
The Role of Microbiota in Primary Sclerosing Cholangitis and Related Biliary Malignancies



Abstract
1. Primary Sclerosing Cholangitis (PSC)—An Overview
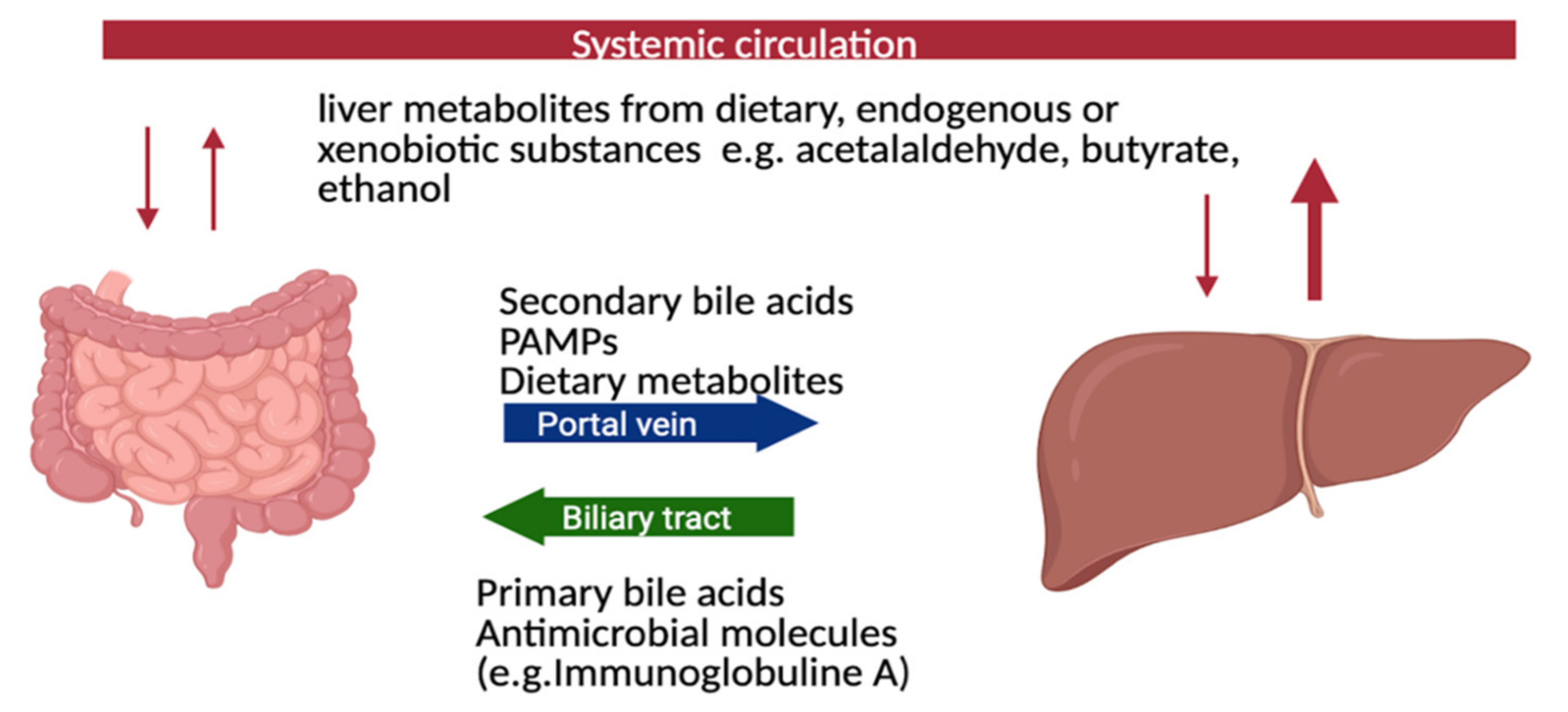
2. Pathogenesis of PSC and the Role of the Gut-Liver Axis
3. Gut Microbiota and PSC Pathogenesis—Mechanistic Insights
4. Gut Microbiota and PSC—Cross-Sectional Studies
4.1. Fecal Analysis of Gut Microbiota in Patients with PSC
4.2. Mucosal Analysis of Gut Microbiota in Patients with PSC
4.3. Gut Virome
5. PSC and Occult Viral Infections
6. Gut Microbiota as a Therapeutic Target and Future Approach
6.1. Antibiotic Treatment
6.2. Fecal Microbiota Transplantation (FMT)
7. Bile Microbiota in PSC
7.1. Bile Acid Pathways and Their Therapeutic Targets
8. PSC and CCA
8.1. PSC and CCA-Overview
8.2. Gut Microbiota in CCA
9. Gallbladder Carcinoma—Overview and Role of Genotoxins in Carcinogenesis
10. Conclusions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| Alkaline phosphatase | ALP |
| Cholangiocarcinoma | CCA |
| Crohn’s disease | CD |
| Endoscopic retrograde cholangiopancreatography | ERCP |
| Extrahepatic CCA | eCCA |
| Fecal microbiota transplantation | FMT |
| Free fatty acids | FFA |
| Helicobacter pylori | HP |
| Inflammatory bowel disease | IBD |
| Intrahepatic CCA | iCCA |
| Lipopolysaccharide-binding protein | LBP |
| Pathogen-associated molecular patterns | PAMPs |
| Peroxisome-proliferator-activated receptor alpha | PPARα |
| Peroxisome-proliferator-activated receptor delta | PPARδ |
| Multidrug resistance 2 | mdr2 |
| Mucosal addressin cell-adhesion molecule 1 | MAdCAM1 |
| Mycophenolate mofetil | MMF |
| Nonalcoholic fatty liver disease | NAFLD |
| Norursodeoxycholic acid | norUDCA |
| Operational Taxonomic Units | OTU |
| Primary sclerosing cholangitis | PSC |
| Short-chain fatty acids | SCFA |
| Ulcerative colitis | UC |
| Ursodeoxycholic acid | UDCA |
| Vascular adhesion protein-1 | VAP-1 |
References
- Lazaridis, K.N.; LaRusso, N.F. Primary Sclerosing Cholangitis. N. Engl. J. Med. 2016, 375, 1161–1170. [Google Scholar] [CrossRef] [PubMed]
- Eaton, J.E.; Talwalkar, J.A.; Lazaridis, K.N.; Gores, G.J.; Lindor, K.D. Pathogenesis of Primary Sclerosing Cholangitis and Advances in Diagnosis and Management. Gastroenterology 2013, 145, 521–536. [Google Scholar] [CrossRef]
- Hirschfield, G.M.; Karlsen, T.H.; Lindor, K.D.; Adams, D. Primary sclerosing cholangitis. Lancet 2013, 382, 1587–1599. [Google Scholar] [CrossRef]
- Molodecky, N.A.; Kareemi, H.; Parab, R.; Barkema, H.; Quan, H.; Myers, R.P.; Kaplan, G.G. Incidence of primary sclerosing cholangitis: A systematic review and meta-analysis. Hepatology 2011, 53, 1590–1599. [Google Scholar] [CrossRef]
- Boonstra, K.; Beuers, U.; Ponsioen, C.Y. Epidemiology of primary sclerosing cholangitis and primary biliary cirrhosis: A systematic review. J. Hepatol. 2012, 56, 1181–1188. [Google Scholar] [CrossRef] [PubMed]
- Karlsen, T.H.; Folseraas, T.; Thorburn, D.; Vesterhus, M. Primary sclerosing cholangitis—A comprehensive review. J. Hepatol. 2017, 67, 1298–1323. [Google Scholar] [CrossRef]
- Staufer, K.; Kivaranovic, D.; Rasoul-Rockenschaub, S.; Soliman, T.; Trauner, M.; Berlakovich, G. Waitlist mortality and post-transplant survival in patients with cholestatic liver disease—Impact of changes in allocation policy. HPB 2018, 20, 916–924. [Google Scholar] [CrossRef] [PubMed]
- Palmela, C.; Peerani, F.; Castaneda, D.; Torres, J.; Itzkowitz, S.H. Inflammatory Bowel Disease and Primary Sclerosing Cholangitis: A Review of the Phenotype and Associated Specific Features. Gut Liver 2018, 12, 17–29. [Google Scholar] [CrossRef] [PubMed]
- Mertz, A.; Nguyen, N.A.; Katsanos, K.H.; Kwok, R.M. Primary sclerosing cholangitis and inflammatory bowel disease comorbidity: An update of the evidence. Ann. Gastroenterol. 2019, 32, 124–133. [Google Scholar] [CrossRef]
- Chapman, R.; Fevery, J.; Kalloo, A.; Nagorney, D.M.; Boberg, K.M.; Shneider, B.; Gores, G.J. Diagnosis and management of primary sclerosing cholangitis. Hepatology 2010, 51, 660–678. [Google Scholar] [CrossRef]
- Lunder, A.K.; Hov, J.R.; Borthne, A.; Gleditsch, J.; Johannesen, G.; Tveit, K.; Viktil, E.; Henriksen, M.; Hovde, Ø.; Huppertz-Hauss, G.; et al. Prevalence of Sclerosing Cholangitis Detected by Magnetic Resonance Cholangiography in Patients With Long-term Inflammatory Bowel Disease. Gastroenterology 2016, 151, 660–669.e4. [Google Scholar] [CrossRef] [PubMed]
- Rühlemann, M.; Liwinski, T.; Heinsen, F.-A.; Bang, C.; Zenouzi, R.; Kummen, M.; Thingholm, L.; Tempel, M.; Lieb, W.; Karlsen, T.; et al. Consistent alterations in faecal microbiomes of patients with primary sclerosing cholangitis independent of associated colitis. Aliment. Pharmacol. Ther. 2019, 50, 580–589. [Google Scholar] [CrossRef]
- Loftus, E.V.; Harewood, G.C.; Loftus, C.G.; Tremaine, W.J.; Harmsen, W.S.; Zinsmeister, A.R.; Jewell, D.A.; Sandborn, W.J. PSC-IBD: A unique form of inflammatory bowel disease associated with primary sclerosing cholangitis. Gut 2005, 54, 91–96. [Google Scholar] [CrossRef] [PubMed]
- De Vries, A.B.; Janse, M.; Blokzijl, H.; Weersma, R.K. Distinctive inflammatory bowel disease phenotype in primary sclerosing cholangitis. World J. Gastroenterol. 2015, 21, 1956–1971. [Google Scholar] [CrossRef]
- Ricciuto, A.; Kamath, B.M.; Griffiths, A.M. The IBD and PSC Phenotypes of PSC-IBD. Curr. Gastroenterol. Rep. 2018, 20, 16. [Google Scholar] [CrossRef]
- Cleveland, N.K.; Rubin, D.T.; Hart, J.; Weber, C.R.; Meckel, K.; Tran, A.L.; Aelvoet, A.S.; Pan, I.; Gonsalves, A.; Gaetano, J.N.; et al. Patients With Ulcerative Colitis and Primary Sclerosing Cholangitis Frequently Have Subclinical Inflammation in the Proximal Colon. Clin. Gastroenterol. Hepatol. 2018, 16, 68–74. [Google Scholar] [CrossRef]
- Vera, A.; Moledina, S.; Gunson, B.; Hubscher, S.; Mirza, D.; Olliff, S.; Neuberger, J. Risk factors for recurrence of primary sclerosing cholangitis of liver allograft. Lancet 2002, 360, 1943–1944. [Google Scholar] [CrossRef]
- Steenstraten, I.C.; Korkmaz, K.S.; Trivedi, P.J.; Inderson, A.; Van Hoek, B.; Girondo, M.D.M.R.; Maljaars, P.W.J. Systematic review with meta-analysis: Risk factors for recurrent primary sclerosing cholangitis after liver transplantation. Aliment. Pharmacol. Ther. 2019, 49, 636–643. [Google Scholar] [CrossRef] [PubMed]
- Kostic, A.; Xavier, R.J.; Gevers, D. The Microbiome in Inflammatory Bowel Disease: Current Status and the Future Ahead. Gastroenterology 2014, 146, 1489–1499. [Google Scholar] [CrossRef]
- Sokol, H.; Pigneur, B.; Watterlot, L.; Lakhdari, O.; Bermúdez-Humarán, L.G.; Gratadoux, J.-J.; Blugeon, S.; Bridonneau, C.; Furet, J.-P.; Corthier, G.; et al. Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc. Natl. Acad. Sci. USA 2008, 105, 16731–16736. [Google Scholar] [CrossRef]
- Joossens, M.; Huys, G.; Cnockaert, M.; De Preter, V.; Verbeke, K.; Rutgeerts, P.; Vandamme, P.; Vermeire, S. Dysbiosis of the faecal microbiota in patients with Crohn’s disease and their unaffected relatives. Gut 2011, 60, 631–637. [Google Scholar] [CrossRef]
- Dignass, A.; Eliakim, R.; Magro, F.; Maaser, C.; Chowers, Y.; Geboes, K.; Mantzaris, G.; Reinisch, W.; Colombel, J.-F.; Vermeire, S.; et al. Second European evidence-based consensus on the diagnosis and management of ulcerative colitis Part 1: Definitions and diagnosis. J. Crohn’s Colitis 2012, 6, 965–990. [Google Scholar] [CrossRef]
- Li, Y.; Tang, R.; Leung, P.S.; Gershwin, M.E.; Ma, X. Bile acids and intestinal microbiota in autoimmune cholestatic liver diseases. Autoimmun. Rev. 2017, 16, 885–896. [Google Scholar] [CrossRef] [PubMed]
- Quraishi, M.N.; Sergeant, M.; Kay, G.; Iqbal, T.; Chan, J.; Constantinidou, C.; Trivedi, P.; Ferguson, J.; Adams, D.; Pallen, M.; et al. The gut-adherent microbiota of PSC–IBD is distinct to that of IBD. Gut 2017, 66, 386–388. [Google Scholar] [CrossRef]
- Bajer, L.; Kverka, M.; Kostovcik, M.; Macinga, P.; Dvorak, J.; Stehlikova, Z.; Brezina, J.; Wohl, P.; Spicak, J.; Drastich, P. Distinct gut microbiota profiles in patients with primary sclerosing cholangitis and ulcerative colitis. World J. Gastroenterol. 2017, 23, 4548–4558. [Google Scholar] [CrossRef] [PubMed]
- Kevans, D.; Tyler, A.D.; Holm, K.; Jorgensen, K.K.; Vatn, M.H.; Karlsen, T.H.; Kaplan, G.; Eksteen, B.; Gevers, D.; Hov, J.; et al. Characterization of Intestinal Microbiota in Ulcerative Colitis Patients with and without Primary Sclerosing Cholangitis. J. Crohn’s Colitis 2016, 10, 330–337. [Google Scholar] [CrossRef] [PubMed]
- Kummen, M.; Holm, K.; Anmarkrud, J.A.; Nygård, S.; Vesterhus, M.; Høivik, M.L.; Trøseid, M.; Marschall, H.-U.; Schrumpf, E.; Moum, B.; et al. The gut microbial profile in patients with primary sclerosing cholangitis is distinct from patients with ulcerative colitis without biliary disease and healthy controls. Gut 2017, 66, 611–619. [Google Scholar] [CrossRef]
- Lazaridis, K.N.; LaRusso, N.F. The Cholangiopathies. Mayo Clin. Proc. 2015, 90, 791–800. [Google Scholar] [CrossRef] [PubMed]
- Turnbaugh, P.J.; Hamady, M.; Yatsunenko, T.; Cantarel, B.L.; Duncan, A.; Ley, R.E.; Sogin, M.L.; Jones, W.J.; Roe, B.A.; Affourtit, J.P.; et al. A core gut microbiome in obese and lean twins. Nature 2009, 457, 480–484. [Google Scholar] [CrossRef] [PubMed]
- Gevers, D.; Kugathasan, S.; Denson, L.A.; Vázquez-Baeza, Y.; Van Treuren, W.; Ren, B.; Schwager, E.; Knights, D.; Song, S.J.; Yassour, M.; et al. The Treatment-Naive Microbiome in New-Onset Crohn’s Disease. Cell Host Microbe 2014, 15, 382–392. [Google Scholar] [CrossRef]
- Qin, N.; Yang, F.; Li, A.; Prifti, E.; Chen, Y.; Shao, L.; Guo, J.; Le Chatelier, E.; Yao, J.; Wu, L.; et al. Alterations of the human gut microbiome in liver cirrhosis. Nature 2014, 513, 59–64. [Google Scholar] [CrossRef]
- Seo, Y.S.; Shah, V.H. The role of gut-liver axis in the pathogenesis of liver cirrhosis and portal hypertension. Clin. Mol. Hepatol. 2012, 18, 337–346. [Google Scholar] [CrossRef]
- Tripathi, A.; Debelius, J.; Brenner, D.A.; Karin, M.; Loomba, R.; Schnabl, B.; Knight, R. The gut–liver axis and the intersection with the microbiome. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 397–411. [Google Scholar] [CrossRef]
- Seki, E.; Schnabl, B. Role of innate immunity and the microbiota in liver fibrosis: Crosstalk between the liver and gut. J. Physiol. 2012, 590, 447–458. [Google Scholar] [CrossRef] [PubMed]
- Anand, G.; Zarrinpar, A.; Loomba, R. Targeting Dysbiosis for the Treatment of Liver Disease. Semin. Liver Dis. 2016, 36, 037–047. [Google Scholar] [CrossRef] [PubMed]
- Lichtman, S.N.; Keku, J.; Clark, R.L.; Schwab, J.H.; Sartor, R.B. Biliary tract disease in rats with experimental small bowel bacterial overgrowth. Hepatology 1991, 13, 766–772. [Google Scholar] [CrossRef]
- Lichtman, S.N.; Okoruwa, E.E.; Keku, J.; Schwab, J.H.; Sartor, R.B. Degradation of endogenous bacterial cell wall polymers by the muralytic enzyme mutanolysin prevents hepatobiliary injury in genetically susceptible rats with experimental intestinal bacterial overgrowth. J. Clin. Investig. 1992, 90, 1313–1322. [Google Scholar] [CrossRef]
- Lee, Y.-M.; Kaplan, M.M. Primary Sclerosing Cholangitis. N. Engl. J. Med. 1995, 332, 924–933. [Google Scholar] [CrossRef] [PubMed]
- Guicciardi, M.E.; Trussoni, C.E.; Krishnan, A.; Bronk, S.F.; Pisarello, M.J.L.; O’Hara, S.P.; Splinter, P.L.; Gao, Y.; Vig, P.; Revzin, A.; et al. Macrophages contribute to the pathogenesis of sclerosing cholangitis in mice. J. Hepatol. 2018, 69, 676–686. [Google Scholar] [CrossRef]
- Lichtman, S.N.; Keku, J.; Schwab, J.H.; Sartor, R. Hepatic injury associated with small bowel bacterial overgrowth in rats is prevented by metronidazole and tetracycline. Gastroenterology 1991, 100, 513–519. [Google Scholar] [CrossRef]
- Dhillon, A.K.; Kummen, M.; Trøseid, M.; Åkra, S.; Liaskou, E.; Moum, B.; Vesterhus, M.; Karlsen, T.H.; Seljeflot, I.; Hov, J.R. Circulating markers of gut barrier function associated with disease severity in primary sclerosing cholangitis. Liver Int. 2018, 39, 371–381. [Google Scholar] [CrossRef]
- Björnsson, E.; Cederborg, A.; Åkvist, A.; Simren, M.; Stotzer, P.-O.; Bjarnason, I. Intestinal permeability and bacterial growth of the small bowel in patients with primary sclerosing cholangitis. Scand. J. Gastroenterol. 2005, 40, 1090–1094. [Google Scholar] [CrossRef]
- Eksteen, B.; Grant, A.J.; Miles, A.; Curbishley, S.M.; Lalor, P.; Hübscher, S.G.; Briskin, M.; Salmon, M.; Adams, D.H. Hepatic Endothelial CCL25 Mediates the Recruitment of CCR9+ Gut-homing Lymphocytes to the Liver in Primary Sclerosing Cholangitis. J. Exp. Med. 2004, 200, 1511–1517. [Google Scholar] [CrossRef] [PubMed]
- Adams, D.H.; Eksteen, B. Aberrant homing of mucosal T cells and extra-intestinal manifestations of inflammatory bowel disease. Nat. Rev. Immunol. 2006, 6, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Terjung, B.; Söhne, J.; Lechtenberg, B.; Gottwein, J.; Muennich, M.; Herzog, V.; Mähler, M.; Sauerbruch, T.; Spengler, U. p-ANCAs in autoimmune liver disorders recognise human -tubulin isotype 5 and cross-react with microbial protein FtsZ. Gut 2009, 59, 808–816. [Google Scholar] [CrossRef]
- Lynch, K.D.; Chapman, R.W.; Keshav, S.; Montano-Loza, A.J.; Mason, A.L.; Kremer, A.E.; Vetter, M.; de Krijger, M.; Ponsioen, C.Y.; Trivedi, P.; et al. Effects of Vedolizumab in Patients With Primary Sclerosing Cholangitis and Inflammatory Bowel Diseases. Clin. Gastroenterol. Hepatol. 2020, 18, 179–187.e6. [Google Scholar] [CrossRef]
- Laborda, T.J.; Ricciuto, A.; Aumar, M.; Carman, N.; DiGuglielmo, M.; Draijer, L.G.; Furuya, K.N.; Gupta, N.; Koot, B.G.; Loomes, K.M.; et al. Vedolizumab Therapy in Children With Primary Sclerosing Cholangitis: Data From the Pediatric Primary Sclerosing Cholangitis Consortium. J. Pediatr. Gastroenterol. Nutr. 2020, 71, 459–464. [Google Scholar] [CrossRef]
- Zuo, T.; Kamm, M.A.; Colombel, J.-F.; Ng, S.C. Urbanization and the gut microbiota in health and inflammatory bowel disease. Nat. Rev. Gastroenterol. Hepatol. 2018, 15, 440–452. [Google Scholar] [CrossRef]
- Schroeder, B.O.; Bäckhed, F. Signals from the gut microbiota to distant organs in physiology and disease. Nat. Med. 2016, 22, 1079–1089. [Google Scholar] [CrossRef] [PubMed]
- Yatsunenko, T.; Rey, F.E.; Manary, M.J.; Trehan, I.; Dominguez-Bello, M.G.; Contreras, M.; Magris, M.; Hidalgo, G.; Baldassano, R.N.; Anokhin, A.P.; et al. Human gut microbiome viewed across age and geography. Nature 2012, 486, 222–227. [Google Scholar] [CrossRef]
- Little, R.; Wine, E.; Kamath, B.M.; Griffiths, A.M.; Ricciuto, A. Gut microbiome in primary sclerosing cholangitis: A review. World J. Gastroenterol. 2020, 26, 2768–2780. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.Z.; Hov, J.R.; Folseraas, T.; Ellinghaus, E.; Rushbrook, S.M.; Doncheva, N.T.; Andreassen, O.A.; Weersma, R.K.; Weismüller, T.J.; Eksteen, B.; et al. Dense genotyping of immune-related disease regions identifies nine new risk loci for primary sclerosing cholangitis. Nat. Genet. 2013, 45, 670–675. [Google Scholar] [CrossRef] [PubMed]
- Rupp, C.; Friedrich, K.; Folseraas, T.; Wannhoff, A.; Bode, K.A.; Weiss, K.-H.; Schirmacher, P.; Sauer, P.; Stremmel, W.; Gotthardt, D.N. Fut2 genotype is a risk factor for dominant stenosis and biliary candida infections in primary sclerosing cholangitis. Aliment. Pharmacol. Ther. 2014, 39, 873–882. [Google Scholar] [CrossRef]
- Wannhoff, A.; Rupp, C.; Friedrich, K.; Brune, M.; Knierim, J.; Flechtenmacher, C.; Sauer, P.; Stremmel, W.; Hov, J.; Schirmacher, P.; et al. Inflammation But Not Biliary Obstruction Is Associated with Carbohydrate Antigen 19-9 Levels in Patients with Primary Sclerosing Cholangitis. Clin. Gastroenterol. Hepatol. 2015, 13, 2372–2379. [Google Scholar] [CrossRef] [PubMed]
- Tabibian, J.H.; O’Hara, S.P.; Trussoni, C.E.; Tietz, P.S.; Splinter, P.L.; Mounajjed, T.; Hagey, L.R.; LaRusso, N.F. Absence of the intestinal microbiota exacerbates hepatobiliary disease in a murine model of primary sclerosing cholangitis. Hepatology 2016, 63, 185–196. [Google Scholar] [CrossRef]
- Nakamoto, N.; Sasaki, N.; Aoki, R.; Miyamoto, K.; Suda, W.; Teratani, T.; Suzuki, T.; Koda, Y.; Chu, P.-S.; Taniki, N.; et al. Gut pathobionts underlie intestinal barrier dysfunction and liver T helper 17 cell immune response in primary sclerosing cholangitis. Nat. Microbiol. 2019, 4, 492–503. [Google Scholar] [CrossRef]
- Patel, M.; Watson, A.J.; Rushbrook, S. A Mechanistic Insight Into the Role of Gut Microbiota in the Pathogenesis of Primary Sclerosing Cholangitis. Gastroenterology 2019, 157, 1686–1688. [Google Scholar] [CrossRef]
- Hov, J.R.; Karlsen, T.H. The Microbiome in Primary Sclerosing Cholangitis: Current Evidence and Potential Concepts. Semin. Liver Dis. 2017, 37, 314–331. [Google Scholar] [CrossRef]
- Sabino, J.; Vieira-Silva, S.; Machiels, K.; Joossens, M.; Falony, G.; Ballet, V.; Ferrante, M.; Van Assche, G.; Van Der Merwe, S.; Vermeire, S.; et al. Primary sclerosing cholangitis is characterised by intestinal dysbiosis independent from IBD. Gut 2016, 65, 1681–1689. [Google Scholar] [CrossRef]
- Iwasawa, K.; Suda, W.; Tsunoda, T.; Oikawa-Kawamoto, M.; Umetsu, S.; Inui, A.; Fujisawa, T.; Morita, H.; Sogo, T.; Hattori, M. Characterisation of the faecal microbiota in Japanese patients with paediatric-onset primary sclerosing cholangitis. Gut 2016, 66, 1344–1346. [Google Scholar] [CrossRef]
- Lemoinne, S.; Kemgang, A.; Ben Belkacem, K.; Straube, M.; Jegou, S.; Corpechot, C.; Chazouillères, O.; Housset, C.; Sokol, H.; Network, S.-A.I. Fungi participate in the dysbiosis of gut microbiota in patients with primary sclerosing cholangitis. Gut 2020, 69, 92–102. [Google Scholar] [CrossRef]
- Kummen, M.; Thingholm, L.B.; Rühlemann, M.C.; Holm, K.; Hansen, S.H.; Moitinho-Silva, L.; Liwinski, T.; Zenouzi, R.; Storm-Larsen, C.; Midttun, Ø.; et al. Altered Gut Microbial Metabolism of Essential Nutrients in Primary Sclerosing Cholangitis. Gastroenterology 2021, 160, 1784–1798. [Google Scholar] [CrossRef] [PubMed]
- Rossen, N.G.; Fuentes, S.; Boonstra, K.; D’Haens, G.R.; Heilig, H.G.; Zoetendal, E.G.; De Vos, W.M.; Ponsioen, C.Y. The Mucosa-associated Microbiota of PSC Patients is Characterized by Low Diversity and Low Abundance of Uncultured Clostridiales II. J. Crohn’s Colitis 2015, 9, 342–348. [Google Scholar] [CrossRef] [PubMed]
- Torres, J.; Bao, X.; Goel, A.; Colombel, J.-F.; Pekow, J.; Jabri, B.; Williams, K.; Castillo, A.; Odin, J.; Meckel, K.; et al. The features of mucosa-associated microbiota in primary sclerosing cholangitis. Aliment. Pharmacol. Ther. 2016, 43, 790–801. [Google Scholar] [CrossRef] [PubMed]
- Quraishi, M.N.; Acharjee, A.; Beggs, A.D.; Horniblow, R.; Tselepis, C.; Gkoutos, G.; Ghosh, S.; Rossiter, A.E.; Loman, N.; Van Schaik, W.; et al. A Pilot Integrative Analysis of Colonic Gene Expression, Gut Microbiota, and Immune Infiltration in Primary Sclerosing Cholangitis-Inflammatory Bowel Disease: Association of Disease With Bile Acid Pathways. J. Crohn’s Colitis 2020, 14, 935–947. [Google Scholar] [CrossRef] [PubMed]
- Liwinski, T.; Zenouzi, R.; John, C.; Ehlken, H.; Rühlemann, M.C.; Bang, C.; Groth, S.; Lieb, W.; Kantowski, M.; Andersen, N.; et al. Alterations of the bile microbiome in primary sclerosing cholangitis. Gut 2020, 69, 665–672. [Google Scholar] [CrossRef]
- Rühlemann, M.; Heinsen, F.-A.; Zenouzi, R.; Lieb, W.; Franke, A.; Schramm, C. Faecal microbiota profiles as diagnostic biomarkers in primary sclerosing cholangitis. Gut 2017, 66, 753–754. [Google Scholar] [CrossRef]
- Bajaj, J.S.; Liu, E.J.; Kheradman, R.; Fagan, A.; Heuman, D.M.; White, M.; Gavis, E.A.; Hylemon, P.; Sikaroodi, M.; Gillevet, P.M. Fungal dysbiosis in cirrhosis. Gut 2017, 67, 1146–1154. [Google Scholar] [CrossRef]
- Trivedi, P.J.; Adams, D.H. Gut–liver immunity. J. Hepatol. 2016, 64, 1187–1189. [Google Scholar] [CrossRef]
- The UniProt Consortium. UniProt: The universal protein knowledgebase in 2021. Nucleic Acids Res. 2021, 49, D480–D489. [Google Scholar] [CrossRef]
- Weston, C.J.; Shepherd, E.L.; Claridge, L.C.; Rantakari, P.; Curbishley, S.M.; Tomlinson, J.W.; Hubscher, S.G.; Reynolds, G.M.; Aalto, K.; Anstee, Q.M.; et al. Vascular adehesion protein 1 promotes liver inflammation and drives hepatic fibrosis. J. Clin. Investig. 2015, 125, 501–520. [Google Scholar] [CrossRef]
- Fodor, A.A.; Klem, E.R.; Gilpin, D.; Elborn, J.; Boucher, R.C.; Tunney, M.; Wolfgang, M.C. The Adult Cystic Fibrosis Airway Microbiota Is Stable over Time and Infection Type, and Highly Resilient to Antibiotic Treatment of Exacerbations. PLoS ONE 2012, 7, e45001. [Google Scholar] [CrossRef]
- Molyneaux, P.L.; Cox, M.J.; Willis-Owen, S.A.G.; Mallia, P.; Russell, K.E.; Russell, A.-M.; Murphy, E.; Johnston, S.L.; Schwartz, D.A.; Wells, A.U.; et al. The role of bacteria in the pathogenesis and progression of idiopathic pulmonary fibrosis. Am. J. Respir. Crit. Care Med. 2014, 190, 906–913. [Google Scholar] [CrossRef]
- De Cruz, P.; Kang, S.; Wagner, J.; Buckley, M.; Sim, W.H.; Prideaux, L.; Lockett, T.; McSweeney, C.; Morrison, M.; Kirkwood, C.D.; et al. Association between specific mucosa-associated microbiota in Crohn’s disease at the time of resection and subsequent disease recurrence: A pilot study. J. Gastroenterol. Hepatol. 2014, 30, 268–278. [Google Scholar] [CrossRef] [PubMed]
- Kugathasan, S.; Denson, L.A.; Walters, T.D.; Kim, M.-O.; Marigorta, U.M.; Schirmer, M.; Mondal, K.; Liu, C.; Griffiths, A.; Noe, J.D.; et al. Prediction of complicated disease course for children newly diagnosed with Crohn’s disease: A multicentre inception cohort study. Lancet 2017, 389, 1710–1718. [Google Scholar] [CrossRef]
- Worthmann, A.; John, C.; Rühlemann, M.C.; Baguhl, M.; Heinsen, F.-A.; Schaltenberg, N.; Heine, M.; Schlein, C.; Evangelakos, I.; Mineo, C.; et al. Cold-induced conversion of cholesterol to bile acids in mice shapes the gut microbiome and promotes adaptive thermogenesis. Nat. Med. 2017, 23, 839–849. [Google Scholar] [CrossRef]
- Sakamoto, M.; Tanaka, Y.; Benno, Y.; Ohkuma, M. Parabacteroides faecis sp. nov., isolated from human faeces. Int. J. Syst. Evol. Microbiol. 2015, 65, 1342–1346. [Google Scholar] [CrossRef] [PubMed]
- Rajilić-Stojanović, M.; De Vos, W.M. The first 1000 cultured species of the human gastrointestinal microbiota. FEMS Microbiol. Rev. 2014, 38, 996–1047. [Google Scholar] [CrossRef]
- Narushima, S.; Itoh, K.; Miyamoto, Y.; Park, S.-H.; Nagata, K.; Kuruma, K.; Uchida, K. Deoxycholic acid formation in gnotobiotic mice associated with human intestinal bacteria. Lipids 2006, 41, 835–843. [Google Scholar] [CrossRef] [PubMed]
- Rosen, R.; Amirault, J.; Liu, H.; Mitchell, P.; Hu, L.; Khatwa, U.; Onderdonk, A. Changes in gastric and lung microflora with acid suppression: Acid suppression and bacterial growth. JAMA Pediatr. 2014, 168, 932–937. [Google Scholar] [CrossRef]
- Oztas, E.; Odemis, B.; Kekilli, M.; Kurt, M.; Dinc, B.M.; Parlak, E.; Kalkanci, A.; Sasmaz, N. Systemic phaeohyphomycosis resembling primary sclerosing cholangitis caused by Exophiala dermatitidis. J. Med. Microbiol. 2009, 58, 1243–1246. [Google Scholar] [CrossRef][Green Version]
- Hong, K.H.; Kim, J.W.; Jang, S.J.; Yu, E.; Kim, E.-C. Liver cirrhosis caused by Exophiala dermatitidis. J. Med. Microbiol. 2009, 58, 674–677. [Google Scholar] [CrossRef]
- Sokol, H.; Leducq, V.; Aschard, H.; Pham, H.-P.; Jegou, S.; Landman, C.; Cohen, D.; Liguori, G.; Bourrier, A.; Nion-Larmurier, I.; et al. Fungal microbiota dysbiosis in IBD. Gut 2016, 66, 1039–1048. [Google Scholar] [CrossRef] [PubMed]
- Ridlon, J.M.; Kang, D.-J.; Hylemon, P.B. Bile salt biotransformations by human intestinal bacteria. J. Lipid Res. 2006, 47, 241–259. [Google Scholar] [CrossRef] [PubMed]
- Hacisalihoglu, A.; Jongejan, J.A.; Duine, J.A. Distribution of amine oxidases and amine dehydrogenases in bacteria grown on primary amines and characterization of the amine oxidase from Klebsiella oxytoca. Microbiology 1997, 143, 505–512. [Google Scholar] [CrossRef]
- Arumugam, M.; Raes, J.; Pelletier, E.; Le Paslier, D.; Yamada, T.; Mende, D.R.; Fernandes, G.R.; Tap, J.; Bruls, T.; Batto, J.M.; et al. Enterotypes of the human gut microbiome. Nature 2011, 473, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Duncan, S.H.; Belenguer, A.; Holtrop, G.; Johnstone, A.M.; Flint, H.J.; Lobley, G.E. Reduced Dietary Intake of Carbohydrates by Obese Subjects Results in Decreased Concentrations of Butyrate and Butyrate-Producing Bacteria in Feces. Appl. Environ. Microbiol. 2007, 73, 1073–1078. [Google Scholar] [CrossRef]
- Pouteau, E.; Nguyen, P.; Ballevre, O.; Krempf, M. Production rates and metabolism of short-chain fatty acids in the colon and whole body using stable isotopes. Proc. Nutr. Soc. 2003, 62, 87–93. [Google Scholar] [CrossRef] [PubMed]
- Venegas, D.P.; De La Fuente, M.K.; Landskron, G.; González, M.J.; Quera, R.; Dijkstra, G.; Harmsen, H.J.M.; Faber, K.N.; Hermoso, M.A. Short Chain Fatty Acids (SCFAs)-Mediated Gut Epithelial and Immune Regulation and Its Relevance for Inflammatory Bowel Diseases. Front. Immunol. 2019, 10, 277. [Google Scholar] [CrossRef]
- Quraishi, M.N.; Shaheen, W.; Oo, Y.H.; Iqbal, T.H. Immunological mechanisms underpinning faecal microbiota transplantation for the treatment of inflammatory bowel disease. Clin. Exp. Immunol. 2020, 199, 24–38. [Google Scholar] [CrossRef]
- Trivedi, P.J.; Tickle, J.; Vesterhus, M.N.; Eddowes, P.; Bruns, T.; Vainio, J.; Parker, R.; Smith, D.; Liaskou, E.; Thorbjørnsen, L.W.; et al. Vascular adhesion protein-1 is elevated in primary sclerosing cholangitis, is predictive of clinical outcome and facilitates recruitment of gut-tropic lymphocytes to liver in a substrate-dependent manner. Gut 2018, 67, 1135–1145. [Google Scholar] [CrossRef] [PubMed]
- Atarashi, K.; Tanoue, T.; Oshima, K.; Suda, W.; Nagano, Y.; Nishikawa, H.; Fukuda, S.; Saito, T.; Narushima, S.; Hase, K.; et al. Treg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 2013, 500, 232–236. [Google Scholar] [CrossRef]
- Pereira, P.; Aho, V.; Arola, J.; Boyd, S.; Jokelainen, K.; Paulin, L.; Auvinen, P.; Färkkilä, M. Bile microbiota in primary sclerosing cholangitis: Impact on disease progression and development of biliary dysplasia. PLoS ONE 2017, 12, e0182924. [Google Scholar] [CrossRef] [PubMed]
- Kakiyama, G.; Pandak, W.M.; Gillevet, P.M.; Hylemon, P.B.; Heuman, D.M.; Daita, K.; Takei, H.; Muto, A.; Nittono, H.; Ridlon, J.M.; et al. Modulation of the fecal bile acid profile by gut microbiota in cirrhosis. J. Hepatol. 2013, 58, 949–955. [Google Scholar] [CrossRef]
- Gurung, M.; Li, Z.; You, H.; Rodrigues, R.; Jump, D.B.; Morgun, A.; Shulzhenko, N. Role of gut microbiota in type 2 diabetes pathophysiology. EBioMedicine 2020, 51, 102590. [Google Scholar] [CrossRef]
- Bouter, K.E.; van Raalte, D.H.; Groen, A.K.; Nieuwdorp, M. Role of the Gut Microbiome in the Pathogenesis of Obesity and Obesity-Related Metabolic Dysfunction. Gastroenterology 2017, 152, 1671–1678. [Google Scholar] [CrossRef] [PubMed]
- Leung, C.; Rivera, L.; Furness, J.B.; Angus, C.L.P.W. The role of the gut microbiota in NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 412–425. [Google Scholar] [CrossRef] [PubMed]
- Tang, R.; Wei, Y.; Li, Y.; Chen, W.; Chen, H.; Wang, Q.; Yang, F.; Miao, Q.; Xiao, X.; Zhang, H.; et al. Gut microbial profile is altered in primary biliary cholangitis and partially restored after UDCA therapy. Gut 2018, 67, 534–541. [Google Scholar] [CrossRef]
- Lv, L.-X.; Fang, D.-Q.; Shi, D.; Chen, D.-Y.; Yan, R.; Zhu, Y.-X.; Chen, Y.-F.; Shao, L.; Guo, F.-F.; Wu, W.-R.; et al. Alterations and correlations of the gut microbiome, metabolism and immunity in patients with primary biliary cirrhosis. Environ. Microbiol. 2016, 18, 2272–2286. [Google Scholar] [CrossRef]
- Norman, J.; Handley, S.A.; Baldridge, M.; Droit, L.; Liu, C.Y.; Keller, B.C.; Kambal, A.; Monaco, C.; Zhao, G.; Fleshner, P.; et al. Disease-Specific Alterations in the Enteric Virome in Inflammatory Bowel Disease. Cell 2015, 160, 447–460. [Google Scholar] [CrossRef]
- Jiang, L.; Lang, S.; Duan, Y.; Zhang, X.; Gao, B.; Chopyk, J.; Schwanemann, L.K.; Ventura-Cots, M.; Bataller, R.; Bosques-Padilla, F.; et al. Intestinal Virome in Patients with Alcoholic Hepatitis. Hepatology 2020, 72, 2182–2196. [Google Scholar] [CrossRef]
- Ponsioen, C.Y.; Defoer, J.; Kate, F.J.W.T.; Weverling, G.J.; Tytgat, G.N.J.; Pannekoek, Y.; Wertheim-Dillen, P.M.E. A survey of infectious agents as risk factors for primary sclerosing cholangitis: Are Chlamydia species involved? Eur. J. Gastroenterol. Hepatol. 2002, 14, 641–648. [Google Scholar] [CrossRef]
- Georgiadou, S.P.; Zachou, K.; Liaskos, C.; Gabeta, S.; Rigopoulou, E.I.; Dalekos, G.N. Occult hepatitis B virus infection in patients with autoimmune liver diseases. Liver Int. 2009, 29, 434–442. [Google Scholar] [CrossRef]
- Kim, S.R.; Imoto, S.; Taniguchi, M.; Kim, K.I.; Sasase, N.; Matsuoka, T.; Maekawa, Y.; Ninomiya, T.; Ando, K.; Mita, K.; et al. Primary Sclerosing Cholangitis and Hepatitis C Virus Infection. Intervirology 2005, 48, 268–272. [Google Scholar] [CrossRef]
- Nayudu, S.K.; Kumbum, K.; Balar, B.; Niazi, M.; Chilimuri, S. Small Duct Primary Sclerosing Cholangitis in Association With Hepatitis C Virus Infection: A Case Report. Gastroenterol. Res. 2011, 4, 39–41. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rahimpour, S.; Nasiri-Toosi, M.; Khalili, H.; Daryani, N.E.; Taromlou, M.K.N.; Azizi, Z. A Triple Blinded, Randomized, Placebo-Controlled Clinical Trial to Evaluate the Efficacy and Safety of Oral Vancomycin in Primary Sclerosing Cholangitis: A Pilot Study. J. Gastrointest. Liver Dis. 2016, 25, 457–464. [Google Scholar] [CrossRef] [PubMed]
- Tabibian, J.H.; Weeding, E.; Jorgensen, R.A.; Petz, J.L.; Keach, J.C.; Talwalkar, J.A.; Lindor, K.D. Randomised clinical trial: Vancomycin or metronidazole in patients with primary sclerosing cholangitis—A pilot study. Aliment. Pharmacol. Ther. 2013, 37, 604–612. [Google Scholar] [CrossRef] [PubMed]
- Färkkilä, M.; Karvonen, A.-L.; Nurmi, H.; Nuutinen, H.; Taavitsainen, M.; Pikkarainen, P.; Kärkkäinen, P. Metronidazole and ursodeoxycholic acid for primary sclerosing cholangitis: A randomized placebo-controlled trial. Hepatology 2004, 40, 1379–1386. [Google Scholar] [CrossRef]
- Tabibian, J.H.; Gossard, A.; El-Youssef, M.; Eaton, J.E.; Petz, J.; Jorgensen, R.; Enders, F.B.; Tabibian, A.; Lindor, K.D. Prospective Clinical Trial of Rifaximin Therapy for Patients With Primary Sclerosing Cholangitis. Am. J. Ther. 2017, 24, e56–e63. [Google Scholar] [CrossRef]
- Silveira, M.G.; Torok, N.J.; Gossard, A.A.; Keach, J.C.; Jorgensen, R.R.A.; Petz, R.J.L.; Lindor, K.D. Minocycline in the Treatment of Patients With Primary Sclerosing Cholangitis: Results of a Pilot Study. Am. J. Gastroenterol. 2009, 104, 83–88. [Google Scholar] [CrossRef] [PubMed]
- Allegretti, J.R.; Kassam, Z.; Carrellas, M.; Mullish, B.; Marchesi, J.R.; Pechlivanis, A.; Smith, M.; Gerardin, Y.; Timberlake, S.; Pratt, D.S.; et al. Fecal Microbiota Transplantation in Patients With Primary Sclerosing Cholangitis: A Pilot Clinical Trial. Am. J. Gastroenterol. 2019, 114, 1071–1079. [Google Scholar] [CrossRef]
- Shah, A.; Macdonald, G.A.; Morrison, M.; Holtmann, G. Targeting the Gut Microbiome as a Treatment for Primary Sclerosing Cholangitis: A Conceptional Framework. Am. J. Gastroenterol. 2020, 115, 814–822. [Google Scholar] [CrossRef] [PubMed]
- Hilscher, M.; Enders, F.B.; Carey, E.J.; Lindor, K.D.; Tabibian, J.H. Alkaline phosphatase normalization is a biomarker of improved survival in primary sclerosing cholangitis. Ann. Hepatol. 2016, 15, 246–253. [Google Scholar] [CrossRef]
- Rupp, C.; Rössler, A.; Halibasic, E.; Sauer, P.; Weiss, K.-H.; Friedrich, K.; Wannhoff, A.; Stiehl, A.; Stremmel, W.; Trauner, M.; et al. Reduction in alkaline phosphatase is associated with longer survival in primary sclerosing cholangitis, independent of dominant stenosis. Aliment. Pharmacol. Ther. 2014, 40, 1292–1301. [Google Scholar] [CrossRef] [PubMed]
- Shah, A.; Crawford, D.; Burger, D.; Martin, N.; Walker, M.; Talley, N.J.; Tallis, C.; Jones, M.; Stuart, K.; Keely, S.; et al. Effects of Antibiotic Therapy in Primary Sclerosing Cholangitis with and without Inflammatory Bowel Disease: A Systematic Review and Meta-Analysis. Semin. Liver Dis. 2019, 39, 432–441. [Google Scholar] [CrossRef] [PubMed]
- Begley, M.; Hill, C.; Gahan, C.G.M. Bile Salt Hydrolase Activity in Probiotics. Appl. Environ. Microbiol. 2006, 72, 1729–1738. [Google Scholar] [CrossRef]
- Kitahara, M.; Sakata, S.; Sakamoto, M.; Benno, Y. Comparison among Fecal Secondary Bile Acid Levels, Fecal Microbiota andClostridium scindensCell Numbers in Japanese. Microbiol. Immunol. 2004, 48, 367–375. [Google Scholar] [CrossRef]
- Taylor, M.R.; Flannigan, K.L.; Rahim, H.; Mohamud, A.; Lewis, I.A.; Hirota, S.A.; Greenway, S.C. Vancomycin relieves mycophenolate mofetil–induced gastrointestinal toxicity by eliminating gut bacterial β-glucuronidase activity. Sci. Adv. 2019, 5, eaax2358. [Google Scholar] [CrossRef]
- Abarbanel, D.N.; Seki, S.M.; Davies, Y.; Marlen, N.; Benavides, J.A.; Cox, K.; Nadeau, K.C.; Cox, K.L. Immunomodulatory Effect of Vancomycin on Treg in Pediatric Inflammatory Bowel Disease and Primary Sclerosing Cholangitis. J. Clin. Immunol. 2013, 33, 397–406. [Google Scholar] [CrossRef] [PubMed]
- Deneau, M.R.; Mack, C.; Mogul, D.; Perito, E.R.; Valentino, P.L.; Amir, A.Z.; DiGuglielmo, M.; Draijer, L.G.; El-Matary, W.; Furuya, K.N.; et al. Oral Vancomycin, Ursodeoxycholic Acid, or No Therapy for Pediatric Primary Sclerosing Cholangitis: A Matched Analysis. Hepatology 2021, 73, 1061–1073. [Google Scholar] [CrossRef] [PubMed]
- Tan, L.-Z.; Reilly, C.R.; Steward-Harrison, L.C.; Balouch, F.; Muir, R.; Lewindon, P.J. Oral vancomycin induces clinical and mucosal remission of colitis in children with primary sclerosing cholangitis–ulcerative colitis. Gut 2019, 68, 1533–1535. [Google Scholar] [CrossRef]
- Bakker, G.J.; Nieuwdorp, M. Fecal Microbiota Transplantation: Therapeutic Potential for a Multitude of Diseases beyond Clostridium difficile. Microbiol. Spectr. 2017, 5, 5. [Google Scholar] [CrossRef]
- Wortelboer, K.; Nieuwdorp, M.; Herrema, H. Fecal microbiota transplantation beyond Clostridioides difficile infections. EBioMedicine 2019, 44, 716–729. [Google Scholar] [CrossRef]
- Wang, J.-W.; Kuo, C.-H.; Kuo, F.-C.; Wang, Y.-K.; Hsu, W.-H.; Yu, F.-J.; Hu, H.-M.; Hsu, P.-I.; Wang, J.-Y.; Wu, D.-C. Fecal microbiota transplantation: Review and update. J. Formos. Med. Assoc. 2019, 118, S23–S31. [Google Scholar] [CrossRef]
- Cammarota, G.; Masucci, L.; Ianiro, G.; Bibbò, S.; Dinoi, G.; Costamagna, G.; Sanguinetti, M.; Gasbarrini, A. Randomised clinical trial: Faecal microbiota transplantation by colonoscopy vs. vancomycin for the treatment of recurrentClostridium difficileinfection. Aliment. Pharmacol. Ther. 2015, 41, 835–843. [Google Scholar] [CrossRef] [PubMed]
- Kelly, C.R.; Khoruts, A.; Staley, C.; Sadowsky, M.J.; Abd, M.; Alani, M.; Bakow, B.; Curran, P.; McKenney, J.; Tisch, A.; et al. Effect of Fecal Microbiota Transplantation on Recurrence in Multiply Recurrent Clostridium difficile Infection. Ann. Intern. Med. 2016, 165, 609–616. [Google Scholar] [CrossRef] [PubMed]
- Hvas, C.L.; Dahl Jørgensen, S.M.; Jørgensen, S.P.; Storgaard, M.; Lemming, L.; Hansen, M.M.; Erikstrup, C.; Dahlerup, J.F. Fecal Microbiota Transplantation Is Superior to Fidaxomicin for Treatment of Recurrent Clostridium difficile Infection. Gastroenterology 2019, 156, 1324–1332.e3. [Google Scholar] [CrossRef] [PubMed]
- Lopez, J.; Grinspan, A. Fecal Microbiota Transplantation for Inflammatory Bowel Disease. Gastroenterol. Hepatol. 2016, 12, 374–379. [Google Scholar]
- Tan, P.; Li, X.; Shen, J.; Feng, Q. Fecal Microbiota Transplantation for the Treatment of Inflammatory Bowel Disease: An Update. Front. Pharmacol. 2020, 11, 574533. [Google Scholar] [CrossRef] [PubMed]
- DeFilipp, Z.; Bloom, P.P.; Soto, M.T.; Mansour, M.K.; Sater, M.; Huntley, M.H.; Turbett, S.; Chung, R.T.; Chen, Y.-B.; Hohmann, E.L. Drug-Resistant E. coli Bacteremia Transmitted by Fecal Microbiota Transplant. N. Engl. J. Med. 2019, 381, 2043–2050. [Google Scholar] [CrossRef] [PubMed]
- Beuers, U.; Hohenester, S.; Wenniger, L.J.M.D.B.; Kremer, A.; Jansen, P.L.M.; Elferink, R.P.J.O. The biliary HCO3− umbrella: A unifying hypothesis on pathogenetic and therapeutic aspects of fibrosing cholangiopathies. Hepatology 2010, 52, 1489–1496. [Google Scholar] [CrossRef]
- Trampert, D.C.; van de Graaf, S.F.; Jongejan, A.; Elferink, R.P.O.; Beuers, U. Hepatobiliary acid-base homeostasis: Insights from analogous secretory epithelia. J. Hepatol. 2021, 74, 428–441. [Google Scholar] [CrossRef]
- Schmucker, D.L.; Ohta, M.; Kanai, S.; Sato, Y.; Kitani, K. Hepatic injury induced by bile salts: Correlation between biochemical and morphological events. Hepatology 1990, 12, 1216–1221. [Google Scholar] [CrossRef]
- Verdier, J.; Luedde, T.; Sellge, G. Biliary Mucosal Barrier and Microbiome. Visc. Med. 2015, 31, 156–161. [Google Scholar] [CrossRef] [PubMed]
- Wu, T.; Zhang, Z.; Liu, B.; Hou, D.; Liang, Y.; Zhang, J.; Shi, P. Gut microbiota dysbiosis and bacterial community assembly associated with cholesterol gallstones in large-scale study. BMC Genom. 2013, 14, 669. [Google Scholar] [CrossRef]
- Jiménez, E.; Sánchez, B.; Farina, A.; Margolles, A.; Rodríguez, J.M. Characterization of the bile and gall bladder microbiota of healthy pigs. Microbiology 2014, 3, 937–949. [Google Scholar] [CrossRef]
- Steck, N.; Hoffmann, M.; Sava, I.G.; Kim, S.C.; Hahne, H.; Tonkonogy, S.L.; Mair, K.; Krueger, D.; Pruteanu, M.; Shanahan, F.; et al. Enterococcus faecalis Metalloprotease Compromises Epithelial Barrier and Contributes to Intestinal Inflammation. Gastroenterology 2011, 141, 959–971. [Google Scholar] [CrossRef]
- Katt, J.; Schwinge, D.; Schoknecht, T.; Quaas, A.; Sobottka, I.; Burandt, E.; Becker, C.; Neurath, M.F.; Lohse, A.W.; Herkel, J.; et al. Increased T helper type 17 response to pathogen stimulation in patients with primary sclerosing cholangitis. Hepatology 2013, 58, 1084–1093. [Google Scholar] [CrossRef]
- Kwon, W.; Jang, J.-Y.; Kim, E.-C.; Park, J.W.; Han, I.W.; Kang, M.J.; Kim, S.-W. Changing trend in bile microbiology and antibiotic susceptibilities: Over 12 years of experience. Infection 2013, 41, 93–102. [Google Scholar] [CrossRef] [PubMed]
- Litvak, Y.; Byndloss, M.; Tsolis, R.M.; Bäumler, A.J. Dysbiotic Proteobacteria expansion: A microbial signature of epithelial dysfunction. Curr. Opin. Microbiol. 2017, 39, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Beuers, U.; Spengler, U.; Kruis, W.; Aydemir, Ü.; Wiebecke, B.; Heldwein, W.; Weinzierl, M.; Pape, G.R.; Sauerbruch, T.; Paumgartner, G. Ursodeoxycholic acid for treatment of primary sclerosing cholangitis: A placebo-controlled trial. Hepatology 1992, 16, 707–714. [Google Scholar] [CrossRef] [PubMed]
- Olsson, R.; Boberg, K.M.; de Muckadell, O.B.S.; Lindgren, S.; Hultcrantz, R.; Folvik, G.; Bell, H.; Gangsøy–Kristiansen, M.; Matre, J.; Rydning, A.; et al. High-Dose Ursodeoxycholic Acid in Primary Sclerosing Cholangitis: A 5-Year Multicenter, Randomized, Controlled Study. Gastroenterology 2005, 129, 1464–1472. [Google Scholar] [CrossRef] [PubMed]
- Lindor, K.D.; Kowdley, K.V.; Luketic, V.A.C.; Harrison, M.E.; McCashland, T.; Befeler, A.S.; Harnois, D.; Jorgensen, R.; Petz, J.; Keach, J.; et al. High-dose ursodeoxycholic acid for the treatment of primary sclerosing cholangitis. Hepatology 2009, 50, 808–814. [Google Scholar] [CrossRef]
- Fickert, P.; Wagner, M. Biliary bile acids in hepatobiliary injury—What is the link? J. Hepatol. 2017, 67, 619–631. [Google Scholar] [CrossRef]
- Kowdley, K.V.; Vuppalanchi, R.; Levy, C.; Floreani, A.; Andreone, P.; LaRusso, N.F.; Shrestha, R.; Trotter, J.; Goldberg, D.; Rushbrook, S.; et al. A randomized, placebo-controlled, phase II study of obeticholic acid for primary sclerosing cholangitis. J. Hepatol. 2020, 73, 94–101. [Google Scholar] [CrossRef]
- Trauner, M.; Gulamhusein, A.; Hameed, B.; Caldwell, S.; Shiffman, M.L.; Landis, C.; Eksteen, B.; Agarwal, K.; Muir, A.; Rushbrook, S.; et al. The Nonsteroidal Farnesoid X Receptor Agonist Cilofexor (GS-9674) Improves Markers of Cholestasis and Liver Injury in Patients With Primary Sclerosing Cholangitis. Hepatology 2019, 70, 788–801. [Google Scholar] [CrossRef]
- Mizuno, S.; Hirano, K.; Tada, M.; Yamamoto, K.; Yashima, Y.; Yagioka, H.; Kawakubo, K.; Ito, Y.; Kogure, H.; Sasaki, T.; et al. Bezafibrate for the treatment of primary sclerosing cholangitis. J. Gastroenterol. 2010, 45, 758–762. [Google Scholar] [CrossRef]
- Mizuno, S.; Hirano, K.; Isayama, H.; Watanabe, T.; Yamamoto, N.; Nakai, Y.; Sasahira, N.; Tada, M.; Omata, M.; Koike, K. Prospective study of bezafibrate for the treatment of primary sclerosing cholangitis. J. Hepato-Biliary-Pancreatic Sci. 2015, 22, 766–770. [Google Scholar] [CrossRef]
- Fickert, P.; Hirschfield, G.; Denk, G.; Marschall, H.-U.; Altorjay, I.; Färkkilä, M.; Schramm, C.; Spengler, U.; Chapman, R.; Bergquist, A.; et al. norUrsodeoxycholic acid improves cholestasis in primary sclerosing cholangitis. J. Hepatol. 2017, 67, 549–558. [Google Scholar] [CrossRef]
- Shoda, J.; Okada, K.; Inada, Y.; Kusama, H.; Utsunomiya, H.; Oda, K.; Yokoi, T.; Yoshizato, K.; Suzuki, H. Bezafibrate induces multidrug-resistance P-Glycoprotein 3 expression in cultured human hepatocytes and humanized livers of chimeric mice. Hepatol. Res. 2007, 37, 548–556. [Google Scholar] [CrossRef] [PubMed]
- Inoue, I.; Noji, S.; Awata, T.; Takahashi, K.; Nakajima, T.; Sonoda, M.; Komoda, T.; Katayama, S. Bezafibrate has an antioxidant effect: Peroxisome proliferator-activated receptor α is associated with Cu2+, Zn2+-superoxide dismutase in the liver. Life Sci. 1998, 63, 135–144. [Google Scholar] [CrossRef]
- Lemoinne, S.; Pares, A.; Reig, A.; Ben Belkacem, K.; Fankem, A.D.K.; Gaouar, F.; Poupon, R.; Housset, C.; Corpechot, C.; Chazouillères, O. Primary sclerosing cholangitis response to the combination of fibrates with ursodeoxycholic acid: French–Spanish experience. Clin. Res. Hepatol. Gastroenterol. 2018, 42, 521–528. [Google Scholar] [CrossRef]
- Callea, F.; Sergi, C.; Fabbretti, G.; Brisigotti, M.; Cozzutto, C.; Medicina, D. Precancerous lesions of the biliary tree. J. Surg. Oncol. 1993, 53, 131–133. [Google Scholar] [CrossRef]
- Bouvier, A.-M.; Remontet, L.; Jougla, E.; Launoy, G.; Grosclaude, P.; Buémi, A.; Tretarre, B.; Velten, M.; Dancourt, V.; Menegoz, F.; et al. Incidence of gastrointestinal cancers in France. Gastroentérologie Clinique et Biologique 2004, 28, 877–881. [Google Scholar] [CrossRef]
- Song, J.; Li, Y.; Bowlus, C.L.; Yang, G.; Leung, P.S.C.; Gershwin, M.E. Cholangiocarcinoma in Patients with Primary Sclerosing Cholangitis (PSC): A Comprehensive Review. Clin. Rev. Allergy Immunol. 2019, 58, 134–149. [Google Scholar] [CrossRef]
- Chung, B.K.; Karlsen, T.H.; Folseraas, T. Cholangiocytes in the pathogenesis of primary sclerosing cholangitis and development of cholangiocarcinoma. Biochim. Biophys. Acta Mol. Basis Dis. 2018, 1864, 1390–1400. [Google Scholar] [CrossRef]
- Ehlken, H.; Schramm, C. Primary Sclerosing Cholangitis and Cholangiocarcinoma: Pathogenesis and Modes of Diagnostics. Dig. Dis. 2013, 31, 118–125. [Google Scholar] [CrossRef] [PubMed]
- Timmer, M.R.; Beuers, U.; Fockens, P.; Ponsioen, C.Y.; Rauws, E.A.; Wang, K.K.; Krishnadath, K.K. Genetic and Epigenetic Abnormalities in Primary Sclerosing Cholangitis-associated Cholangiocarcinoma. Inflamm. Bowel Dis. 2013, 19, 1789–1797. [Google Scholar] [CrossRef]
- Burak, K.W.; Angulo, P.; Pasha, T.M.; Egan, K.M.; Petz, J.; Lindor, K.D. Incidence and Risk Factors for Cholangiocarcinoma in Primary Sclerosing Cholangitis. Am. J. Gastroenterol. 2004, 99, 523–526. [Google Scholar] [CrossRef]
- Yamada, D.; Rizvi, S.; Razumilava, N.; Bronk, S.F.; Davila, J.I.; Champion, M.D.; Borad, M.J.; Bezerra, J.A.; Chen, X.; Gores, G.J. IL-33 facilitates oncogene-induced cholangiocarcinoma in mice by an interleukin-6-sensitive mechanism. Hepatology 2015, 61, 1627–1642. [Google Scholar] [CrossRef] [PubMed]
- Chalasani, N.; Baluyut, A.; Ismail, A.; Zaman, A.; Sood, G.; Ghalib, R.; McCashland, T.M.; Reddy, K.R.; Zervos, X.; Anbari, M.A.; et al. Cholangiocarcinoma in patients with primary sclerosing cholangitis: A multicenter case-control study. Hepatology 2000, 31, 7–11. [Google Scholar] [CrossRef] [PubMed]
- Saab, M.; Mestivier, D.; Sohrabi, M.; Rodriguez, C.; Khonsari, M.R.; Faraji, A.; Sobhani, I. Characterization of biliary microbiota dysbiosis in extrahepatic cholangiocarcinoma. PLoS ONE 2021, 16, e0247798. [Google Scholar] [CrossRef] [PubMed]
- Carpino, G.; Cardinale, V.; Renzi, A.; Hov, J.R.; Berloco, P.B.; Rossi, M.; Karlsen, T.H.; Alvaro, D.; Gaudio, E. Activation of biliary tree stem cells within peribiliary glands in primary sclerosing cholangitis. J. Hepatol. 2015, 63, 1220–1228. [Google Scholar] [CrossRef] [PubMed]
- Moeini, A.; Haber, P.K.; Sia, D. Cell of origin in biliary tract cancers and clinical implications. JHEP Rep. 2021, 3, 100226. [Google Scholar] [CrossRef] [PubMed]
- Boyd, S.; Tenca, A.; Jokelainen, K.; Mustonen, H.; Krogerus, L.; Arola, J.; Färkkilä, M. Screening primary sclerosing cholangitis and biliary dysplasia with endoscopic retrograde cholangiography and brush cytology: Risk factors for biliary neoplasia. Endoscopy 2016, 48, 432–439. [Google Scholar] [CrossRef]
- Fevery, J.; Verslype, C.; Lai, G.; Aerts, R.; Van Steenbergen, W. Incidence, Diagnosis, and Therapy of Cholangiocarcinoma in Patients with Primary Sclerosing Cholangitis. Dig. Dis. Sci. 2007, 52, 3123–3135. [Google Scholar] [CrossRef]
- Thanan, R.; Pairojkul, C.; Pinlaor, S.; Khuntikeo, N.; Wongkham, C.; Sripa, B.; Ma, N.; Vaeteewoottacharn, K.; Furukawa, A.; Kobayashi, H.; et al. Inflammation-related DNA damage and expression of CD133 and Oct3/4 in cholangiocarcinoma patients with poor prognosis. Free. Radic. Biol. Med. 2013, 65, 1464–1472. [Google Scholar] [CrossRef]
- Kerr, S.E.; Fritcher, E.G.B.; Campion, M.B.; Voss, J.S.; Kipp, B.R.; Halling, K.C.; Lewis, J.T. Biliary dysplasia in primary sclerosing cholangitis harbors cytogenetic abnormalities similar to cholangiocarcinoma. Hum. Pathol. 2014, 45, 1797–1804. [Google Scholar] [CrossRef] [PubMed]
- Cadamuro, M.; Nardo, G.; Indraccolo, S.; Dall’Olmo, L.; Sambado, L.; Moserle, L.; Franceschet, I.; Colledan, M.; Massani, M.; Stecca, T.; et al. Platelet-derived growth factor-D and Rho GTPases regulate recruitment of cancer-associated fibroblasts in cholangiocarcinoma. Hepatology 2013, 58, 1042–1053. [Google Scholar] [CrossRef]
- Shen, H.; Ye, F.; Xie, L.; Yang, J.; Li, Z.; Xu, P.; Meng, F.; Li, L.; Xiaochen, B.; Bo, X.; et al. Metagenomic sequencing of bile from gallstone patients to identify different microbial community patterns and novel biliary bacteria. Sci. Rep. 2015, 5, 17450. [Google Scholar] [CrossRef] [PubMed]
- Ye, F.; Shen, H.; Li, Z.; Meng, F.; Li, L.; Yang, J.; Chen, Y.; Bo, X.; Zhang, X.; Ni, M. Influence of the Biliary System on Biliary Bacteria Revealed by Bacterial Communities of the Human Biliary and Upper Digestive Tracts. PLoS ONE 2016, 11, e0150519. [Google Scholar] [CrossRef]
- Hiramatsu, K.; Harada, K.; Tsuneyama, K.; Sasaki, M.; Fujita, S.; Hashimoto, T.; Kaneko, S.; Kobayashi, K.; Nakanuma, Y. Amplification and sequence analysis of partial bacterial 16S ribosomal RNA gene in gallbladder bile from patients with primary biliary cirrhosis. J. Hepatol. 2000, 33, 9–18. [Google Scholar] [CrossRef]
- Jia, X.; Lu, S.; Zeng, Z.; Liu, Q.; Dong, Z.; Chen, Y.; Zhu, Z.; Hong, Z.; Zhang, T.; Du, G.; et al. Characterization of Gut Microbiota, Bile Acid Metabolism, and Cytokines in Intrahepatic Cholangiocarcinoma. Hepatology 2020, 71, 893–906. [Google Scholar] [CrossRef] [PubMed]
- Jusakul, A.; Khuntikeo, N.; Haigh, W.G.; Kuver, R.; Ioannou, G.N.; Loilome, W.; Namwat, N.; Bhudhisawasdi, V.; Pugkhem, A.; Pairojkul, C.; et al. Identification of biliary bile acids in patients with benign biliary diseases, hepatocellular carcinoma and cholangiocarcinoma. Asian Pac. J. Cancer Prev. 2012, 13. [Google Scholar]
- Jiménez, F.A.; Guitron, A.; Segura-Lopez, F.K.; Méndez-Tenorio, A.; Iwai, S.; Hernández-Guerrero, A.; Torres, J. Microbiota studies in the bile duct strongly suggest a role for Helicobacter pylori in extrahepatic cholangiocarcinoma. Clin. Microbiol. Infect. 2016, 22, 178-e11–178-e22. [Google Scholar] [CrossRef]
- Gupta, A.; Dhakan, D.B.; Maji, A.; Saxena, R.; Prasoodanan, V.P.K.; Mahajan, S.; Pulikkan, J.; Kurian, J.; Gomez, A.M.; Scaria, J.; et al. Association of Flavonifractor plautii, a Flavonoid-Degrading Bacterium, with the Gut Microbiome of Colorectal Cancer Patients in India. MSystems 2019, 4. [Google Scholar] [CrossRef]
- Sobhani, I.; Tap, J.; Roudot-Thoraval, F.; Roperch, J.P.; Letulle, S.; Langella, P.; Corthier, G.; Van Nhieu, J.T.; Furet, J.-P. Microbial Dysbiosis in Colorectal Cancer (CRC) Patients. PLoS ONE 2011, 6, e16393. [Google Scholar] [CrossRef]
- Molinero, N.; Ruiz, L.; Milani, C.; Gutiérrez-Díaz, I.; Sánchez, B.; Mangifesta, M.; Segura, J.; Cambero, I.; Campelo, A.B.; Garcia, J.I.R.; et al. The human gallbladder microbiome is related to the physiological state and the biliary metabolic profile. Microbiome 2019, 7, 1–17. [Google Scholar] [CrossRef]
- Rath, H.C.; Herfarth, H.H.; Ikeda, J.S.; Grenther, W.B.; Hamm, T.E.; Balish, E.; Taurog, J.D.; Hammer, R.E.; Wilson, K.H.; Sartor, R.B. Normal luminal bacteria, especially Bacteroides species, mediate chronic colitis, gastritis, and arthritis in HLA-B27/human beta2 microglobulin transgenic rats. J. Clin. Investig. 1996, 98, 945–953. [Google Scholar] [CrossRef]
- Nanjundappa, R.H.; Ronchi, F.; Wang, J.; Clemente-Casares, X.; Yamanouchi, J.; Umeshappa, C.S.; Yang, Y.; Blanco, J.; Bassolas-Molina, H.; Salas, A.; et al. A Gut Microbial Mimic that Hijacks Diabetogenic Autoreactivity to Suppress Colitis. Cell 2017, 171, 655–667.e17. [Google Scholar] [CrossRef]
- Lazaridis, K.N.; Gores, G.J. Primary Sclerosing Cholangitis and Cholangiocarcinoma. Semin. Liver Dis. 2006, 26, 042–051. [Google Scholar] [CrossRef] [PubMed]
- Randi, G.; Franceschi, S.; La Vecchia, C. Gallbladder cancer worldwide: Geographical distribution and risk factors. Int. J. Cancer 2006, 118, 1591–1602. [Google Scholar] [CrossRef] [PubMed]
- Crawford, R.W.; Gibson, D.L.; Kay, W.W.; Gunn, J.S. Identification of a Bile-Induced Exopolysaccharide Required for Salmonella Biofilm Formation on Gallstone Surfaces. Infect. Immun. 2008, 76, 5341–5349. [Google Scholar] [CrossRef] [PubMed]
- Crawford, R.W.; Rosales-Reyes, R.; Ramirez-Aguilar, M.D.L.L.; Chapa-Azuela, O.; Alpuche-Aranda, C.; Gunn, J.S. Gallstones play a significant role in Salmonella spp. gallbladder colonization and carriage. Proc. Natl. Acad. Sci. USA 2010, 107, 4353–4358. [Google Scholar] [CrossRef]
- Gonzalez-Escobedo, G.; Marshall, J.M.; Gunn, J.S. Chronic and acute infection of the gall bladder by Salmonella Typhi: Understanding the carrier state. Nat. Rev. Genet. 2010, 9, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Nagaraja, V.; Eslick, G.D. Systematic review with meta-analysis: The relationship between chronicSalmonella typhicarrier status and gall-bladder cancer. Aliment. Pharmacol. Ther. 2014, 39, 745–750. [Google Scholar] [CrossRef] [PubMed]
- Sepe, L.P.; Hartl, K.; Iftekhar, A.; Berger, H.; Kumar, N.; Goosmann, C.; Chopra, S.; Schmidt, S.C.; Gurumurthy, R.K.; Meyer, T.F.; et al. Genotoxic Effect of Salmonella Paratyphi A Infection on Human Primary Gallblader Cells. MBio. 2020, 11. [Google Scholar] [CrossRef]
- Chumduri, C.; Gurumurthy, R.K.; Zietlow, R.; Meyer, T.F. Subversion of host genome integrity by bacterial pathogens. Nat. Rev. Mol. Cell Biol. 2016, 17, 659–673. [Google Scholar] [CrossRef]
- Koeppel, M.; Garcia-Alcalde, F.; Glowinski, F.; Schlaermann, P.; Meyer, T.F. Helicobacter pylori Infection Causes Characteristic DNA Damage Patterns in Human Cells. Cell Rep. 2015, 11, 1703–1713. [Google Scholar] [CrossRef] [PubMed]
- Toller, I.M.; Neelsen, K.J.; Steger, M.; Hartung, M.L.; Hottiger, M.; Stucki, M.; Kalali, B.; Gerhard, M.; Sartori, A.; Lopes, M.; et al. Carcinogenic bacterial pathogen Helicobacter pylori triggers DNA double-strand breaks and a DNA damage response in its host cells. Proc. Natl. Acad. Sci. USA 2011, 108, 14944–14949. [Google Scholar] [CrossRef]
- Hartl, K.; Sigal, M. Microbe-Driven Genotoxicity in Gastrointestinal Carcinogenesis. Int. J. Mol. Sci. 2020, 21, 7439. [Google Scholar] [CrossRef] [PubMed]
- Sigal, M.; Meyer, T.F. Coevolution between the Human Microbiota and the Epithelial Immune System. Dig. Dis. 2016, 34, 190–193. [Google Scholar] [CrossRef] [PubMed]
- Faïs, T.; Delmas, J.; Barnich, N.; Bonnet, R.; Dalmasso, G. Colibactin: More Than a New Bacterial Toxin. Toxins 2018, 10, 151. [Google Scholar] [CrossRef]
- Dziubańska-Kusibab, P.J.; Berger, H.; Battistini, F.; Bouwman, B.A.M.; Iftekhar, A.; Katainen, R.; Cajuso, T.; Crosetto, N.; Orozco, M.; Aaltonen, L.A.; et al. Colibactin DNA-damage signature indicates mutational impact in colorectal cancer. Nat. Med. 2020, 26, 1063–1069. [Google Scholar] [CrossRef]
- Pleguezuelos-Manzano, C.; Puschhof, J.; Huber, A.R.; Van Hoeck, A.; Wood, H.M.; Nomburg, J.; Gurjao, C.; Manders, F.; Dalmasso, G.; Stege, P.B.; et al. Mutational signature in colorectal cancer caused by genotoxic pks+ E. coli. Nature 2020, 580, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Iftekhar, A.; Berger, H.; Bouznad, N.; Heuberger, J.; Boccellato, F.; Dobrindt, U.; Hermeking, H.; Sigal, M.; Meyer, T.F. Genomic aberrations after short-term exposure to colibactin-producing E. coli transform primary colon epithelial cells. Nat. Commun. 2021, 12, 1–15. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Reference | Sample Origin | No. of PSC Patients with and without IBD (n=) | Microbiology Assessment | Major Findings with Regard to the Gut Microbiome |
|---|---|---|---|---|
| Sabino et al., 2016 [59] | Stool | 66 | 16sRNA gene sequencing | Significant over-representation of Enterococcus, Fusobacterium, and Lactobacillus genera |
| Kummen et al., 2017 [27] | Stool | 85 | 16sRNA gene sequencing | Enrichment of the Veillonella genus |
| Bajer et al., 2017 [25] | Stool | 43 | 16sRNA gene sequencing | Rothia, Enterococcus, Streptococcus, Veillonella were overrepresented in PSC regardless of concomitant IBD Decreased abundance of Adlercreutzia equolifaciens and Prevotella copri |
| Iwasawa et al., 2017 [60] | Stool | 27 | 16sRNA gene sequencing | Abundance of Enterococcus (E. faecalis especially), Streptococcus (with prevalence of Streptococcus parasanguinis) and Veillonella species Parabacteroides overrepresented |
| Rühlemann et al., 2019 [12] | Stool | 137 | 16sRNA gene sequencing | Increased abundance in eight genera Veillonella, Streptococcus, Lactobacillus, Enterococcus; Proteobacteria, Lactobacillales (Bacilli), Parabacteroides, and Bacteroides |
| Lemoinne et al., 2020 [61] | Stool | 49 | 16sRNA gene sequencing | Fungal dysbiosis: increased proportion of Exophiala, decreased proportion of Saccharomyces cerevisiae Bacterial dysbiosis: Firmicutes decreased (except Veillonella increased) and Proteobacteria increased |
| Kummen et al., 2021 [62] | Stool | 136 | metagenomic shotgun sequencing | Increased abundance of Clostridiales species Depletion of Eubacterium, Ruminococcus obeum |
| Rossen et al., 2015 [63] | Ileum mucosa | 12 | 16sRNA gene sequencing | Abundance of uncultured Clostridiales II |
| Kevans et al., 2016 [26] | Colon mucosa | 31 | 16sRNA gene sequencing | No changes |
| Torres et al., 2016 [64] | Ileum, colon mucosa | 20 | 16sRNA gene sequencing | significant increase in Barnesiellaceae and Blautia |
| Quraishi et al., 2017 [24] | Colon mucosa | 11 | 16sRNA gene sequencing | significant increase in Escherichia, Megasphera and Lachnospiraceae |
| Quraishi et al., 2020 [65] | Sigmoid mucosa | 20 | 16sRNA gene sequencing | significantly higher abundance of the genera Bacteroides fragilis, Roseburia, Shewanella, and Clostridium ramosum in patients with PSC-IBD compared to patients with UC |
| Liwinski et al., 2020 [66] | Duodenal and oral fluids, duodenal mucosa, bile | 46 | 16sRNA gene sequencing | Duodenal mucosa biopsy: overrepresentation of Escherichia coli and Veillonella dispar Bile fluid: overrepresentation of Enterococcus (especially Enterococcus faecalis), Proteobacteria Staphylococcus, Neisseria, Veillonella dispar |
| References | Therapeutic Agents | Study Type | PSC Patients (N=) | Treatment Duration | Primary Endpoint | Results |
|---|---|---|---|---|---|---|
| Rahimpour et al., 2016 [106] | Vancomycin | RCT | 8 | 12 weeks | Decrease in MRS at week 12 | Significant reduction in mean MRS and ALP levels |
| Tabibian et al., 2013 [107] | Vancomycin vs.Metronidazole | RCT | 9 9 | 12 weeks | Decrease in ALP levels at week 12 | Non-dose dependent significant ALP reduction (for low and high dose vancomycin) |
| Färkkilä et al., 2004 [108] | Metronidazole (and UDCA) | RCT | 39 | 36 months | Decrease in ALP or other liver enzymes, MRS, symptoms or histology at week 36 | Significant ALP level and MRS reduction, improvement (of stage and grade) in histology |
| Tabibian et al., 2017 [109] | Rifaximin | Open-label study | 16 | 12 weeks | Decrease in ALP levels at months 3 | No significant reduction |
| Silveira et al., 2009 [110] | Minocycline | Open-label study | 16 | 1 year | Decrease in ALP levels at month 12 | Significant ALP reduction and mean MRS |
| Allegretti et al., 2019 [111] | Fecal transplantation | Open-label pilot | 10 | 24 weeks | ≥50% ALP reduction at week 24 | Significant reduction in ALP levels in 3/10 |
| References | Therapeutic Agents | Target Receptor/ Mechanism | Study Type | N | Treatment Duration | Primary Endpoint | Results |
|---|---|---|---|---|---|---|---|
| Beuers et al., 1992 [141] | UDCA 13–15 mg/kgper day | Choleretic effect, unknown receptor | RCT | 5 | 12 months | Significant ALP reduction | |
| Olsson et al., 2005 [142] | UDCA 17–23 mg/kgper day | Choleretic effect, unknown receptor | RCT | 110 | 5 years | Occurrence of liver transplantation | Significant improvement in ALP levels, no impact on survival |
| Lindor et al., 2009 [143] | UDCA 28–30 mg/kgper day | Choleretic effect, unknown receptor | RCT | 76 | 6 years | Changes in ALP and other liver enzymes | Significant improvement in ALP levels, no impact on survival, higher doses are associated with a higher rate of serious adverse events |
| Fickert et al., 2017 [144] | NorUDCA | Choleretic effect, unknown receptor | RCT | 161 | 12 weeks | Mean relative change in ALP levels | Significant dose-dependent ALP reductions |
| Kowdley et al., 2020 [145] | OCA | Endogenous FXR agonist | RCT | 76 | 24 weeks | Change in ALP at week 24 | Significant reduction in ALP levels (only for use of OCA 5–10 mg) |
| Trauner et al., 2019 [146] | Cilofexor | Nonsteroidal FXR agonist | RCT | 42 | 12 weeks | Change in ALP and other liver enzymes at week 12 | Significant reduction in ALP levels and other liver enzymes |
| Mizuno et al., 2010 [147] | Bezafibrate | PPARa-agonist | Open-label pilot | 7 | 6 months | Change in ALP levels at month 6 | Significant ALP reduction |
| Mizuno et al., 2015 [148] | Bezafibrate | PPARa-agonist | Open-label pilot | 11 | 12 weeks | Improvement in liver function tests | Significant ALP reduction |
| NCT04024813 | Seladelpar | Selective PPARδ agonist | RCT | 24 weeks | Change in ALP at week 24 | Ongoing |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Özdirik, B.; Müller, T.; Wree, A.; Tacke, F.; Sigal, M. The Role of Microbiota in Primary Sclerosing Cholangitis and Related Biliary Malignancies. Int. J. Mol. Sci. 2021, 22, 6975. https://doi.org/10.3390/ijms22136975
Özdirik B, Müller T, Wree A, Tacke F, Sigal M. The Role of Microbiota in Primary Sclerosing Cholangitis and Related Biliary Malignancies. International Journal of Molecular Sciences. 2021; 22(13):6975. https://doi.org/10.3390/ijms22136975
Chicago/Turabian StyleÖzdirik, Burcin, Tobias Müller, Alexander Wree, Frank Tacke, and Michael Sigal. 2021. "The Role of Microbiota in Primary Sclerosing Cholangitis and Related Biliary Malignancies" International Journal of Molecular Sciences 22, no. 13: 6975. https://doi.org/10.3390/ijms22136975
APA StyleÖzdirik, B., Müller, T., Wree, A., Tacke, F., & Sigal, M. (2021). The Role of Microbiota in Primary Sclerosing Cholangitis and Related Biliary Malignancies. International Journal of Molecular Sciences, 22(13), 6975. https://doi.org/10.3390/ijms22136975