
UHPLC-ESI-MS/MS Quantification of Relevant Substrates and Metabolites of the Kynurenine Pathway Present in Serum and Peritoneal Fluid from Gastric Cancer Patients—Method Development and Validation

 ,
,  , and
, and 
Abstract
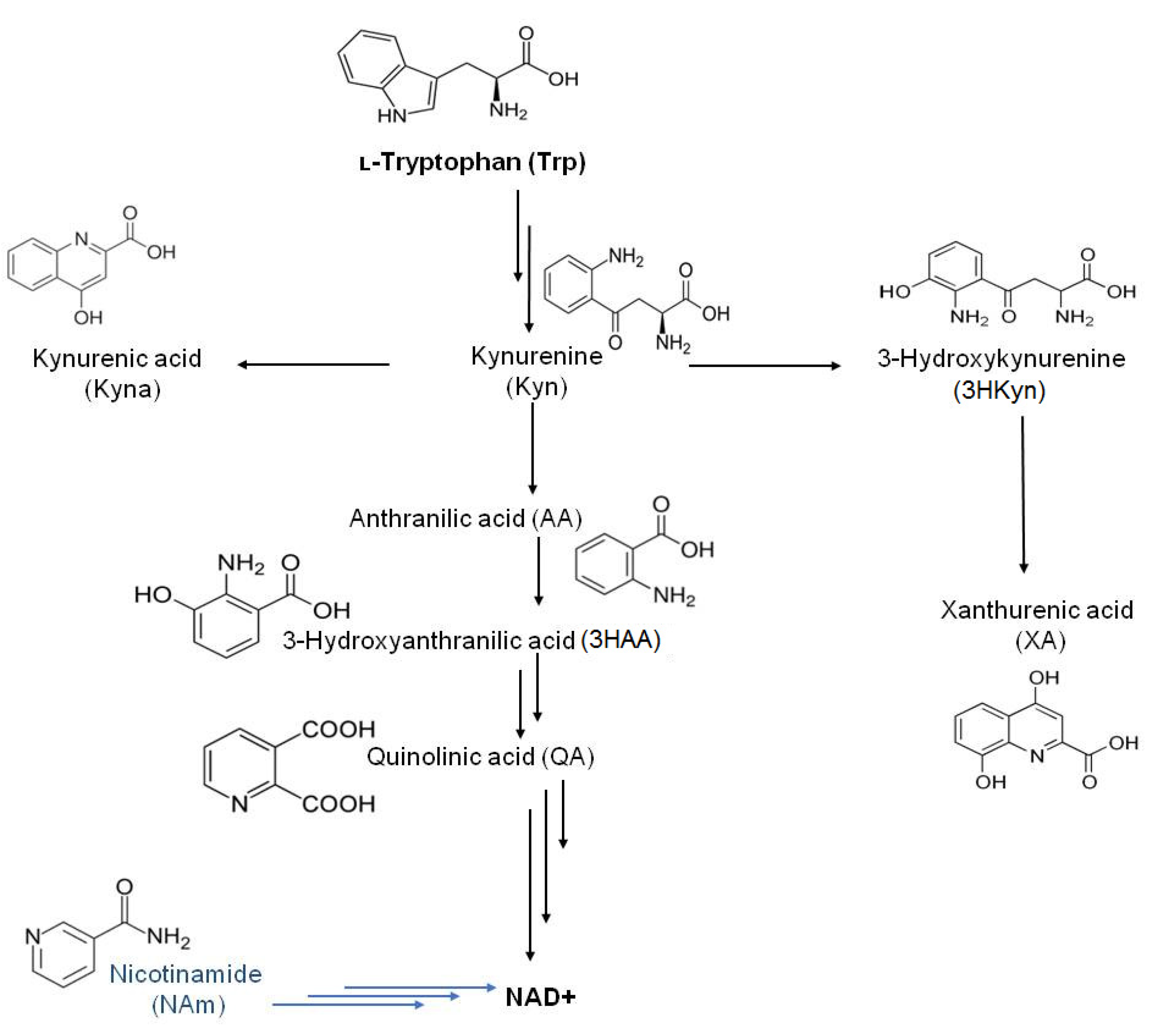
:1. Introduction
2. Results
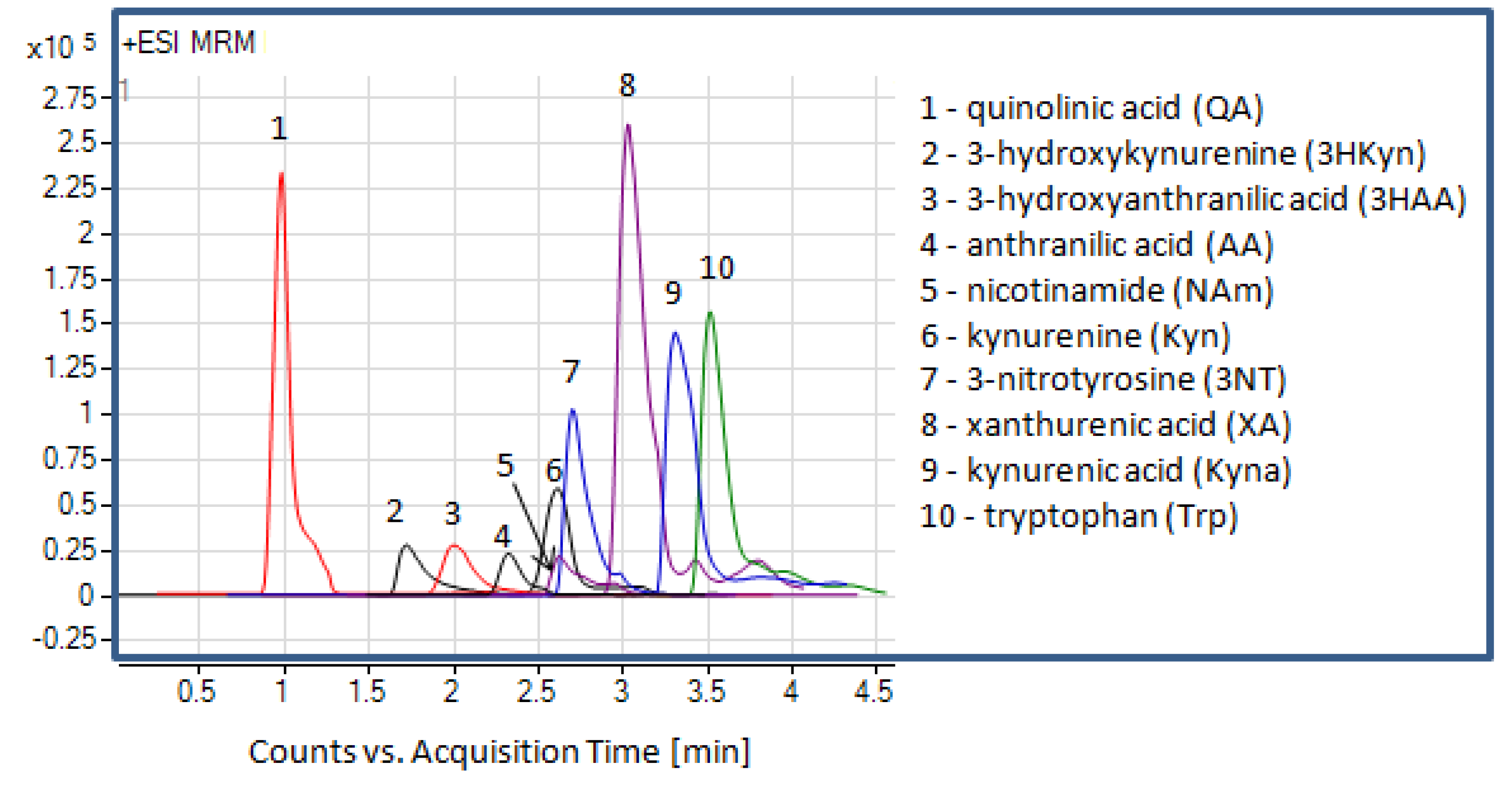
2.1. Liquid Chromatography and Mass Spectrometry
2.2. Optimization of the Protocol for Serum Preparation
2.3. Development of the Protocol for Peritoneal Fluid Preparation
2.4. Method Validation
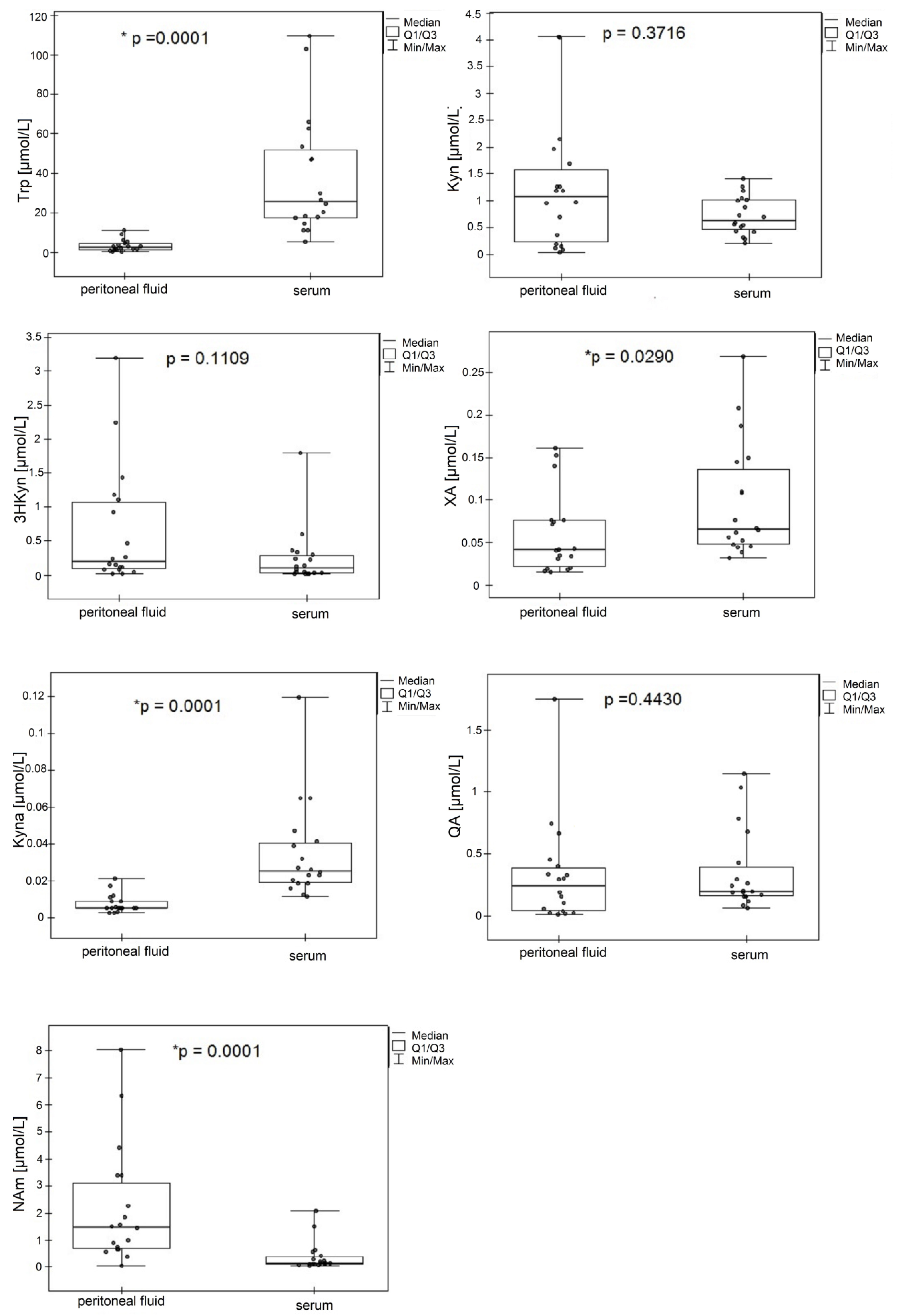
2.5. Application to Clinical Samples
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Instrumentation
4.3. Patient Characteristics
4.4. Preparation of Serum Samples
4.5. Preparation of Peritoneal Fluid
4.6. Preparation of the Charcoal-Pretreated Serum and Peritoneal Fluid
4.7. Preparation of the Matrix-Matched Calibration Standards
4.8. Method Validation
4.8.1. Limit of Detection, Limit of Quantification, and Linearity
4.8.2. Precision and Accuracy
4.8.3. Recovery, Matrix Effects, and Stability
4.9. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- González, C.A.; Agudo, A. Carcinogenesis, prevention and early detection of gastric cancer: Where we are and where we should go. Int. J. Cancer 2012, 130, 745–753. [Google Scholar] [CrossRef]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Dyba, T.; Randi, G.; Bettio, M.; Gavin, A.; Visser, O.; Bray, F. Cancer incidence and mortality patterns in Europe: Estimates for 40 countries and 25 major cancers in 2018. Eur. J. Cancer 2018, 103, 356–387. [Google Scholar] [CrossRef]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rawla, P.; Barsouk, A. Epidemiology of gastric cancer: Global trends, risk factors and prevention. Prz. Gastroenterol. 2019, 14, 26–38. [Google Scholar] [CrossRef] [PubMed]
- Santhanam, S.; Alvarado, D.M.; Ciorba, M.A. Therapeutic targeting of inflammation and tryptophan metabolism in colon and gastrointestinal cancer. Transl. Res. 2016, 167, 67–79. [Google Scholar] [CrossRef] [Green Version]
- Fukunaga, M.; Yamamoto, Y.; Kawasoe, M.; Arioka, Y.; Murakami, Y.; Hoshi, M.; Saito, K. Studies on tissue and cellular distribution of indoleamine 2,3-dioxygenase 2: The absence of IDO1 upregulates IDO2 expression in the epididymis. J. Histochem. Cytochem. 2012, 60, 854–860. [Google Scholar] [CrossRef] [Green Version]
- Karanikas, V.; Zamanakou, M.; Kerenidi, T.; Dahabreh, J.; Hevas, A.; Nakou, M.; Gourgoulianis, K.I.; Germenis, A.E. Indoleamine 2,3-dioxygenase (IDO) expression in lung cancer. Cancer Biol. Ther. 2007, 6, 1258–1262. [Google Scholar] [CrossRef] [Green Version]
- Merlo, L.M.F.; Mandik-Nayak, L. IDO2: A Pathogenic Mediator of Inflammatory Autoimmunity. Clin. Med. Insights Pathol. 2016, 9, 21–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Savitz, J. The kynurenine pathway: A finger in every pie. Mol. Psychiatry 2020, 25, 131–147. [Google Scholar] [CrossRef]
- Opitz, C.A.; Litzenburger, U.M.; Sahm, F.; Ott, M.; Tritschler, I.; Trump, S.; Schumacher, T.; Jestaedt, L.; Schrenk, D.; Weller, M.; et al. An endogenous tumour-promoting ligand of the human aryl hydrocarbon receptor. Nature 2011, 478, 197–203. [Google Scholar] [CrossRef]
- Frumento, G.; Rotondo, R.; Tonetti, M.; Damonte, G.; Benatti, U.; Ferrara, G.B. Tryptophan-derived catabolites are responsible for inhibition of T and natural killer cell proliferation induced by indoleamine 2,3-dioxygenase. J. Exp. Med. 2002, 196, 459–468. [Google Scholar] [CrossRef] [Green Version]
- Ramírez-Ortega, D.; Ramiro-Salazar, A.; González-Esquivel, D.; Ríos, C.; Pineda, B.; Pérez De La Cruz, V. 3-hydroxykynurenine and 3-hydroxyanthranilic acid enhance the toxicity induced by copper in rat astrocyte culture. Oxid. Med. Cell. Longev. 2017, 2017, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Sathyasaikumar, K.V.; Tararina, M.; Wu, H.Q.; Neale, S.A.; Weisz, F.; Salt, T.E.; Schwarcz, R. Xanthurenic Acid Formation from 3-Hydroxykynurenine in the Mammalian Brain: Neurochemical Characterization and Physiological Effects. Neuroscience 2017, 367, 85–97. [Google Scholar] [CrossRef] [Green Version]
- Malina, H.; Richter, C.; Frueh, B.; Hess, O.M. Lens epithelial cell apoptosis and intracellular Ca2+increasein the presence of xanthurenic acid. BMC Ophthalmol. 2002, 2, 1–7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guillemin, G.J. Quinolinic acid, the inescapable neurotoxin. FEBS J. 2012, 279, 1325–1365. [Google Scholar] [CrossRef]
- Çatak, J.; Yaman, M. Determination of Nicotinic Acid and Nicotinamide Forms of Vitamin B3 (Niacin) in Fruits and Vegetables by HPLC Using Postcolumn Derivatization System. Pak. J. Nutr. 2019, 18, 563–570. [Google Scholar] [CrossRef]
- Badawy, A.A.B. Kynurenine pathway of tryptophan metabolism: Regulatory and functional aspects. Int. J. Tryptophan Res. 2017, 10, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Shen, Z.; Wang, Z.; Wang, X.; Zhang, H.; Qin, J.; Qin, X.; Xu, J.; Sun, Y. Increased expression of IDO associates with poor postoperative clinical outcome of patients with gastric adenocarcinoma. Sci. Rep. 2016, 6, 21319–21327. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.W.; Nam, K.H.; Ahn, S.H.; Park, D.J.; Kim, H.H.; Kim, S.H.; Chang, H.; Lee, J.O.; Kim, Y.J.; Lee, H.S.; et al. Prognostic implications of immunosuppressive protein expression in tumors as well as immune cell infiltration within the tumor microenvironment in gastric cancer. Gastric Cancer 2016, 19, 42–52. [Google Scholar] [CrossRef] [Green Version]
- Li, F.; Sun, Y.; Huang, J.; Xu, W.; Liu, J.; Yuan, Z. CD4/CD 8 + T cells, DC subsets, Foxp3, and IDO expression are predictive indictors of gastric cancer prognosis. Cancer Med. 2019, 8, 7330–7344. [Google Scholar] [CrossRef]
- Xiang, Z.; Li, J.; Song, S.; Wang, J.; Cai, W.; Hu, W.; Ji, J.; Zhu, Z.; Zang, L.; Yan, R.; et al. A positive feedback between IDO1 metabolite and COL12A1 via MAPK pathway to promote gastric cancer metastasis. J. Exp. Clin. Cancer Res. 2019, 38, 314–325. [Google Scholar] [CrossRef] [Green Version]
- Kuligowski, J.; Sanjuan-Herráez, D.; Vázquez-Sánchez, M.A.; Brunet-Vega, A.; Pericay, C.; Ramírez-Lázaro, M.J.; Lario, S.; Gombau, L.; Junquera, F.; Calvet, X.; et al. Metabolomic analysis of gastric cancer progression within the Correa’s cascade using ultraperformance liquid chromatography-mass spectrometry. J. Proteome Res. 2016, 15, 2729–2738. [Google Scholar] [CrossRef]
- Engin, A.B.; Karahalil, B.; Karakaya, A.E.; Engin, A. Exposure to helicobacter pylori and serum kynurenine to tryptophan ratio in patients with gastric cancer. Pteridines 2010, 21, 110–120. [Google Scholar] [CrossRef]
- Choi, J.M.; Park, W.S.; Song, K.Y.; Lee, H.J.; Jung, B.H. Development of simultaneous analysis of tryptophan metabolites in serum and gastric juice—An investigation towards establishing a biomarker test for gastric cancer diagnosis. Biomed. Chromatogr. 2016, 30, 1963–1974. [Google Scholar] [CrossRef] [PubMed]
- Sadok, I.; Gamian, A.; Staniszewska, M.M. Chromatographic analysis of tryptophan metabolites. J. Sep. Sci. 2017, 40, 3020–3045. [Google Scholar] [CrossRef]
- Anesi, A.; Rubert, J.; Oluwagbemigun, K.; Orozco-Ruiz, X.; Nöthlings, U.; Breteler, M.M.B.; Mattivi, F. Metabolic profiling of human plasma and urine, targeting tryptophan, tyrosine and branched chain amino acid pathways. Metabolites 2019, 9, 261. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.S.; Zhang, M.D.; Tao, X.; Zhou, Y.F.; Liu, X.M.; Pan, R.L.; Liao, Y.H.; Chang, Q. LC-MS/MS-based quantification of tryptophan metabolites and neurotransmitters in the serum and brain of mice. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2019, 1112, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Whiley, L.; Nye, L.C.; Grant, I.; Andreas, N.; Chappell, K.E.; Sarafian, M.H.; Misra, R.; Plumb, R.S.; Lewis, M.R.; Nicholson, J.K.; et al. Ultrahigh-Performance Liquid Chromatography Tandem Mass Spectrometry with Electrospray Ionization Quantification of Tryptophan Metabolites and Markers of Gut Health in Serum and Plasma—Application to Clinical and Epidemiology Cohorts. Anal. Chem. 2019, 91, 5207–5216. [Google Scholar] [CrossRef] [Green Version]
- Galla, Z.; Rajda, C.; Rácz, G.; Grecsó, N.; Baráth, Á.; Vécsei, L.; Bereczki, C.; Monostori, P. Simultaneous determination of 30 neurologically and metabolically important molecules: A sensitive and selective way to measure tyrosine and tryptophan pathway metabolites and other biomarkers in human serum and cerebrospinal fluid. J. Chromatogr. A 2021, 1635, 461775. [Google Scholar] [CrossRef]
- Sadok, I.; Rachwał, K.; Staniszewska, M. Application of the optimized and validated LC-MS method for simultaneous quantification of tryptophan metabolites in culture medium from cancer cells. J. Pharm. Biomed. Anal. 2019, 176, 112805–112815. [Google Scholar] [CrossRef]
- Food and Drug Administration. Bioanalytical method validation guidance. J. Chromatogr. B Anal. Technol. Biomed. Life Sci. 2018, 1043, 25. [Google Scholar]
- Munn, D.H.; Mellor, A.L. Indoleamine 2,3-dioxygenase and tumor-induced tolerance. J. Clin. Investig. 2007, 117, 1147–1154. [Google Scholar] [CrossRef] [Green Version]
- Fallarino, F.; Grohmann, U.; Vacca, C.; Bianchi, R.; Orabona, C.; Spreca, A.; Fioretti, M.C.; Puccetti, P. T cell apoptosis by tryptophan catabolism. Cell Death Differ. 2002, 9, 1069–1077. [Google Scholar] [CrossRef]
- Della Chiesa, M.; Carlomagno, S.; Frumento, G.; Balsamo, M.; Cantoni, C.; Conte, R.; Moretta, L.; Moretta, A.; Vitale, M. The tryptophan catabolite L-kynurenine inhibits the surface expression of NKp46- and NKG2D-activating receptors and regulates NK-cell function. Blood 2006, 108, 4118–4125. [Google Scholar] [CrossRef]
- Weber, W.P.; Feder-Mengus, C.; Chiarugi, A.; Rosenthal, R.; Reschner, A.; Schumacher, R.; Zajac, P.; Misteli, H.; Frey, D.M.; Oertli, D.; et al. Differential effects of the trytophan metabolite 3-hydroxyanthranilic acid on the proliferation of human CD8 T cells induced by TCR triggering or homeostatic cytokines. Eur. J. Immunol. 2006, 36, 296–304. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.J.; Chung, W.C.; Choi, S.; Jung, Y.D.; Lee, J.; Chae, S.Y.; Jun, K.H.; Chin, H.M. The Detection of Messenger RNA for Carcinoembryonic Antigen and Cytokeratin 20 in Peritoneal Washing Fluid in Patients with Advanced Gastric Cancer. Korean J. Gastroenterol. 2017, 69, 220–225. [Google Scholar] [CrossRef] [Green Version]
- Kanda, M.; Kodera, Y. Molecular mechanisms of peritoneal dissemination in gastric cancer. World J. Gastroenterol. 2016, 22, 6829–6840. [Google Scholar] [CrossRef]
- Wang, Z.; Chen, J.; Liu, J.; Tian, L. Issues on peritoneal metastasis of gastric cancer: An update. World J. Surg. Oncol. 2019, 17, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Smyth, E.C.; Nilsson, M.; Grabsch, H.I.; Grieken, N.C.; van Lordic, F. Gastric cancer. Lancet 2020, 396, 635–648. [Google Scholar] [CrossRef]
- Lefèvre, A.; Mavel, S.; Nadal-Desbarats, L.; Galineau, L.; Attucci, S.; Dufour, D.; Sokol, H.; Emond, P. Validation of a global quantitative analysis methodology of tryptophan metabolites in mice using LC-MS. Talanta 2019, 195, 593–598. [Google Scholar] [CrossRef]
- Kelton, J.G.; Ulan, R.; Stiller, C.; Holmes, E. Comparison of chemical composition of peritoneal fluid and serum. A method for monitoring dialysis patients and a tool for assessing binding to serum proteins in vivo. Ann. Intern. Med. 1978, 89, 67–70. [Google Scholar] [CrossRef] [PubMed]
- Gika, H.G.; Michopoulos, F.; Divanis, D.; Metalidis, S.; Nikolaidis, P.; Theodoridis, G.A. Daptomycin determination by liquid chromatography-mass spectrometry in peritoneal fluid, blood plasma, and urine of clinical patients receiving peritoneal dialysis treatment. Anal. Bioanal. Chem. 2010, 397, 2191–2197. [Google Scholar] [CrossRef]
- Pan, G.; Ma, Y.; Suo, J.; Li, W.; Zhang, Y.; Qin, S.; Jiao, Y.; Zhang, S.; Li, S.; Kong, Y.; et al. Discovering biomarkers in peritoneal metastasis of gastric cancer by metabolomics. Onco. Targets. Ther. 2020, 13, 7199–7211. [Google Scholar] [CrossRef] [PubMed]
- Sadok, I.; Rachwał, K.; Staniszewska, M. Simultaneous quantification of selected kynurenines analyzed by liquid chromatography-mass spectrometry in medium collected from cancer cell cultures. J. Vis. Exp. 2020, 159, e61031–e61040. [Google Scholar] [CrossRef]
- Boulet, L.; Faure, P.; Flore, P.; Montérémal, J.; Ducros, V. Simultaneous determination of tryptophan and 8 metabolites in human plasma by liquid chromatography/tandem mass spectrometry. J. Chromatogr. B 2017, 1054, 36–43. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Analyte | Precursor ion (m/z) | Product ion (m/z) | FV (V) | CE (eV) | RT (min) | ΔRT | Polarity |
|---|---|---|---|---|---|---|---|
| QA | 168.0 | 150.0 | 60 | 8 | 1.10 | 1.50 | ESI+ |
| 124.0 | 60 | 8 | |||||
| 3HKyn | 225.0 | 208.0 | 80 | 8 | 1.72 | 1.50 | ESI+ |
| 162.0 | 80 | 10 | |||||
| 3HAA * | 154.0 | 136.0 | 100 | 8 | 1.20 | 1.50 | ESI+ |
| 103.0 | 100 | 20 | |||||
| AA * | 138.0 | 120.0 | 100 | 4 | 2.32 | 1.50 | ESI+ |
| 91.5 | 100 | 16 | |||||
| NAm | 123.0 | 96.0 | 110 | 20 | 2.58 | 2.00 | ESI+ |
| 106.0 | 110 | 20 | |||||
| Kyn | 209.0 | 192.0 | 100 | 8 | 2.64 | 2.00 | ESI+ |
| 174.0 | 100 | 10 | |||||
| 3NT ** | 227.0 | 181.0 | 80 | 8 | 2.80 | 2.00 | ESI+ |
| 117.0 | 80 | 20 | |||||
| XA | 206.0 | 160.0 | 110 | 20 | 3.03 | 2.00 | ESI+ |
| 188.0 | 110 | 8 | |||||
| Kyna | 190.0 | 144.0 | 100 | 20 | 3.32 | 2.00 | ESI+ |
| 172.0 | 100 | 10 | |||||
| Trp | 205.0 | 188.0 | 110 | 8 | 3.52 | 2.00 | ESI+ |
| 159.0 | 110 | 10 |
| Serum | Peritoneal Fluid | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Analyte | Calibration Range (µmol/L) | LOD (nmol/L) | LOQ (nmol/L) | Regression Parameters | Calibration Range (µmol/L) | LOD (nmol/L) | LOQ (nmol/L) | Regression Parameters | ||||
| Slope | Intercept | R2 | Slope | Intercept | R2 | |||||||
| QA | 0.01–2.99 | 3.95 | 11.96 | 0.117 | −0.004 | 0.998 | 0.02–11.96 | 5.92 | 17.94 | 0.017 | 0.001 | 0.993 |
| 3HKyn | 0.02–2.23 | 7.59 | 23.00 | 0.045 | −0.001 | 0.999 | 0.02–9.20 | 7.59 | 23.00 | 0.004 | 0.001 | 0.999 |
| 3HAA | n.s. | n.s. | n.s. | n.s. | n.s. | n.s. | 0.03–13.06 | 10.77 | 32.65 | 0.013 | 0.001 | 0.999 |
| AA | n.s. | n.s. | n.s. | n.s. | n.s. | n.s. | 0.0–14.58 | 12.03 | 36.46 | 0.067 | −0.018 | 0.994 |
| NAm | 8.19 *–4.10 | 2.70 | 8.19 | 0.118 | −0.001 | 0.998 | 8.19 *–16.38 | 2.70 | 8.19 | 0.013 | 0.008 | 0.996 |
| Kyn | 4.80 *–2.40 | 1.58 | 4.80 | 1.225 | 0.013 | 0.999 | 4.80 *–9.60 | 1.58 | 4.80 | 0.096 | 0.030 | 0.997 |
| XA | 0.01–1.47 | 3.23 | 9.80 | 3.599 | 0.029 | 0.995 | 0.01–9.50 | 4.70 | 14.25 | 0.704 | 0.001 | 0.999 |
| Kyna | 5.30 *–2.65 | 1.75 | 5.30 | 10.732 | 0.012 | 0.999 | 5.32 *–10.64 | 1.76 | 5.32 | 1.572 | 0.230 | 0.998 |
| Trp | 2.45 *–2.45 | 0.81 | 2.45 | 3.257 | 0.002 | 0.998 | 2.45 *–9.80 | 0.81 | 2.45 | 0.461 | 0.353 | 0.998 |
| Serum | Peritoneal Fluid | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Analyte | Spiked (µmol/L) | Intraday (n = 6) | Interday (n = 18) | Spiked (µmol/L) | Intraday (n = 6) | Interday (n = 18) | ||||||||
| Precision | Accuracy (%) | Precision | Accuracy (%) | Precision | Accuracy (%) | Precision | Accuracy (%) | |||||||
| Found ± SD (µmol/L) | CV (%) | Found ± SD (µmol/L) | CV (%) | Found ± SD (µmol/L) | CV (%) | Found ± SD (µmol/L) | CV (%) | |||||||
| QA | 11.96 * | 12.96 ± 0.36 * | 2.90 | 108.35 | 11.99 ± 1.57 * | 13.11 | 100.25 | 17.94 * | 21.10 ± 2.79 * | 13.19 | 117.95 | 20.51 ± 3.20 * | 15.60 | 114.33 |
| 35.88 * | 37.71 ± 2.15 * | 5.71 | 105.11 | 37.4 ± 2.11 * | 5.92 | 104.39 | 53.82 * | 58.4 ± 7.66 * | 13.10 | 108.68 | 57.93 ± 7.85 * | 13.56 | 107.64 | |
| 1.50 | 1.56 ± 0.08 | 4.98 | 104.23 | 1.45 ± 0.18 | 12.10 | 96.69 | 5.98 | 6.42 ± 0.20 | 3.12 | 107.37 | 6.21 ± 0.83 | 13.33 | 103.84 | |
| 2.99 | 2.74 ± 0.06 | 2.05 | 91.71 | 2.69 ± 0.37 | 13.62 | 89.84 | 11.96 | 12.2 ± 0.34 | 2.76 | 102.11 | 11.60 ± 0.70 | 6.05 | 96.97 | |
| 3HKyn | 23.00 * | 20.00 ± 2.36 * | 11.78 | 86.95 | 19.8 ± 3.12 * | 15.72 | 86.24 | 23.00 * | 20.8 ± 3.41 * | 16.37 | 90.47 | 20.59 ± 2.95 * | 14.31 | 89.51 |
| 111.50 * | 98.02 ± 6.24 * | 6.36 | 87.91 | 97.5 ± 9.84 * | 10.08 | 87.52 | 69.00 * | 61.8 ± 5.23 * | 8.46 | 89.58 | 62.71 ± 6.67 * | 10.64 | 90.89 | |
| 1.12 | 1.06 ± 0.06 | 5.28 | 94.72 | 1.04 ± 0.13 | 12.58 | 92.83 | 2.30 | 2.20 ± 0.27 | 12.37 | 95.67 | 2.15 ± 0.24 | 11.36 | 93.57 | |
| 2.23 | 2.31 ± 0.16 | 7.07 | 103.78 | 2.25 ± 0.26 | 11.33 | 100.96 | 9.20 | 8.07 ± 0.26 | 3.19 | 87.55 | 7.95 ± 0.48 | 6.08 | 86.37 | |
| 3HAA | n.s. | n.s. | n.s. | n.s. | n.s. | n.s. | 32.65 * | 36.80 ± 4.01 * | 10.88 | 112.94 | 34.77 ± 4.41 * | 12.64 | 106.49 | |
| n.s. | n.s. | n.s. | n.s. | n.s. | n.s. | 97.95 * | 97.60 ± 7.57 * | 7.76 | 99.64 | 98.11 ± 8.46 * | 8.62 | 100.16 | ||
| n.s. | n.s. | n.s. | n.s. | n.s. | n.s. | 3.27 | 3.41 ± 0.37 | 10.98 | 104.40 | 3.27 ± 0.47 | 14.39 | 99.23 | ||
| n.s. | n.s. | n.s. | n.s. | n.s. | n.s. | 13.06 | 13.90 ± 0.73 | 5.26 | 106.54 | 14.36 ± 0.89 | 6.20 | 109.25 | ||
| AA | n.s. | n.s. | n.s. | n.s. | n.s. | n.s. | 36.46 * | 30.60 ± 1.60 * | 5.21 | 83.90 | 30.81 ± 2.62 * | 8.50 | 84.41 | |
| n.s. | n.s. | n.s. | n.s. | n.s. | n.s. | 109.40 * | 104.78 ± 4.62 * | 4.41 | 95.77 | 109.40 ± 10.25 * | 9.37 | 100.03 | ||
| n.s. | n.s. | n.s. | n.s. | n.s. | n.s. | 3.65 | 3.66 ± 0.27 | 7.24 | 100.31 | 3.77 ± 0.24 | 6.27 | 103.30 | ||
| n.s. | n.s. | n.s. | n.s. | n.s. | n.s. | 14.58 | 15.03 ± 0.47 | 3.09 | 103.06 | 4.45 ± 1.31 | 9.06 | 99.06 | ||
| NAm | 8.19 * | 7.13 ± 1.04 * | 14.58 | 87.11 | 7.61 ± 1.36 * | 17.81 | 92.96 | 8.19 * | 8.13 ± 1.12 * | 13.73 | 99.22 | 8.62 ± 1.26 * | 14.57 | 105.29 |
| 24.60 * | 23.86 ± 1.29 * | 5.41 | 96.98 | 24.5 ± 1.47 * | 5.98 | 99.97 | 24.60 * | 26.75 ± 2.61 * | 9.76 | 108.88 | 25.64 ± 2.96 * | 11.54 | 104.35 | |
| 2.05 | 2.27 ± 0.05 | 2.19 | 110.77 | 2.35 ± 0.12 | 5.25 | 114.44 | 4.10 | 4.02 ± 0.60 | 14.95 | 98.26 | 3.89 ± 0.37 | 9.57 | 94.99 | |
| 4.10 | 4.65 ± 0.25 | 5.31 | 113.34 | 4.67 ± 0.21 | 4.45 | 113.80 | 16.38 | 16.16 ± 0.68 | 4.21 | 98.66 | 16.35 ± 1.46 | 8.92 | 99.84 | |
| Kyn | 4.80 * | 4.60 ± 0.38 * | 8.28 | 95.86 | 4.57 ± 0.77 * | 16.92 | 95.30 | 4.80 * | 4.48 ± 0.64 * | 14.34 | 93.36 | 4.66 ± 0.50 * | 10.66 | 97.11 |
| 14.40 * | 15.14 ± 0.79 * | 5.21 | 105.13 | 14.5 ± 1.30 * | 8.89 | 101.23 | 14.40 * | 14.28 ± 1.24 * | 8.66 | 99.18 | 14.11 ± 1.37 * | 9.69 | 97.96 | |
| 1.20 | 1.25 ± 0.03 | 2.60 | 103.96 | 1.34 ± 0.20 | 14.86 | 111.81 | 2.40 | 2.35 ± 0.08 | 3.38 | 97.80 | 2.29 ± 0.14 | 6.19 | 95.58 | |
| 2.40 | 2.50 ± 0.09 | 3.78 | 104.26 | 2.74 ± 0.22 | 8.19 | 114.11 | 9.60 | 8.84 ± 0.36 | 4.07 | 92.03 | 9.22 ± 0.59 | 6.42 | 96.37 | |
| XA | 9.80 * | 10.62 ± 0.58 * | 5.48 | 108.32 | 10.4 ± 0.73 * | 6.98 | 106.39 | 14.25 * | 15.36 ± 1.11 * | 7.25 | 107.79 | 15.13 ± 1.33 * | 8.77 | 106.16 |
| 29.40 * | 33.22 ± 1.49 * | 4.49 | 112.98 | 33.7 ± 2.47 * | 7.32 | 114.68 | 42.75 * | 44.52 ± 5.27 * | 11.84 | 104.14 | 42.33 ± 5.58 * | 13.18 | 99.03 | |
| 0.74 | 0.83 ± 0.03 | 3.21 | 112.57 | 0.77 ± 0.10 | 13.03 | 104.53 | 2.38 | 2.22 ± 0.12 | 5.64 | 93.17 | 2.25 ± 0.22 | 9.97 | 94.57 | |
| 1.47 | 1.63 ± 0.04 | 2.28 | 110.67 | 1.58 ± 0.18 | 11.31 | 107.47 | 9.50 | 10.51 ± 0.45 | 4.24 | 110.67 | 9.54 ± 1.25 | 13.11 | 100.46 | |
| Kyna | 5.30 * | 6.03 ± 0.19 * | 3.08 | 113.76 | 6.09 ± 0.18 * | 3.04 | 114.84 | 5.32 * | 4.95 ± 0.54 * | 10.98 | 93.03 | 5.31 ± 0.76 * | 14.24 | 99.75 |
| 31.80 * | 34.79 ± 2.05 * | 5.90 | 109.40 | 34.6 ± 2.35 * | 6.77 | 109.03 | 15.96 * | 16.93 ± 1.49 * | 8.79 | 106.06 | 16.69 ± 1.98 * | 11.88 | 104.60 | |
| 1.33 | 1.37 ± 0.03 | 2.11 | 103.05 | 1.43 ± 0.08 | 5.87 | 107.72 | 2.66 | 2.44 ± 0.15 | 6.24 | 91.63 | 2.65 ± 0.19 | 7.14 | 99.68 | |
| 2.65 | 3.01 ± 0.11 | 3.75 | 113.72 | 3.04 ± 0.09 | 2.97 | 114.76 | 10.64 | 11.50 ± 0.52 | 4.54 | 108.05 | 10.49 ± 0.88 | 8.38 | 98.61 | |
| Trp | 2.45 * | 2.30 ± 0.23 * | 10.09 | 94.04 | 2.34 ± 0.25 * | 10.82 | 95.57 | 2.45 * | 2.45 ± 0.25 * | 9.88 | 101.03 | 2.45 ± 0.38 * | 15.53 | 100.15 |
| 7.35 * | 8.29 ± 0.54 * | 6.53 | 112.84 | 8.19 ± 0.99 * | 12.05 | 111.40 | 7.35 * | 7.19 ± 0.56 * | 7.81 | 97.88 | 7.93 ± 0.80 * | 10.05 | 107.84 | |
| 1.23 | 1.38 ± 0.06 | 4.55 | 113.72 | 1.40 ± 0.05 | 3.47 | 113.73 | 2.45 | 2.28 ± 0.22 | 9.83 | 93.23 | 2.42 ± 0.27 | 11.08 | 98.68 | |
| 2.45 | 2.61 ± 0.08 | 3.02 | 106.41 | 2.74 ± 0.11 | 4.09 | 111.94 | 9.80 | 8.92 ± 0.34 | 3.77 | 91.06 | 8.94 ± 0.42 | 4.68 | 91.21 | |
| Serum | Peritoneal Fluid | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Analyte | Spiked (µmol/L) | RE ± SD (%) | ME ± SD (%) | Stability ** ± SD (%) | FT Stability ± SD (%) | Spiked (µmol/L) | RE± SD (%) | ME ± SD (%) | Stability ** ± SD (%) | FT Stability ± SD (%) |
| QA | 11.96 * | 83.75 ± 2.44 | 129.56 ± 5.64 | 95.45 ± 9.78 | 84.16 ± 3.68 | 17.94 * | 82.97 ± 3.76 | 73.90 ± 3.96 | 96.02 ± 12.45 | 97.99 ± 9.91 |
| 35.88 * | 95.16 ± 3.14 | 104.40 ± 6.46 | 92.75 ± 12.26 | 93.00 ± 7.13 | 53.82 * | 87.94 ± 1.66 | 86.12 ± 4.23 | 92.40 ± 8.55 | 101.09 ± 9.15 | |
| 1.50 | 86.50 ± 3.36 | 97.39 ± 6.94 | 98.41 ± 1.03 | 121.28 ± 1.27 | 5.98 | 89.58 ± 2.52 | 84.23 ± 7.67 | 96.14 ± 7.86 | 99.01 ± 10.52 | |
| 2.99 | 94.86 ± 1.40 | 109.93 ± 1.36 | 102.33 ± 1.12 | 105.38 ± 1.50 | 11.96 | 90.06 ± 4.02 | 83.74 ± 8.22 | 98.71 ± 8.39 | 100.16 ± 9.37 | |
| 3HKyn | 23.00 * | 92.90 ± 7.35 | 80.92 ± 10.84 | 76.28 ± 12.62 | 79.62 ± 5.28 | 23.00 * | 49.39 ± 5.29 | 89.90 ± 2.86 | 87.87 ± 6.64 | 70.60 ± 9.90 |
| 111.50 * | 88.25 ± 8.00 | 109.19 ± 11.57 | 53.74 ± 10.14 | 73.73 ± 5.16 | 0.07 | 50.40 ± 2.76 | 81.81 ± 2.28 | 87.42 ± 6.69 | 69.10 ± 8.74 | |
| 1.12 | 87.95 ± 7.31 | 87.15 ± 7.04 | 65.51 ± 7.14 | 70.90 ± 12.56 | 2.30 | 50.99 ± 2.06 | 76.59 ± 3.59 | 83.35 ± 4.25 | 64.28 ± 4.95 | |
| 2.23 | 100.35 ± 6.45 | 94.17 ± 8.72 | 64.22 ± 6.92 | 76.46 ± 13.13 | 9.20 | 57.89 ± 4.41 | 77.43 ± 8.77 | 83.70 ± 8.71 | 64.87 ± 6.41 | |
| 3HAA | n.s. | n.s. | n.s. | n.s. | 32.65 * | 91.84 ± 2.74 | 80.40 ± 8.95 | 101.12 ± 13.82 | 94.02 ± 6.12 | |
| n.s. | n.s. | n.s. | n.s. | 97.95 * | 97.18 ± 9.22 | 85.88 ± 7.10 | 101.15 ± 7.27 | 95.61 ± 3.21 | ||
| n.s. | n.s. | n.s. | n.s. | 3.27 | 108.89 ± 4.89 | 85.10 ± 3.54 | 67.71 ± 7.28 | 90.39 ± 8.15 | ||
| n.s. | n.s. | n.s. | n.s. | 13.06 | 112.70 ± 3.63 | 88.10 ± 2.78 | 66.82 ± 5.35 | 93.15 ± 4.99 | ||
| AA | n.s. | n.s. | n.s. | n.s. | 36.46 * | 95.66 ± 9.13 | 77.37 ± 6.44 | 97.89 ± 9.93 | 94.42 ± 3.99 | |
| n.s. | n.s. | n.s. | n.s. | 109.40 * | 96.89 ± 5.37 | 80.58 ± 2.77 | 104.84 ± 7.46 | 105.06 ± 5.63 | ||
| n.s. | n.s. | n.s. | n.s. | 3.65 | 89.98 ± 3.40 | 80.80 ± 9.16 | 92.88 ± 2.18 | 100.43 ± 12.91 | ||
| n.s. | n.s. | n.s. | n.s. | 14.58 | 90.42 ± 2.02 | 87.32 ± 4.10 | 88.95 ± 2.76 | 97.18 ± 4.49 | ||
| NAm | 8.19 * | 91.92 ± 8.56 | 113.53 ± 7.41 | 105.99 ± 9.19 | 76.83 ± 12.19 | 8.19 * | 83.64 ± 7.05 | 126.55 ± 6.53 | 140.51 ± 15.84 | 100.07 ± 7.40 |
| 24.60 * | 93.73 ± 5.63 | 104.40 ± 6.46 | 107.76 ± 9.21 | 108.47 ± 7.48 | 24.60 * | 85.47 ± 6.10 | 114.41 ± 9.97 | 128.61 ± 16.89 | 97.60 ± 6.57 | |
| 2.05 | 97.61 ± 3.52 | 121.20 ± 3.49 | 102.18 ± 1.58 | 98.89 ± 2.51 | 4.10 | 85.89 ± 3.46 | 128.01 ± 1.75 | 104.55 ± 6.60 | 103.33 ± 5.93 | |
| 4.10 | 96.98 ± 3.89 | 154.05 ± 10.24 | 102.56 ± 1.90 | 99.62 ± 0.86 | 16.38 | 85.45 ± 2.08 | 107.11 ± 6.94 | 107.93 ± 4.79 | 104.57 ± 9.49 | |
| Kyn | 4.80 * | 93.61 ± 5.75 | 100.75 ± 3.88 | 105.73 ± 1.85 | 90.93 ± 3.35 | 4.80 * | 85.53 ± 3.33 | 123.75 ± 8.17 | 112.48 ± 10.58 | 98.15 ± 9.62 |
| 14.40 * | 103.62 ± 7.72 | 109.87 ± 6.52 | 108.02 ± 11.62 | 107.62 ± 2.91 | 14.40 * | 86.33 ± 6.10 | 122.74 ± 9.64 | 109.01 ± 12.78 | 98.01 ± 3.71 | |
| 1.20 | 91.11 ± 3.65 | 125.61 ± 4.40 | 110.11 ± 3.18 | 108.58 ± 0.52 | 2.40 | 88.78 ± 1.27 | 121.38 ± 11.30 | 91.92 ± 4.47 | 105.19 ± 6.35 | |
| 2.40 | 96.40 ± 1.75 | 133.20 ± 9.63 | 102.17 ± 0.79 | 109.16 ± 0.84 | 9.60 | 94.43 ± 3.38 | 122.84 ± 10.86 | 97.62 ± 6.84 | 101.01 ± 5.10 | |
| XA | 9.80 * | 81.44 ± 3.58 | 84.55 ± 7.12 | 124.49 ± 7.22 | 126.20 ± 7.64 | 14.25 * | 103.43 ± 8.60 | 113.55 ± 5.38 | 92.28 ± 6.76 | 83.76 ± 6.29 |
| 29.40 * | 87.09 ± 7.31 | 72.26 ± 2.98 | 90.99 ± 6.38 | 93.19 ± 5.58 | 42.75 * | 109.06 ± 8.95 | 115.42 ± 1.22 | 103.87 ± 4.20 | 90.31 ± 8.12 | |
| 0.74 | 85.17 ± 4.16 | 78.61 ± 8.25 | 103.26 ± 1.06 | 99.01 ± 5.82 | 2.38 | 110.33 ± 8.66 | 97.24 ± 4.25 | 82.58 ± 5.39 | 87.07 ± 7.58 | |
| 1.47 | 86.13 ± 3.62 | 79.80 ± 4.61 | 99.10 ± 5.12 | 99.63 ± 1.71 | 9.50 | 111.00 ± 4.33 | 100.46 ± 1.84 | 84.56 ± 5.26 | 84.43 ± 8.02 | |
| Kyna | 5.30 * | 91.82 ± 2.73 | 83.76 ± 1.44 | 98.64 ± 2.46 | 111.62 ± 4.28 | 5.32 * | 87.88 ± 5.18 | 119.91 ± 10.49 | 140.90 ± 14.55 | 92.32 ± 6.14 |
| 31.80 * | 98.41 ± 6.58 | 95.35 ± 2.27 | 107.32 ± 6.25 | 109.01 ± 7.80 | 15.96 * | 91.94 ± 7.02 | 113.44 ± 11.32 | 131.72 ± 14.84 | 97.08 ± 3.90 | |
| 1.33 | 99.59 ± 2.79 | 91.74 ± 7.54 | 103.74 ± 1.88 | 107.67 ± 0.79 | 2.66 | 108.88 ± 4.60 | 105.65 ± 0.98 | 98.89 ± 3.61 | 103.82 ± 4.67 | |
| 2.65 | 101.85 ± 1.32 | 90.71 ± 0.34 | 102.10 ± 1.12 | 106.99 ± 0.30 | 10.64 | 113.62 ± 3.33 | 103.96 ± 1.97 | 92.33 ± 3.58 | 96.53 ± 1.58 | |
| Trp | 2.45 * | 88.27 ± 2.92 | 86.76 ± 2.61 | 92.76 ± 4.05 | 97.55 ± 3.02 | 2.45 * | 95.29 ± 6.37 | 114.70 ± 11.78 | 123.18 ± 6.03 | 98.17 ± 3.72 |
| 7.35 * | 88.10 ± 7.12 | 66.07 ± 6.20 | 88.21 ± 10.57 | 89.20 ± 3.45 | 7.35 * | 94.51 ± 2.85 | 97.32 ± 11.01 | 118.11 ± 5.61 | 98.86 ± 6.53 | |
| 1.23 | 88.96 ± 2.61 | 71.47 ± 4.16 | 87.70 ± 3.24 | 99.79 ± 2.31 | 2.45 | 99.12 ± 1.46 | 93.09 ± 3.63 | 96.63 ± 8.19 | 104.80 ± 6.49 | |
| 2.45 | 87.51 ± 2.48 | 74.50 ± 1.15 | 89.94 ± 3.18 | 99.44 ± 0.15 | 9.80 | 115.35 ± 3.83 | 101.14 ± 3.95 | 104.82 ± 4.51 | 109.17 ± 9.42 | |
| 3NT | 2.21 | 98.23 ± 2.55 | 101.82 ± 2.27 | 101.15 ± 5.33 | 98.20 ± 1.70 | 55.25 | 104.97 ± 2.51 | 97.19 ± 4.85 | 81.14 ± 4.75 | 89.54 ± 4.01 |
| Analyte | Serum (µmol/L) | Peritoneal Fluid (µmol/L) | ||||
|---|---|---|---|---|---|---|
| Min | Median | Max | Min | Median | Max | |
| Trp * | 5.15 | 25.64 | 109.56 | 0.34 | 2.35 | 11.22 |
| Kyn | 0.20 | 0.64 | 1.41 | 0.04 | 1.07 | 4.06 |
| 3HKyn | 0.02 | 0.11 | 1.80 | 0.02 | 0.20 | 3.20 |
| XA * | 0.03 | 0.07 | 0.27 | 0.02 | 0.04 | 0.16 |
| Kyna * | 0.01 | 0.03 | 0.12 | 2.52 ** | 5.23 ** | 21.04 ** |
| AA | n.s. | n.s. | n.s. | 0.02 | 0.04 | 0.43 |
| 3HAA | n.s. | n.s. | n.s. | 0.16 | 0.43 | 2.03 |
| QA | 0.07 | 0.20 | 0.15 | 0.01 | 0.24 | 1.75 |
| NAm * | 0.04 | 0.13 | 2.09 | 0.04 | 1.47 | 8.04 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sadok, I.; Jędruchniewicz, K.; Rawicz-Pruszyński, K.; Staniszewska, M. UHPLC-ESI-MS/MS Quantification of Relevant Substrates and Metabolites of the Kynurenine Pathway Present in Serum and Peritoneal Fluid from Gastric Cancer Patients—Method Development and Validation. Int. J. Mol. Sci. 2021, 22, 6972. https://doi.org/10.3390/ijms22136972
Sadok I, Jędruchniewicz K, Rawicz-Pruszyński K, Staniszewska M. UHPLC-ESI-MS/MS Quantification of Relevant Substrates and Metabolites of the Kynurenine Pathway Present in Serum and Peritoneal Fluid from Gastric Cancer Patients—Method Development and Validation. International Journal of Molecular Sciences. 2021; 22(13):6972. https://doi.org/10.3390/ijms22136972
Chicago/Turabian StyleSadok, Ilona, Katarzyna Jędruchniewicz, Karol Rawicz-Pruszyński, and Magdalena Staniszewska. 2021. "UHPLC-ESI-MS/MS Quantification of Relevant Substrates and Metabolites of the Kynurenine Pathway Present in Serum and Peritoneal Fluid from Gastric Cancer Patients—Method Development and Validation" International Journal of Molecular Sciences 22, no. 13: 6972. https://doi.org/10.3390/ijms22136972
APA StyleSadok, I., Jędruchniewicz, K., Rawicz-Pruszyński, K., & Staniszewska, M. (2021). UHPLC-ESI-MS/MS Quantification of Relevant Substrates and Metabolites of the Kynurenine Pathway Present in Serum and Peritoneal Fluid from Gastric Cancer Patients—Method Development and Validation. International Journal of Molecular Sciences, 22(13), 6972. https://doi.org/10.3390/ijms22136972