
Transcriptional Profiling of Cardiac Cells Links Age-Dependent Changes in Acetyl-CoA Signaling to Chromatin Modifications



Abstract
:1. Introduction
2. Results
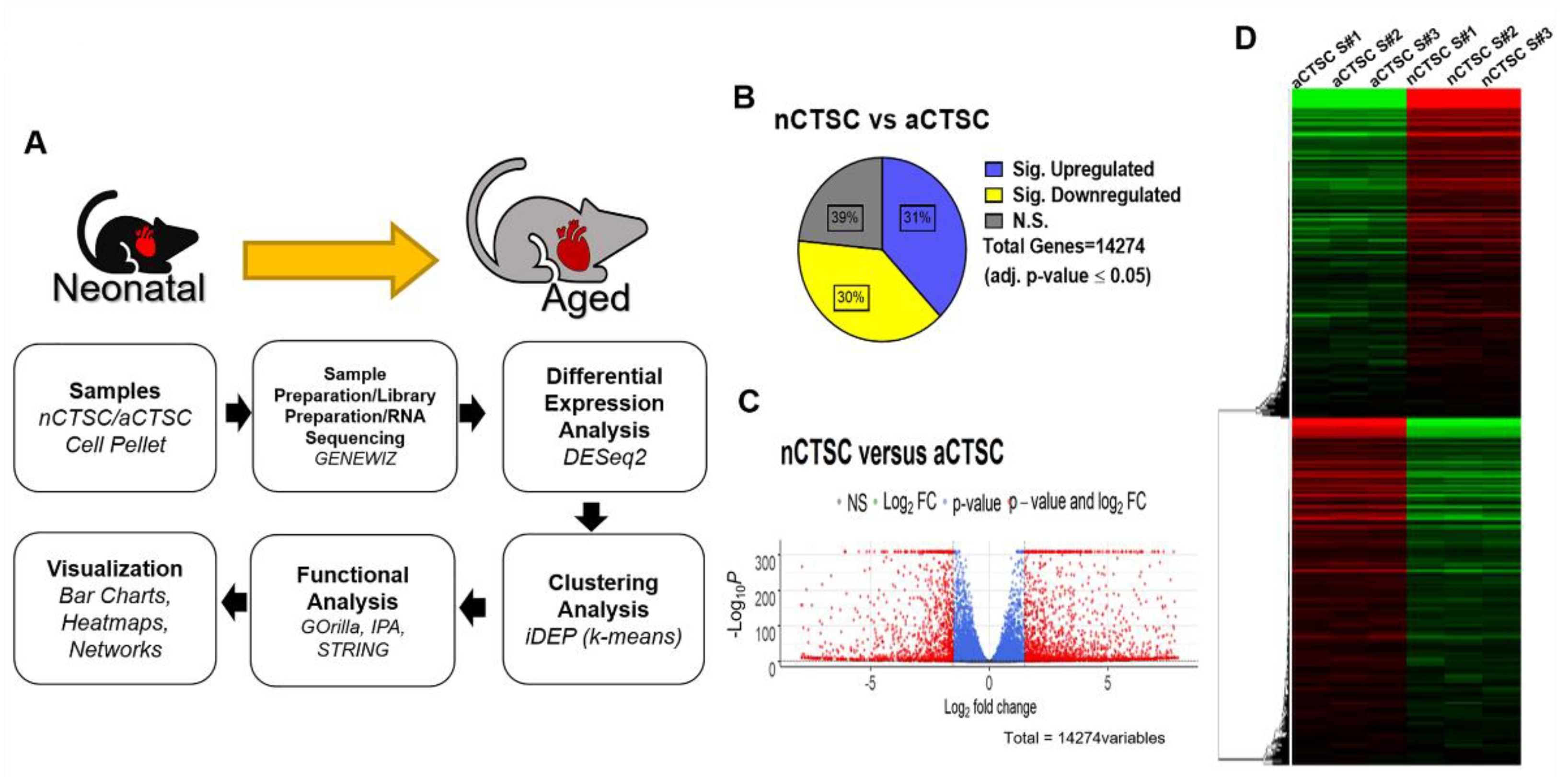
2.1. Distinct CTSC Profiles in Response to Aging as Revealed by Transcriptomic Analysis
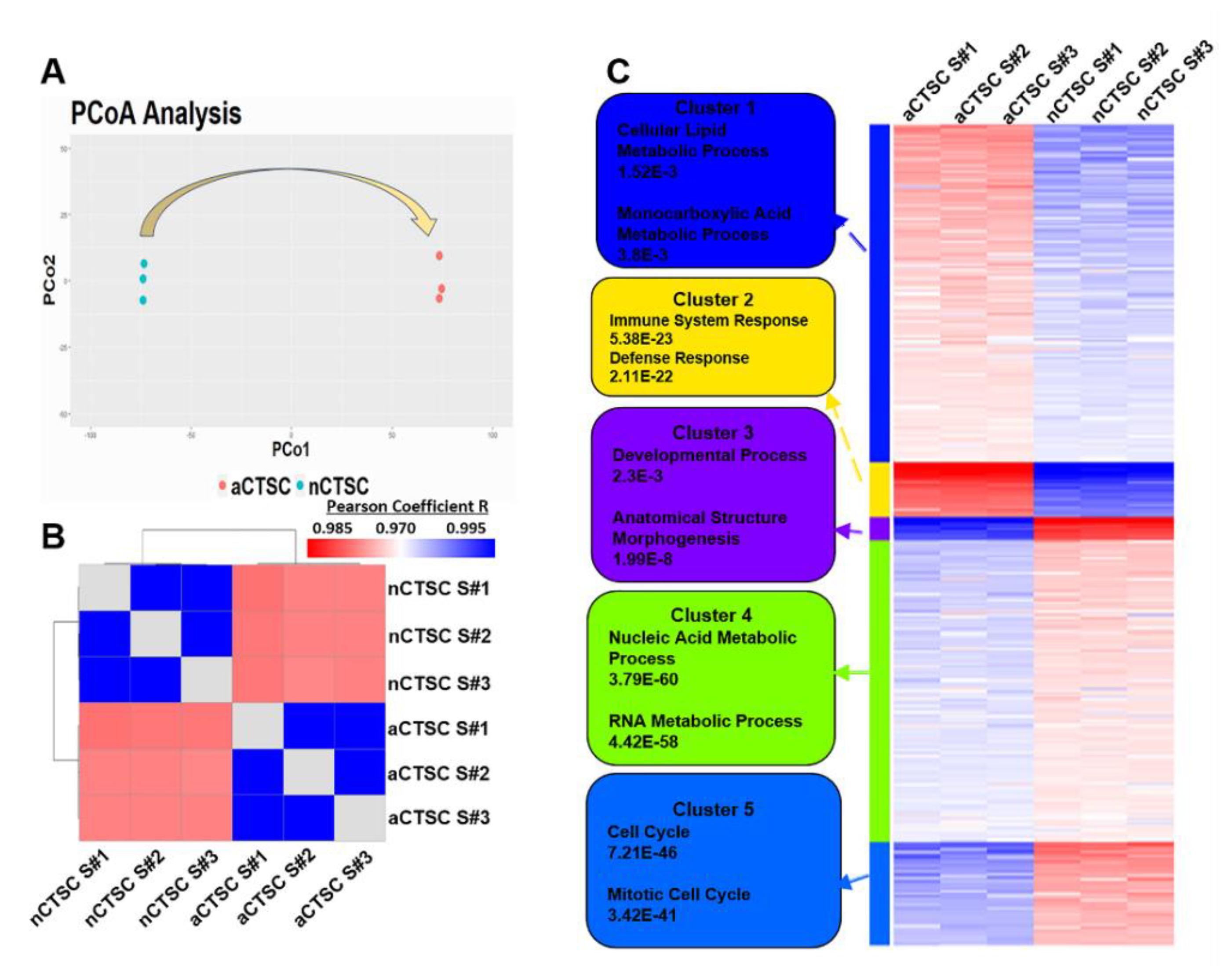
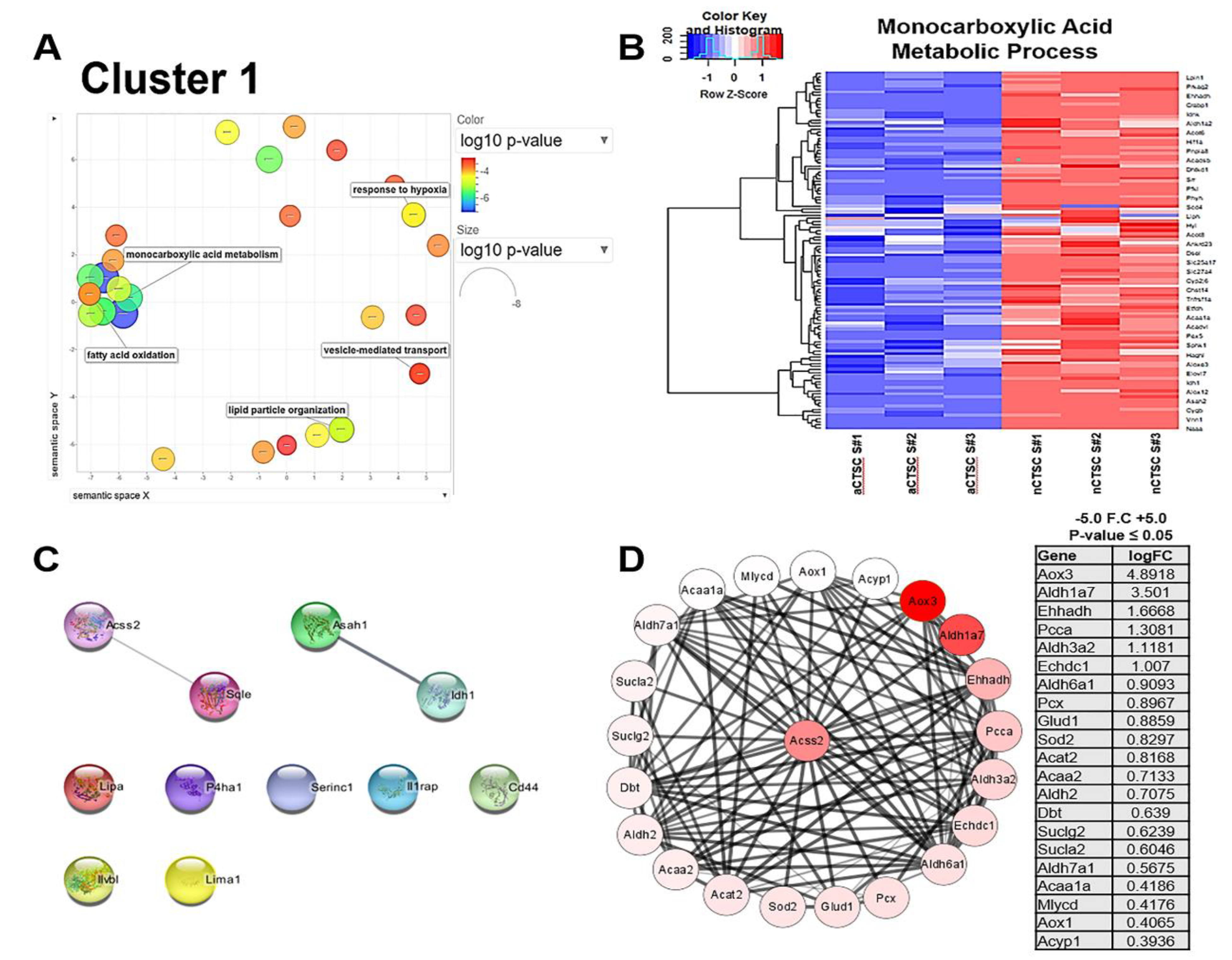
2.2. Metabolic Processes Are Upregulated in nCTSC as Shown by Clustering Analysis
2.3. ACSS2 Is Highly Enriched in nCTSCs Compared to aCTSCs
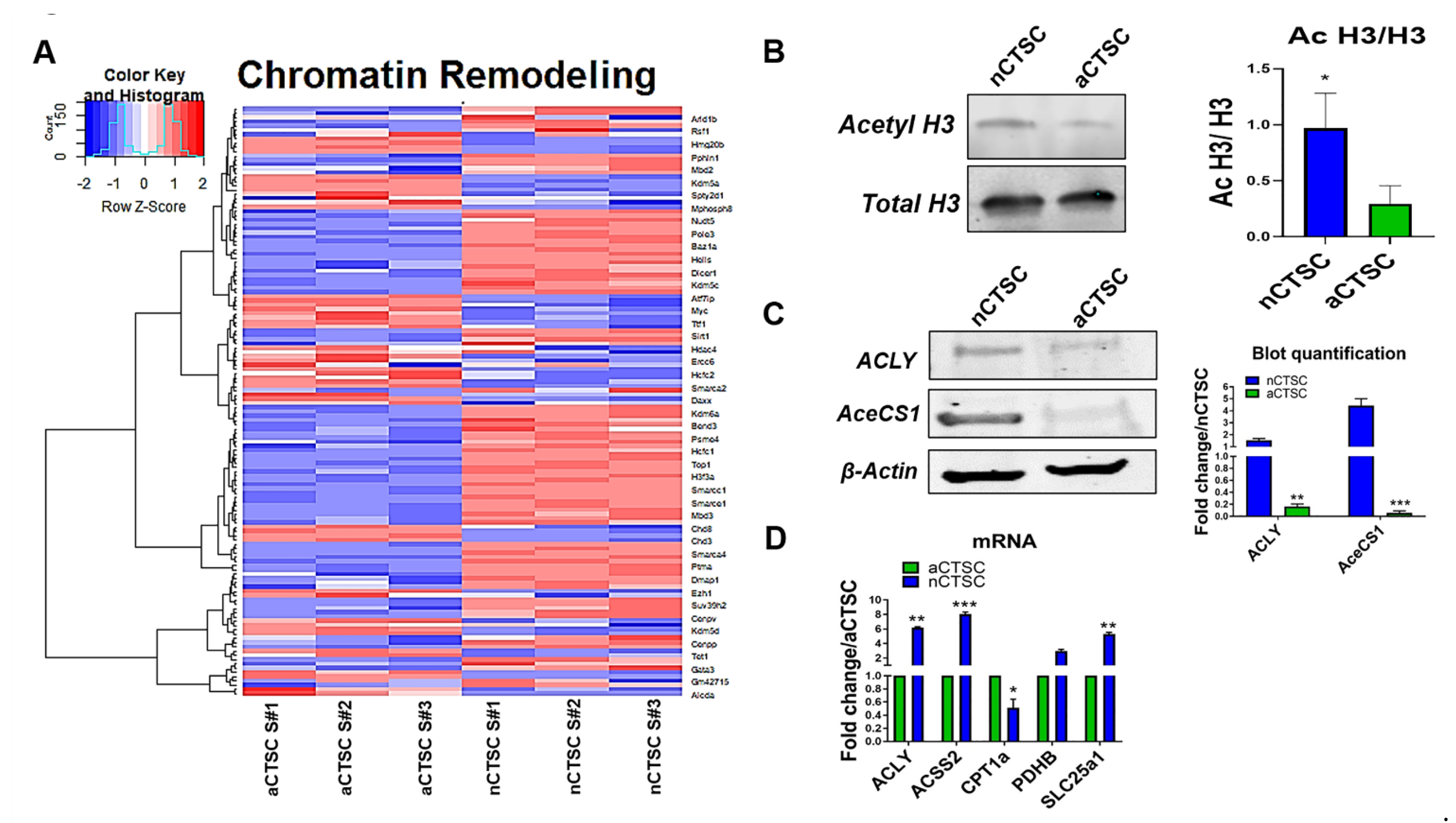
2.4. Altered ACSS2 Signaling Pathway and Chromatin Modifications in nCTSCs
3. Discussion
4. Materials and Methods
4.1. Cell Isolation and Culture
4.2. RNA Sequencing
4.3. Bioinformatics Analysis
4.4. Functional Analysis
4.5. Reverse Transcriptase Polymerase Chain Reaction
4.6. Western Blot
4.7. Statistical Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mohsin, S.; Khan, M.; Toko, H.; Bailey, B.; Cottage, C.T.; Wallach, K.; Nag, D.; Lee, A.; Siddiqi, S.; Lan, F.; et al. Human cardiac progenitor cells engineered with Pim-I kinase enhance myocardial repair. J. Am. Coll. Cardiol. 2012, 60, 1278–1287. [Google Scholar] [CrossRef] [Green Version]
- Haider, H.; Jiang, S.; Idris, N.M.; Ashraf, M. IGF-1-overexpressing mesenchymal stem cells accelerate bone marrow stem cell mobilization via paracrine activation of SDF-1alpha/CXCR4 signaling to promote myocardial repair. Circ. Res. 2008, 103, 1300–1308. [Google Scholar] [CrossRef] [Green Version]
- Bolli, R.; Mitrani, R.D.; Hare, J.M.; Pepine, C.J.; Perin, E.C.; Willerson, J.T.; Traverse, J.H.; Henry, T.D.; Yang, P.C.; Murphy, M.P.; et al. Cardiovascular Cell Therapy Research, A Phase II study of autologous mesenchymal stromal cells and c-kit positive cardiac cells, alone or in combination, in patients with ischaemic heart failure: The CCTRN CONCERT-HF trial. Eur. J. Heart Fail. 2021, 23, 661–674. [Google Scholar] [CrossRef] [PubMed]
- Mohsin, S.; Siddiqi, S.; Collins, B.; Sussman, M.A. Empowering adult stem cells for myocardial regeneration. Circ. Res. 2011, 109, 1415–1428. [Google Scholar] [CrossRef] [Green Version]
- Makkar, R.R.; Smith, R.R.; Cheng, K.; Malliaras, K.; Thomson, L.E.; Berman, D.; Czer, L.S.; Marban, L.; Mendizabal, A.; Johnston, P.V.; et al. Intracoronary cardiosphere-derived cells for heart regeneration after myocardial infarction (CADUCEUS): A prospective, randomised phase 1 trial. Lancet 2012, 379, 895–904. [Google Scholar] [CrossRef] [Green Version]
- Kurian, J.; Yuko, A.E.; Kasatkin, N.; Rigaud, V.O.C.; Busch, K.; Harlamova, D.; Wagner, M.; Recchia, F.A.; Wang, H.; Mohsin, S.; et al. Uncoupling protein 2-mediated metabolic adaptations define cardiac cell function in the heart during transition from young to old age. Stem Cells Transl. Med. 2020. [CrossRef]
- Moussaieff, A.; Rouleau, M.; Kitsberg, D.; Cohen, M.; Levy, G.; Barasch, D.; Nemirovski, A.; Shen-Orr, S.; Laevsky, I.; Amit, M.; et al. Glycolysis-mediated changes in acetyl-CoA and histone acetylation control the early differentiation of embryonic stem cells. Cell Metab. 2015, 21, 392–402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, K.; Suda, T. Metabolic requirements for the maintenance of self-renewing stem cells. Nat. Rev. Mol. Cell Biol. 2014, 15, 243–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, S.L.; Wellen, K.E. Metabolic Signaling to the Nucleus in Cancer. Mol. Cell 2018, 71, 398–408. [Google Scholar] [CrossRef] [Green Version]
- Trefely, S.; Lovell, C.D.; Snyder, N.W.; Wellen, K.E. Compartmentalised acyl-CoA metabolism and roles in chromatin regulation. Mol. Metab. 2020, 38, 100941. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, H.; McCaffery, J.M.; Irizarry, R.A.; Boeke, J.D. Nucleocytosolic acetyl-coenzyme a synthetase is required for histone acetylation and global transcription. Mol. Cell 2006, 23, 207–217. [Google Scholar] [CrossRef] [PubMed]
- Wellen, K.E.; Hatzivassiliou, G.; Sachdeva, U.M.; Bui, T.V.; Cross, J.R.; Thompson, C.B. ATP-citrate lyase links cellular metabolism to histone acetylation. Science 2009, 324, 1076–1080. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choudhary, C.; Weinert, B.T.; Nishida, Y.; Verdin, E.; Mann, M. The growing landscape of lysine acetylation links metabolism and cell signaling. Nat. Rev. Mol. Cell Biol. 2009, 15, 536–550. [Google Scholar] [CrossRef] [PubMed]
- Pietrocola, F.; Galluzzi, L.; Bravo-San Pedro, J.M.; Madeo, F.; Kroemer, G. Acetyl coenzyme A: A central metabolite and second messenger. Cell Metab. 2015, 21, 805–821. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gilsbach, R.; Schwaderer, M.; Preissl, S.; Gruning, B.A.; Kranzhofer, D.; Schneider, P.; Nuhrenberg, T.G.; Mulero-Navarro, S.; Weichenhan, D.; Braun, C.; et al. Distinct epigenetic programs regulate cardiac myocyte development and disease in the human heart in vivo. Nat. Commun. 2018, 9, 391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, P.; Hang, C.T.; Yang, J.; Chang, C.P. Chromatin remodeling in cardiovascular development and physiology. Circ. Res. 2011, 108, 378–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dow, J.; Simkhovich, B.Z.; Kedes, L.; Kloner, R.A. Washout of transplanted cells from the heart: A potential new hurdle for cell transplantation therapy. Cardiovasc. Res. 2005, 67, 301–307. [Google Scholar] [CrossRef]
- Hong, K.U.; Li, Q.H.; Guo, Y.; Patton, N.S.; Moktar, A.; Bhatnagar, A.; Bolli, R. A highly sensitive and accurate method to quantify absolute numbers of c-kit+ cardiac stem cells following transplantation in mice. Basic Res. Cardiol. 2013, 108, 346. [Google Scholar] [CrossRef] [PubMed]
- Simpson, D.L.; Mishra, R.; Sharma, S.; Goh, S.K.; Deshmukh, S.; Kaushal, S. A strong regenerative ability of cardiac stem cells derived from neonatal hearts. Circulation 2012, 126 (Suppl. 1), S46–S53. [Google Scholar] [CrossRef] [Green Version]
- Castaldi, A.; Dodia, R.M.; Orogo, A.M.; Zambrano, C.M.; Najor, R.H.; Gustafsson, A.B.; Heller Brown, J.; Purcell, N.H. Decline in cellular function of aged mouse c-kit(+) cardiac progenitor cells. J. Physiol. 2017, 595, 6249–6262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, W.E.; Yang, D.; Li, L.; Wang, W.; Peng, Y.; Chen, C.; Chen, P.; Xia, X.; Wang, H.; Jiang, J.; et al. Prolyl hydroxylase domain protein 2 silencing enhances the survival and paracrine function of transplanted adipose-derived stem cells in infarcted myocardium. Circ. Res. 2013, 113, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Wu, Q.; Liu, Q.; Zhan, J.; Wang, Q.; Zhang, D.; He, S.; Pu, S.; Zhou, Z. Cited2 regulates proliferation and survival in young and old mouse cardiac stem cells. BMC Mol. Cell Biol. 2019, 20, 25. [Google Scholar] [CrossRef] [Green Version]
- Shyh-Chang, N.; Ng, H.H. The metabolic programming of stem cells. Genes Dev. 2017, 31, 336–346. [Google Scholar] [CrossRef] [Green Version]
- Kimura, W.; Sadek, H.A. The cardiac hypoxic niche: Emerging role of hypoxic microenvironment in cardiac progenitors. Cardiovasc. Diagn. Ther. 2012, 2, 278–289. [Google Scholar]
- Khan, M.; Mohsin, S.; Khan, S.N.; Riazuddin, S. Repair of senescent myocardium by mesenchymal stem cells is dependent on the age of donor mice. J. Cell Mol. Med. 2011, 15, 1515–1527. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beegle, J.; Lakatos, K.; Kalomoiris, S.; Stewart, H.; Isseroff, R.R.; Nolta, J.A.; Fierro, F.A. Hypoxic preconditioning of mesenchymal stromal cells induces metabolic changes, enhances survival, and promotes cell retention in vivo. Stem Cells 2015, 33, 1818–1828. [Google Scholar] [CrossRef]
- Derlet, A.; Rasper, T.; Roy Choudhury, A.; Bothur, S.; Rieger, M.A.; Namgaladze, D.; Fischer, A.; Schurmann, C.; Brandes, R.P.; Tschulena, U.; et al. Metabolism Regulates Cellular Functions of Bone Marrow-Derived Cells used for Cardiac Therapy. Stem Cells 2016, 34, 2236–2248. [Google Scholar] [CrossRef] [PubMed]
- Salazar-Noratto, G.E.; Luo, G.; Denoeud, C.; Padrona, M.; Moya, A.; Bensidhoum, M.; Bizios, R.; Potier, E.; Logeart-Avramoglou, D.; Petite, H. Understanding and leveraging cell metabolism to enhance mesenchymal stem cell transplantation survival in tissue engineering and regenerative medicine applications. Stem Cells 2020, 38, 22–33. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Nuebel, E.; Daley, G.Q.; Koehler, C.M.; Teitell, M.A. Metabolic regulation in pluripotent stem cells during reprogramming and self-renewal. Cell Stem Cell 2012, 11, 589–595. [Google Scholar] [CrossRef] [Green Version]
- Newman, J.C.; He, W.; Verdin, E. Mitochondrial protein acylation and intermediary metabolism: Regulation by sirtuins and implications for metabolic disease. J. Biol. Chem. 2012, 287, 42436–42443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pillai, V.B.; Sundaresan, N.R.; Gupta, M.P. Regulation of Akt signaling by sirtuins: Its implication in cardiac hypertrophy and aging. Circ. Res. 2014, 114, 368–378. [Google Scholar] [CrossRef] [Green Version]
- Borden, A.; Kurian, J.; Nickoloff, E.; Yang, Y.; Troupes, C.D.; Ibetti, J.; Lucchese, A.M.; Gao, E.; Mohsin, S.; Koch, W.J.; et al. Transient Introduction of miR-294 in the Heart Promotes Cardiomyocyte Cell Cycle Reentry After Injury. Circ. Res. 2019, 125, 14–25. [Google Scholar] [CrossRef] [PubMed]
- Khan, M.; Nickoloff, E.; Abramova, T.; Johnson, J.; Verma, S.K.; Krishnamurthy, P.; Mackie, A.R.; Vaughan, E.; Garikipati, V.N.; Benedict, C.; et al. Embryonic stem cell-derived exosomes promote endogenous repair mechanisms and enhance cardiac function following myocardial infarction. Circ. Res. 2015, 117, 52–64. [Google Scholar] [CrossRef] [Green Version]


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Cluster 1 | Metabolism | GO Description | Top Gene | LogFC | adj. p-Value |
|---|---|---|---|---|---|
| GO:0044255 | Cellular lipid Metabolic process | Lipa | 1.880428797 | 8.3778 × 10−265 | |
| GO:0006629 | Lipid metabolic process | Lima1 | 1.386559217 | 4.9641 × 10−287 | |
| GO:0055114 | oxidation-reduction process | P4ha1 | 1.216435527 | 4.0584 × 10−292 | |
| GO:0032787 | monocarboxylic acid metabolic process | Idh1 | 1.584898068 | 1.4915 × 10−241 | |
| GO:0044242 | cellular lipid catabolic process | Asah1 | 0.963340936 | 1.8342 × 10−167 | |
| GO:0051092 | positive regulation of NF-kappaB transcription factor activity | Il1rap | 2.253245162 | 4.1379 × 10−303 | |
| GO:0008610 | lipid biosynthetic process | Serinc1 | 0.986106666 | 7.161 × 10−242 | |
| GO:0008610 | lipid biosynthetic process | Serinc1 | 0.986106666 | 7.161 × 10−242 | |
| GO:0072329 | monocarboxylic acid catabolic process | Ilvbl | 2.185676922 | 1.048 × 10−151 | |
| GO:0034389 | lipid droplet organization | Sqle | 0.920963676 | 4.2693 × 10−134 | |
| GO:0044281 | small molecule metabolic process | Cd44 | 0.996198493 | 5.32 × 10−285 | |
| Cluster 2 | Immune | GO Description | Top Gene | logFC | adj. p-Value |
| GO:0006952 | defense response | Rtp4 | 4.132592871 | 1.8172 × 10−298 | |
| GO:0051707 | response to other organism | Usp18 | 6.2573426 | 1.7065 × 10−301 | |
| GO:0043207 | response to external biotic stimulus | Stat1 | 2.589408409 | 1.0021 × 10−269 | |
| GO:0002376 | immune system process | Ifi44 | 6.77996421 | 8.9746 × 10−266 | |
| GO:0009607 | response to biotic stimulus | Mlkl | 4.440672777 | 7.6562 × 10−261 | |
| GO:0006955 | immune response | Cyba | 6.401161196 | 2.3696 × 10−247 | |
| GO:0051704 | multi-organism process | Ifit2 | 2.868875789 | 7.9652 × 10−245 | |
| GO:0009605 | response to external stimulus | Gbp3 | 4.860447879 | 4.3027 × 10−236 | |
| GO:0009615 | response to virus | Oasl2 | 7.648377266 | 2.6227 × 10−227 | |
| GO:0098542 | defense response to other organism | Irgm2 | 4.930420913 | 4.1127 × 10−205 | |
| Cluster 3 | Development | GO Description | Top Gene | logFC | adj. p-Value |
| GO:0032502 | developmental process | Slc6a17 | −3.493587034 | 1.5326 × 10−295 | |
| GO:0048869 | cellular developmental process | Kazald1 | −4.33489428 | 1.468 × 10−281 | |
| GO:0051239 | regulation of multicellular organismal process | Fam19a5 | −4.123121971 | 4.1061 × 10−272 | |
| GO:0032501 | multicellular organismal process | Plxnb1 | −4.447505569 | 3.2518 × 10−270 | |
| GO:0050793 | regulation of developmental process | Tenm3 | −7.891281718 | 2.4011 × 10−266 | |
| GO:0009653 | anatomical structure morphogenesis | Jag1 | −3.608377716 | 3.508 × 10−228 | |
| GO:2000026 | regulation of multicellular organismal development | Prdm16 | −3.568565321 | 9.015 × 10−253 | |
| GO:0051094 | positive regulation of developmental process | P2rx7 | −6.008024776 | 1.4826 × 10−187 | |
| GO:0007155 | cell adhesion | Sele | −5.033733739 | 4.4538 × 10−253 | |
| GO:0022610 | biological adhesion | Col12a1 | −3.822295006 | 9.6969 × 10−249 | |
| Cluster 4 | Gene Expression | GO Description | Top Gene | logFC | adj. p-Value |
| GO:0090304 | nucleic acid metabolic process | Set | −1.108981655 | 1.9585 × 10−289 | |
| GO:0016070 | RNA metabolic process | Ddx3x | −1.081698642 | 3.464 × 10−268 | |
| GO:0016070 | RNA metabolic process | Ddx3x | −1.081698642 | 3.464 × 10−268 | |
| GO:0006139 | nucleobase-containing compound metabolic process | Ran | −1.018798075 | 1.1 × 10−244 | |
| GO:0006396 | RNA processing | Tardbp | −1.265502477 | 5.7847 × 10−261 | |
| GO:0046483 | heterocycle metabolic process | Dhx9 | −1.062768239 | 1.0373 × 10−192 | |
| GO:0006725 | cellular aromatic compound metabolic process | Gspt1 | −1.02188382 | 1.6288 × 10−189 | |
| GO:0034641 | cellular nitrogen compound metabolic process | Mbnl1 | −0.97759175 | 6.1882 × 10−188 | |
| GO:1901360 | organic cyclic compound metabolic process | Smc1a | −1.255023232 | 1.3984 × 10−187 | |
| GO:0034660 | ncRNA metabolic process | Rpl11 | −0.834339298 | 5.4663 × 10−171 | |
| GO:0043170 | macromolecule metabolic process | Srsf3 | −1.232300645 | 7.5139 × 10−239 | |
| Cluster 5 | Cell Cycle | GO Description | Top Gene | logFC | adj. p-Value |
| GO:0007049 | cell cycle | Birc5 | −2.60656371 | 2.1503 × 10−303 | |
| GO:1903047 | mitotic cell cycle process | Tgfb1 | −1.695288695 | 5.3792 × 10−306 | |
| GO:0022402 | cell cycle process | Prc1 | −1.763009143 | 7.0517 × 10−300 | |
| GO:0006260 | DNA replication | Ccdc88a | −1.596474351 | 1.4062 × 10−302 | |
| GO:0006259 | DNA metabolic process | Nasp | −2.48862378 | 1.2227 × 10−294 | |
| GO:0051301 | cell division | Cad | −1.549902684 | 3.5803 × 10−285 | |
| GO:0051276 | chromosome organization | Cenpe | −1.775921663 | 6.4099 × 10−272 | |
| GO:0010564 | regulation of cell cycle process | Ranbp1 | −0.391960617 | 1.1679 × 10−295 | |
| GO:0051983 | regulation of chromosome segregation | Racgap1 | −1.68593893 | 8.6581 × 10−262 | |
| GO:0051726 | regulation of cell cycle | Cenpf | −2.099842197 | 1.7343 × 10−268 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kurian, J.; Bohl, V.; Behanan, M.; Mohsin, S.; Khan, M. Transcriptional Profiling of Cardiac Cells Links Age-Dependent Changes in Acetyl-CoA Signaling to Chromatin Modifications. Int. J. Mol. Sci. 2021, 22, 6987. https://doi.org/10.3390/ijms22136987
Kurian J, Bohl V, Behanan M, Mohsin S, Khan M. Transcriptional Profiling of Cardiac Cells Links Age-Dependent Changes in Acetyl-CoA Signaling to Chromatin Modifications. International Journal of Molecular Sciences. 2021; 22(13):6987. https://doi.org/10.3390/ijms22136987
Chicago/Turabian StyleKurian, Justin, Veronica Bohl, Michael Behanan, Sadia Mohsin, and Mohsin Khan. 2021. "Transcriptional Profiling of Cardiac Cells Links Age-Dependent Changes in Acetyl-CoA Signaling to Chromatin Modifications" International Journal of Molecular Sciences 22, no. 13: 6987. https://doi.org/10.3390/ijms22136987