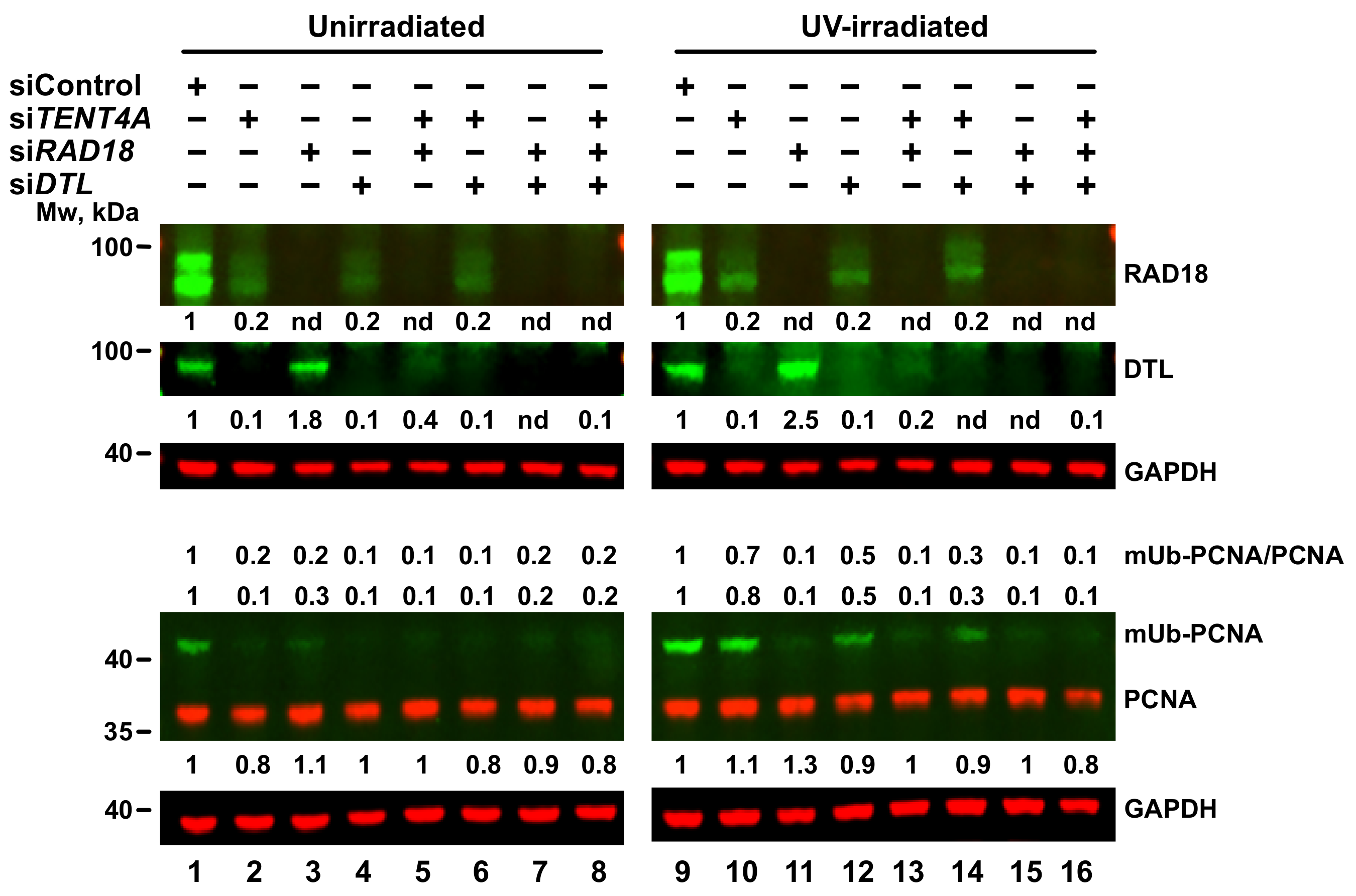
2.5. Regulation of PCNA Monoubiquitination by TENT4A
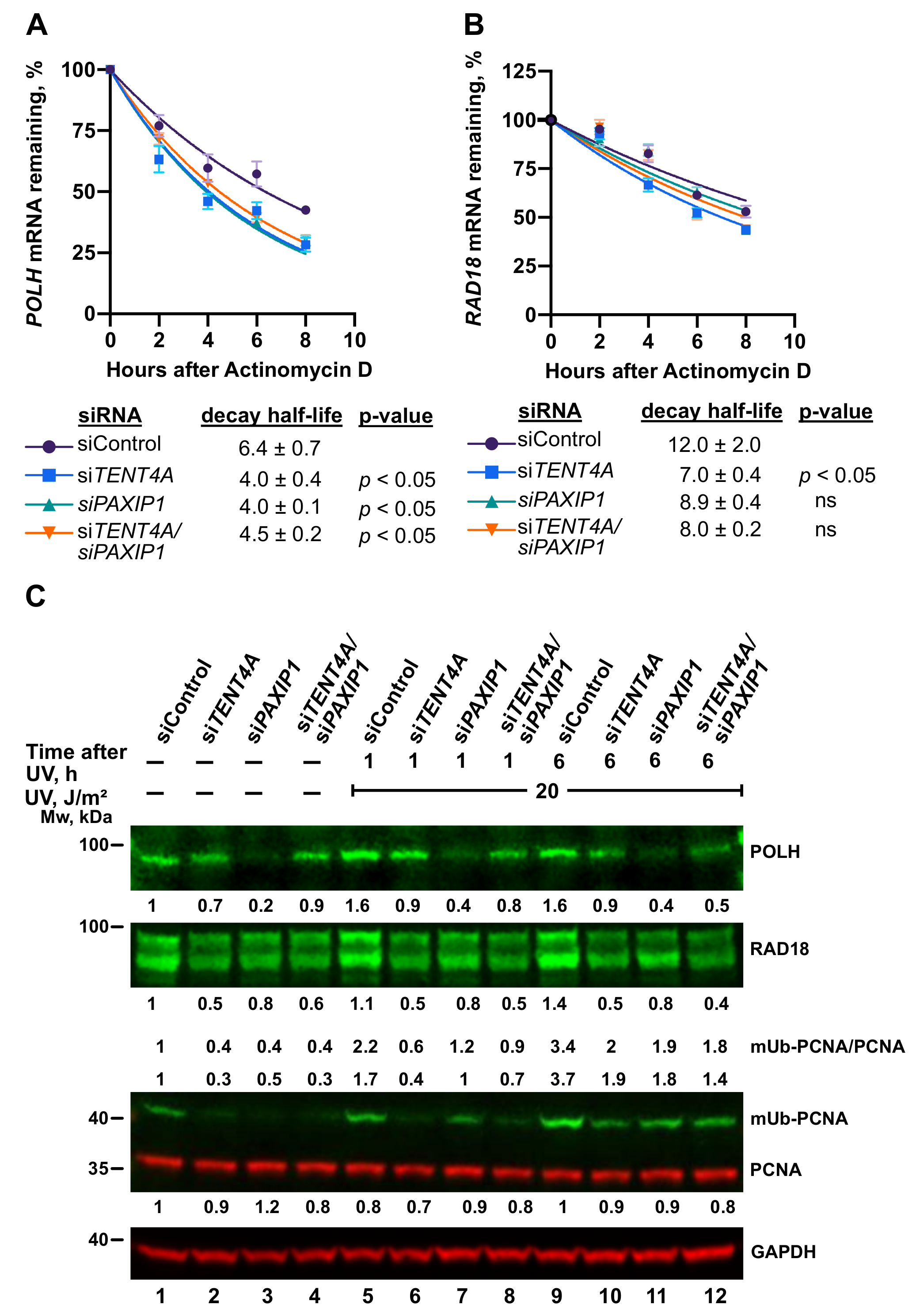
2.6. TENT4A-Regulated CYLD Regulates RAD18, DTL and POLH
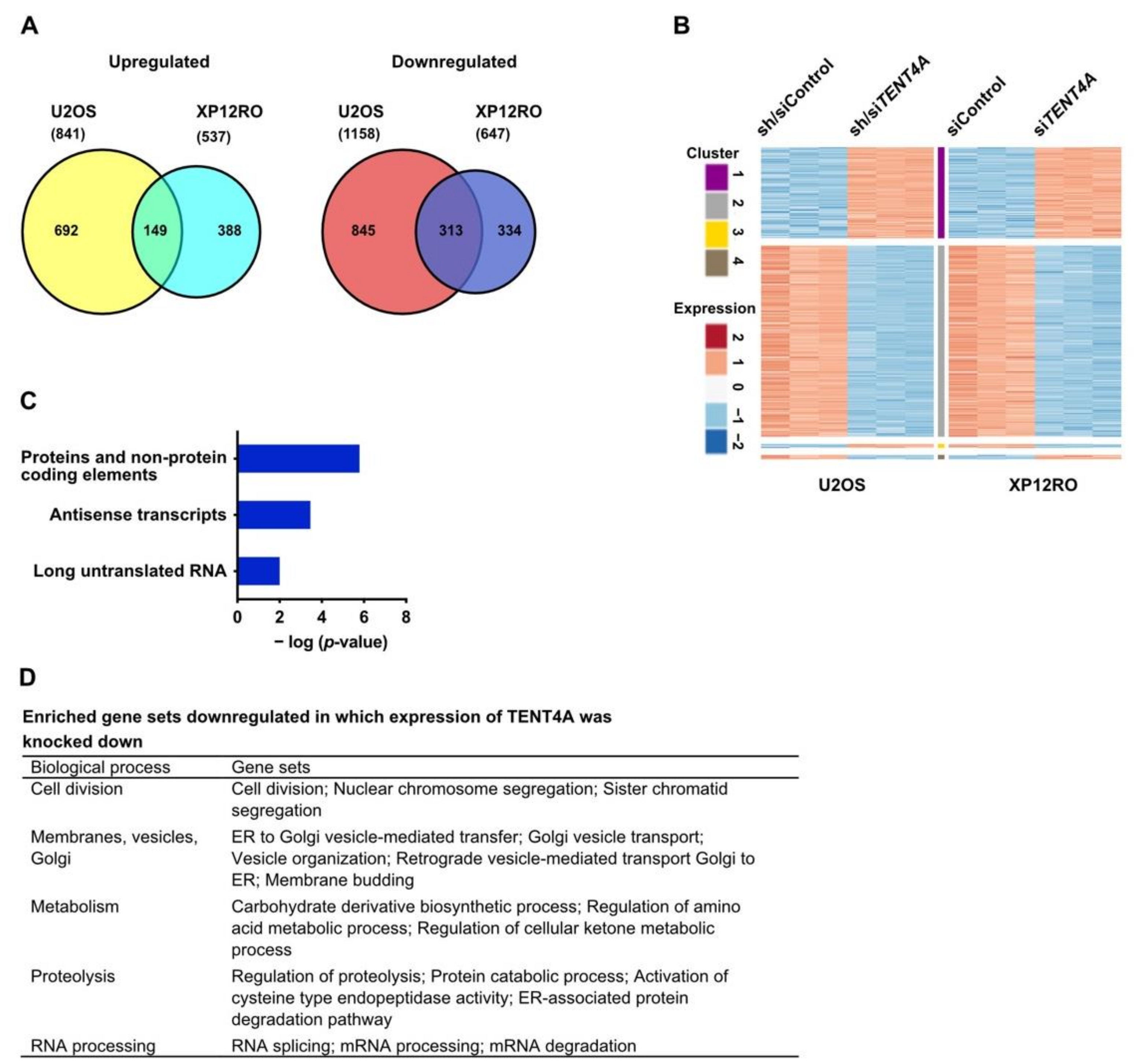
2.7. TENT4A Regulates Multiple Genes and Pathways, Including Long Non-Coding RNAs and Antisense Transcripts
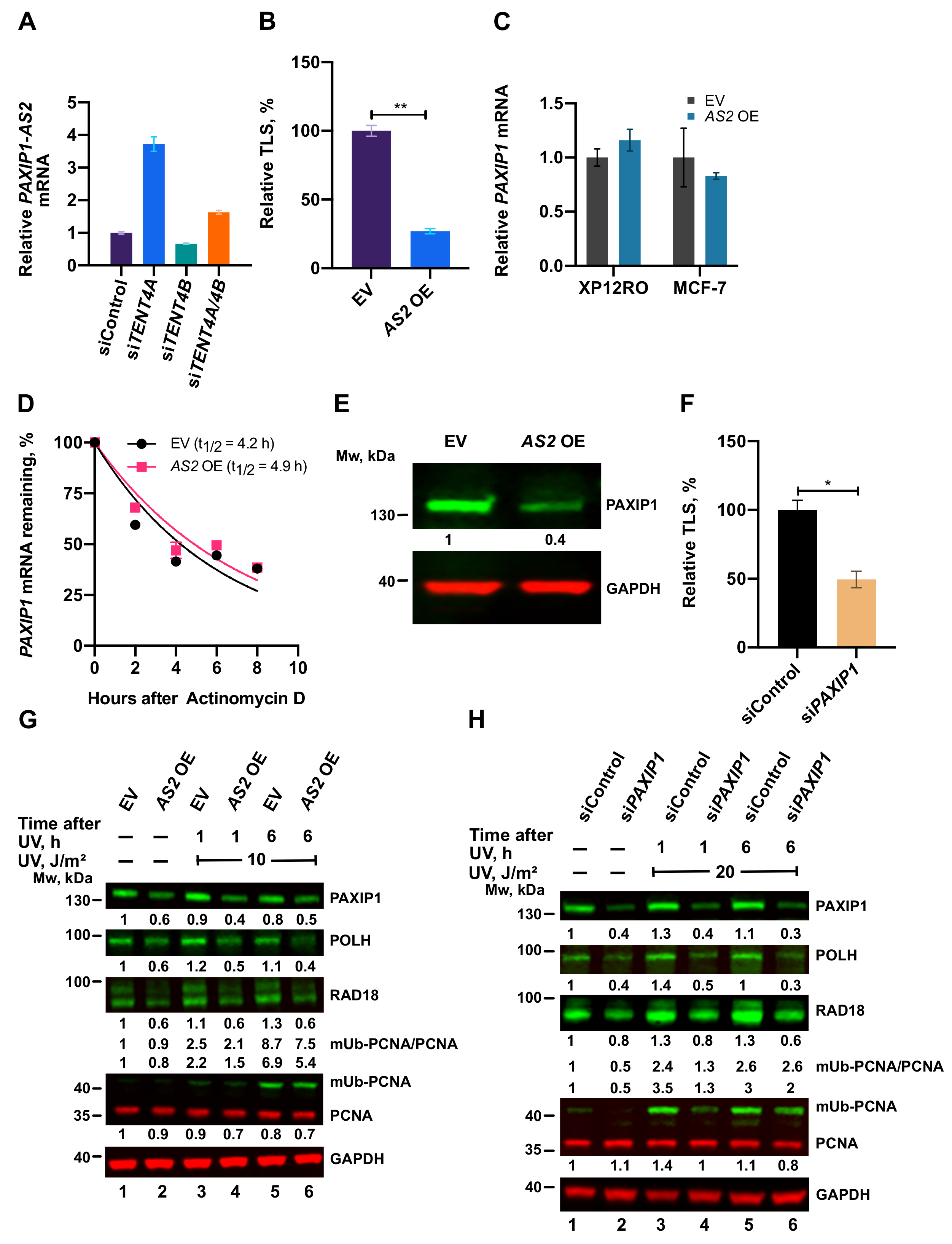
2.8. A TENT4A-Regulated Long Non-Coding Antisense RNA to the PAXIP1 (PTIP) Gene Regulates RAD18, POLH and mUb-PCNA
2.9. TENT4A and TLS Genes Are Frequently Mutated in Endometrial Cancer
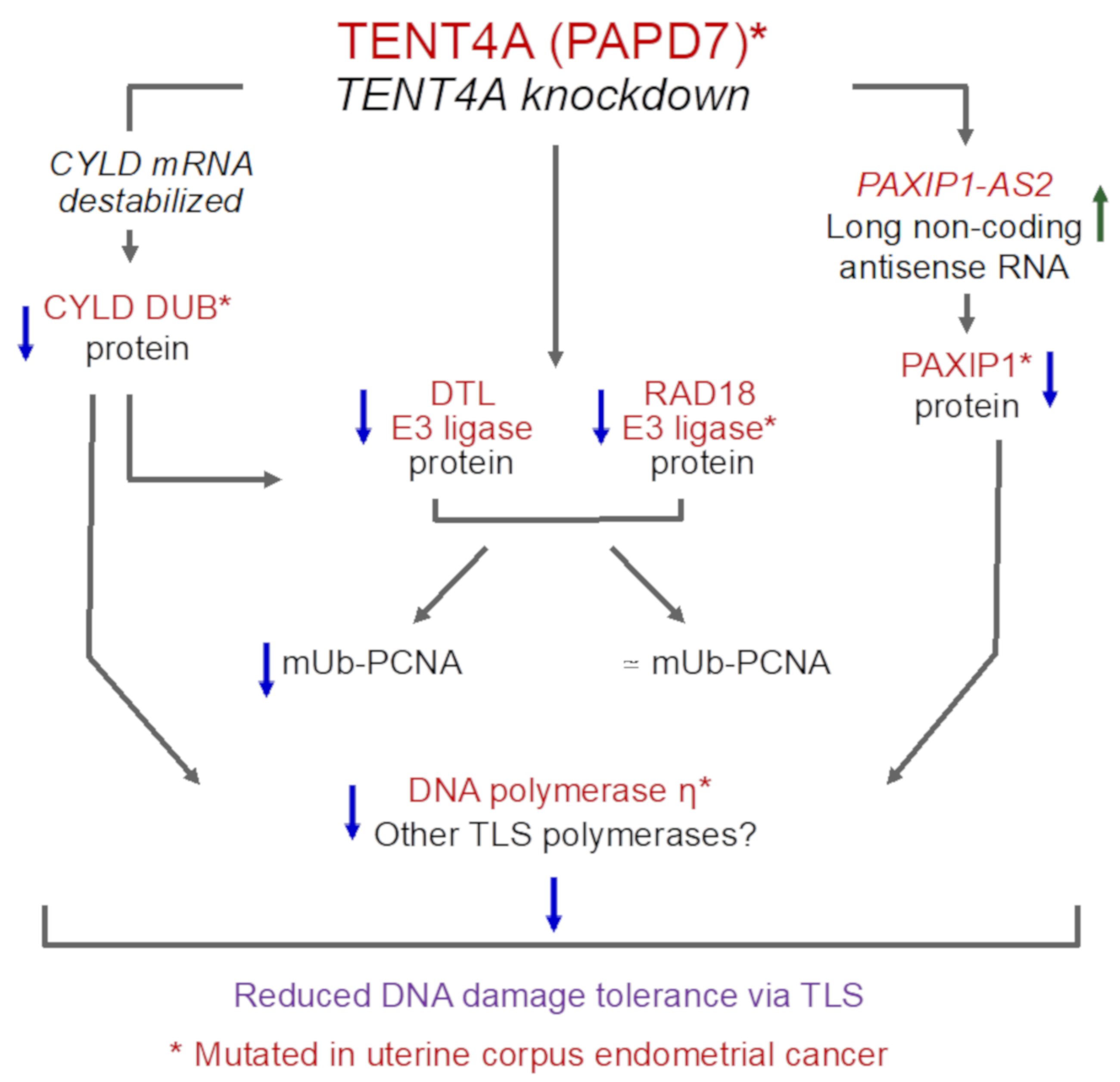
3. Discussion
4. Materials and Methods
4.1. Cell Cultures and Transfections
4.2. Generation of Lentiviral Stable Cell Lines
4.3. TLS Assay in Cultured Mammalian Cells
4.4. RNA Isolation and qPCR
4.5. mRNA Stability Assay
4.6. RNA-Immunoprecipitation (RNA-IP) and Extension Poly(A) Test (ePAT)
4.7. UV-Irradiation, Whole-Cell Extracts and Western Blotting
4.8. Antibodies
4.9. RNA-Seq and Bioinformatics
4.10. Gene Set Enrichment Analysis (GSEA)
4.11. TCGA Database Mutation Analysis
4.12. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Errol, C.F.; Roger, A.S.; Wolfram, S.; Graham, C.W.; Tom, E.; Richard, D.W. DNA Repair and Mutagenesis, 2nd ed.; American Society of Microbiology: Washington, DC, USA, 2006. [Google Scholar]
- Marteijn, J.A.; Lans, H.; Vermeulen, W.; Hoeijmakers, J.H. Understanding nucleotide excision repair and its roles in cancer and ageing. Nat. Rev. Mol. Cell Biol. 2014, 15, 465–481. [Google Scholar] [CrossRef]
- Modrich, P. Mismatch repair, genetic stability, and cancer. Science 1994, 266, 1959–1960. [Google Scholar] [CrossRef] [PubMed]
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef]
- Li, Z.; Woo, C.J.; Iglesias-Ussel, M.D.; Ronai, D.; Scharff, M.D. The generation of antibody diversity through somatic hypermutation and class switch recombination. Genes Dev. 2004, 18, 1–11. [Google Scholar] [CrossRef]
- Akbari, M.; Morevati, M.; Croteau, D.; Bohr, V.A. The role of DNA base excision repair in brain homeostasis and disease. DNA Repair 2015, 32, 172–179. [Google Scholar] [CrossRef]
- Branzei, D.; Foiani, M. Maintaining genome stability at the replication fork. Nat. Rev. Mol. Cell Biol. 2010, 11, 208–219. [Google Scholar] [CrossRef]
- Friedberg, E.C. Suffering in silence: The tolerance of DNA damage. Nat. Rev. Mol. Cell Biol. 2005, 6, 943–953. [Google Scholar] [CrossRef] [PubMed]
- Livneh, Z.; Cohen, I.S.; Paz-Elizur, T.; Davidovsky, D.; Carmi, D.; Swain, U.; Mirlas-Neisberg, N. High-resolution genomic assays provide insight into the division of labor between TLS and HDR in mammalian replication of damaged DNA. DNA Repair 2016, 44, 59–67. [Google Scholar] [CrossRef]
- Sale, J.E.; Lehmann, A.R.; Woodgate, R. Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nat. Rev. Mol. Cell Biol. 2012, 13, 141–152. [Google Scholar] [CrossRef]
- Livneh, Z. Keeping mammalian mutation load in check: Regulation of the activity of error-prone DNA polymerases by p53 and p21. Cell Cycle 2006, 5, 1918–1922. [Google Scholar] [CrossRef] [PubMed]
- Hendel, A.; Ziv, O.; Gueranger, Q.; Geacintov, N.; Livneh, Z. Reduced efficiency and increased mutagenicity of translesion DNA synthesis across a TT cyclobutane pyrimidine dimer, but not a TT 6-4 photoproduct, in human cells lacking DNA polymerase eta. DNA Repair 2008, 7, 1636–1646. [Google Scholar] [CrossRef] [PubMed]
- Cohen, I.S.; Bar, C.; Paz-Elizur, T.; Ainbinder, E.; Leopold, K.; de Wind, N.; Geacintov, N.; Livneh, Z. DNA lesion identity drives choice of damage tolerance pathway in murine cell chromosomes. Nucleic Acids Res. 2015, 43, 1637–1645. [Google Scholar] [CrossRef] [PubMed]
- Avkin, S.; Sevilya, Z.; Toube, L.; Geacintov, N.; Chaney, S.G.; Oren, M.; Livneh, Z. p53 and p21 regulate error-prone DNA repair to yield a lower mutation load. Mol. Cell 2006, 22, 407–413. [Google Scholar] [CrossRef] [PubMed]
- Hoege, C.; Pfander, B.; Moldovan, G.L.; Pyrowolakis, G.; Jentsch, S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 2002, 419, 135–141. [Google Scholar] [CrossRef]
- Kannouche, P.L.; Wing, J.; Lehmann, A.R. Interaction of Human DNA Polymerase η with Monoubiquitinated PCNA. Mol. Cell 2004, 14, 491–500. [Google Scholar] [CrossRef]
- Hendel, A.; Krijger, P.H.; Diamant, N.; Goren, Z.; Langerak, P.; Kim, J.; Reissner, T.; Lee, K.Y.; Geacintov, N.E.; Carell, T.; et al. PCNA ubiquitination is important, but not essential for translesion DNA synthesis in mammalian cells. PLoS Genet. 2011, 7, e1002262. [Google Scholar] [CrossRef]
- Ziv, O.; Zeisel, A.; Mirlas-Neisberg, N.; Swain, U.; Nevo, R.; Ben-Chetrit, N.; Martelli, M.P.; Rossi, R.; Schiesser, S.; Canman, C.E.; et al. Identification of novel DNA-damage tolerance genes reveals regulation of translesion DNA synthesis by nucleophosmin. Nat. Commun 2014, 5, 5437. [Google Scholar] [CrossRef]
- Wang, Z.; Castano, I.B.; De Las Penas, A.; Adams, C.; Christman, M.F. Pol kappa: A DNA polymerase required for sister chromatid cohesion. Science 2000, 289, 774–779. [Google Scholar] [CrossRef]
- Haracska, L.; Johnson, R.E.; Prakash, L.; Prakash, S. Trf4 and Trf5 proteins of Saccharomyces cerevisiae exhibit poly(A) RNA polymerase activity but no DNA polymerase activity. Mol. Cell Biol. 2005, 25, 10183–10189. [Google Scholar] [CrossRef]
- Ogami, K.; Cho, R.; Hoshino, S. Molecular cloning and characterization of a novel isoform of the non-canonical poly(A) polymerase PAPD7. Biochem. Biophys. Res. Commun. 2013, 432, 135–140. [Google Scholar] [CrossRef]
- Lim, J.; Kim, D.; Lee, Y.S.; Ha, M.; Lee, M.; Yeo, J.; Chang, H.; Song, J.; Ahn, K.; Kim, V.N. Mixed tailing by TENT4A and TENT4B shields mRNA from rapid deadenylation. Science 2018, 361, 701–704. [Google Scholar] [CrossRef] [PubMed]
- Mueller, H.; Lopez, A.; Tropberger, P.; Wildum, S.; Schmaler, J.; Pedersen, L.; Han, X.; Wang, Y.; Ottosen, S.; Yang, S.; et al. PAPD5/7 Are Host Factors That Are Required for Hepatitis B Virus RNA Stabilization. Hepatology 2019, 69, 1398–1411. [Google Scholar] [CrossRef] [PubMed]
- Shachar, S.; Ziv, O.; Avkin, S.; Adar, S.; Wittschieben, J.; Reissner, T.; Chaney, S.; Friedberg, E.C.; Wang, Z.; Carell, T.; et al. Two-polymerase mechanisms dictate error-free and error-prone translesion DNA synthesis in mammals. EMBO J. 2009, 28, 383–393. [Google Scholar] [CrossRef]
- Watanabe, K.; Tateishi, S.; Kawasuji, M.; Tsurimoto, T.; Inoue, H.; Yamaizumi, M. Rad18 guides pol eta to replication stalling sites through physical interaction and PCNA monoubiquitination. EMBO J. 2004, 23, 3886–3896. [Google Scholar] [CrossRef] [PubMed]
- Janicke, A.; Vancuylenberg, J.; Boag, P.R.; Traven, A.; Beilharz, T.H. ePAT: A simple method to tag adenylated RNA to measure poly(A)-tail length and other 3′ RACE applications. RNA 2012, 18, 1289–1295. [Google Scholar] [CrossRef]
- Terai, K.; Abbas, T.; Jazaeri, A.A.; Dutta, A. CRL4(Cdt2) E3 ubiquitin ligase monoubiquitinates PCNA to promote translesion DNA synthesis. Mol. Cell 2010, 37, 143–149. [Google Scholar] [CrossRef] [PubMed]
- Gohler, T.; Munoz, I.M.; Rouse, J.; Blow, J.J. PTIP/Swift is required for efficient PCNA ubiquitination in response to DNA damage. DNA Repair 2008, 7, 775–787. [Google Scholar] [CrossRef]
- Schwab, K.R.; Smith, G.D.; Dressler, G.R. Arrested spermatogenesis and evidence for DNA damage in PTIP mutant testes. Dev. Biol. 2013, 373, 64–71. [Google Scholar] [CrossRef]
- Callen, E.; Faryabi, R.B.; Luckey, M.; Hao, B.; Daniel, J.A.; Yang, W.; Sun, H.W.; Dressler, G.; Peng, W.; Chi, H.; et al. The DNA damage- and transcription-associated protein paxip1 controls thymocyte development and emigration. Immunity 2012, 37, 971–985. [Google Scholar] [CrossRef]
- Wang, J.; Aroumougame, A.; Lobrich, M.; Li, Y.; Chen, D.; Chen, J.; Gong, Z. PTIP associates with Artemis to dictate DNA repair pathway choice. Genes Dev. 2014, 28, 2693–2698. [Google Scholar] [CrossRef]
- Mijic, S.; Zellweger, R.; Chappidi, N.; Berti, M.; Jacobs, K.; Mutreja, K.; Ursich, S.; Ray Chaudhuri, A.; Nussenzweig, A.; Janscak, P.; et al. Replication fork reversal triggers fork degradation in BRCA2-defective cells. Nat. Commun. 2017, 8, 859. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Tian, Y.; Zhang, J.; Tong, X.; Huang, H.; Li, S.; Zhao, H.; Tang, Y.; Yuan, C.; Wang, K.; et al. In vivo CRISPR screening unveils histone demethylase UTX as an important epigenetic regulator in lung tumorigenesis. Proc. Natl. Acad. Sci. USA 2018, 115, E3978–E3986. [Google Scholar] [CrossRef]
- Jhuraney, A.; Woods, N.T.; Wright, G.; Rix, L.; Kinose, F.; Kroeger, J.L.; Remily-Wood, E.; Cress, W.D.; Koomen, J.M.; Brantley, S.G.; et al. PAXIP1 Potentiates the Combination of WEE1 Inhibitor AZD1775 and Platinum Agents in Lung Cancer. Mol. Cancer Ther. 2016, 15, 1669–1681. [Google Scholar] [CrossRef] [PubMed]
- Carrieri, C.; Cimatti, L.; Biagioli, M.; Beugnet, A.; Zucchelli, S.; Fedele, S.; Pesce, E.; Ferrer, I.; Collavin, L.; Santoro, C.; et al. Long non-coding antisense RNA controls Uchl1 translation through an embedded SINEB2 repeat. Nature 2012, 491, 454–457. [Google Scholar] [CrossRef]
- Huang, M.; Zhou, B.; Gong, J.; Xing, L.; Ma, X.; Wang, F.; Wu, W.; Shen, H.; Sun, C.; Zhu, X.; et al. RNA-splicing factor SART3 regulates translesion DNA synthesis. Nucleic Acids Res. 2018, 46, 4560–4574. [Google Scholar] [CrossRef]
- Durando, M.; Tateishi, S.; Vaziri, C. A non-catalytic role of DNA polymerase eta in recruiting Rad18 and promoting PCNA monoubiquitination at stalled replication forks. Nucleic Acids Res. 2013, 41, 3079–3093. [Google Scholar] [CrossRef]
- Casali, P.; Pal, Z.; Xu, Z.; Zan, H. DNA repair in antibody somatic hypermutation. Trends Immunol. 2006, 27, 313–321. [Google Scholar] [CrossRef][Green Version]
- Wittschieben, J.P.; Reshmi, S.C.; Gollin, S.M.; Wood, R.D. Loss of DNA polymerase zeta causes chromosomal instability in mammalian cells. Cancer Res. 2006, 66, 134–142. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network; Kandoth, C.; Schultz, N.; Cherniack, A.D.; Akbani, R.; Liu, Y.; Shen, H.; Robertson, A.G.; Pashtan, I.; Shen, R.; et al. Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar] [CrossRef]
- Suhaimi, S.S.; Ab Mutalib, N.S.; Jamal, R. Understanding Molecular Landscape of Endometrial Cancer through Next Generation Sequencing: What We Have Learned so Far? Front. Pharmacol. 2016, 7, 409. [Google Scholar] [CrossRef]
- Zafar, M.K.; Eoff, R.L. Translesion DNA Synthesis in Cancer: Molecular Mechanisms and Therapeutic Opportunities. Chem. Res. Toxicol. 2017, 30, 1942–1955. [Google Scholar] [CrossRef] [PubMed]
- Gallo, D.; Brown, G.W. Post-replication repair: Rad5/HLTF regulation, activity on undamaged templates, and relationship to cancer. Crit. Rev. Biochem. Mol. Biol. 2019, 54, 301–332. [Google Scholar] [CrossRef] [PubMed]
- Diamant, N.; Hendel, A.; Vered, I.; Carell, T.; Reissner, T.; de Wind, N.; Geacinov, N.; Livneh, Z. DNA damage bypass operates in the S and G2 phases of the cell cycle and exhibits differential mutagenicity. Nucleic Acids Res. 2012, 40, 170–180. [Google Scholar] [CrossRef] [PubMed]
- Ziv, O.; Diamant, N.; Shachar, S.; Hendel, A.; Livneh, Z. Quantitative Measurement of Translesion DNA Synthesis in Mammalian Cells. In DNA Repair Protocols; Bjergbæk, L., Ed.; Humana Press: Totowa, NJ, USA, 2012; pp. 529–542. [Google Scholar]
- Keene, J.D.; Komisarow, J.M.; Friedersdorf, M.B. RIP-Chip: The isolation and identification of mRNAs, microRNAs and protein components of ribonucleoprotein complexes from cell extracts. Nat. Protoc. 2006, 1, 302–307. [Google Scholar] [CrossRef] [PubMed]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Colaprico, A.; Silva, T.C.; Olsen, C.; Garofano, L.; Cava, C.; Garolini, D.; Sabedot, T.S.; Malta, T.M.; Pagnotta, S.M.; Castiglioni, I.; et al. TCGAbiolinks: An R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 2016, 44, e71. [Google Scholar] [CrossRef]
- Mayakonda, A.; Lin, D.C.; Assenov, Y.; Plass, C.; Koeffler, H.P. Maftools: Efficient and comprehensive analysis of somatic variants in cancer. Genome Res. 2018, 28, 1747–1756. [Google Scholar] [CrossRef]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| (A) Number of samples with mutations in TLS-related vs. random genes in top-ranking cancers a. | |||||
| Cancer type b | Samples, n c | Samples with mutations, n | P-total f | p-value g Chi-squared | |
| TLS-related genes d | Random genes, Ave e | ||||
| LAML | 144 | 21 | 5.03 | <0.001 | <0.001 |
| THYM | 123 | 9 | 3.30 | 0.001 | 0.08 |
| UCEC | 530 | 196 | 153.92 | 0.029 | 0.006 |
| (B) Fraction of cancer samples with mutations in TLS-related genes. | |||||
| Cancer type b: | LAML | UCEC | Cancer type b: | LAML | UCEC |
| Gene | Samples with mutations | Gene | Samples with mutations | ||
| CYLD | 1% | 11% | PRIMPOL | 1% | 6% |
| NPM1 | 13% | 3% | RAD18 | 0% | 6% |
| PAXIP1 | 1% | 11% | REV1 | 0% | 11% |
| PCNA | 0% | 3% | REV3L | 2% | 18% |
| POLH | 0% | 6% | TENT4A | 0% | 11% |
| POLI | 1% | 7% | TENT4B | 1% | 15% |
| POLK | 0% | 8% | USP1 | 0% | 7% |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Swain, U.; Friedlander, G.; Sehrawat, U.; Sarusi-Portuguez, A.; Rotkopf, R.; Ebert, C.; Paz-Elizur, T.; Dikstein, R.; Carell, T.; Geacintov, N.E.; et al. TENT4A Non-Canonical Poly(A) Polymerase Regulates DNA-Damage Tolerance via Multiple Pathways That Are Mutated in Endometrial Cancer. Int. J. Mol. Sci. 2021, 22, 6957. https://doi.org/10.3390/ijms22136957
Swain U, Friedlander G, Sehrawat U, Sarusi-Portuguez A, Rotkopf R, Ebert C, Paz-Elizur T, Dikstein R, Carell T, Geacintov NE, et al. TENT4A Non-Canonical Poly(A) Polymerase Regulates DNA-Damage Tolerance via Multiple Pathways That Are Mutated in Endometrial Cancer. International Journal of Molecular Sciences. 2021; 22(13):6957. https://doi.org/10.3390/ijms22136957
Chicago/Turabian StyleSwain, Umakanta, Gilgi Friedlander, Urmila Sehrawat, Avital Sarusi-Portuguez, Ron Rotkopf, Charlotte Ebert, Tamar Paz-Elizur, Rivka Dikstein, Thomas Carell, Nicholas E. Geacintov, and et al. 2021. "TENT4A Non-Canonical Poly(A) Polymerase Regulates DNA-Damage Tolerance via Multiple Pathways That Are Mutated in Endometrial Cancer" International Journal of Molecular Sciences 22, no. 13: 6957. https://doi.org/10.3390/ijms22136957
APA StyleSwain, U., Friedlander, G., Sehrawat, U., Sarusi-Portuguez, A., Rotkopf, R., Ebert, C., Paz-Elizur, T., Dikstein, R., Carell, T., Geacintov, N. E., & Livneh, Z. (2021). TENT4A Non-Canonical Poly(A) Polymerase Regulates DNA-Damage Tolerance via Multiple Pathways That Are Mutated in Endometrial Cancer. International Journal of Molecular Sciences, 22(13), 6957. https://doi.org/10.3390/ijms22136957