
Bifunctional Role of CrkL during Bone Remodeling

 , , , , ,
, , , , , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
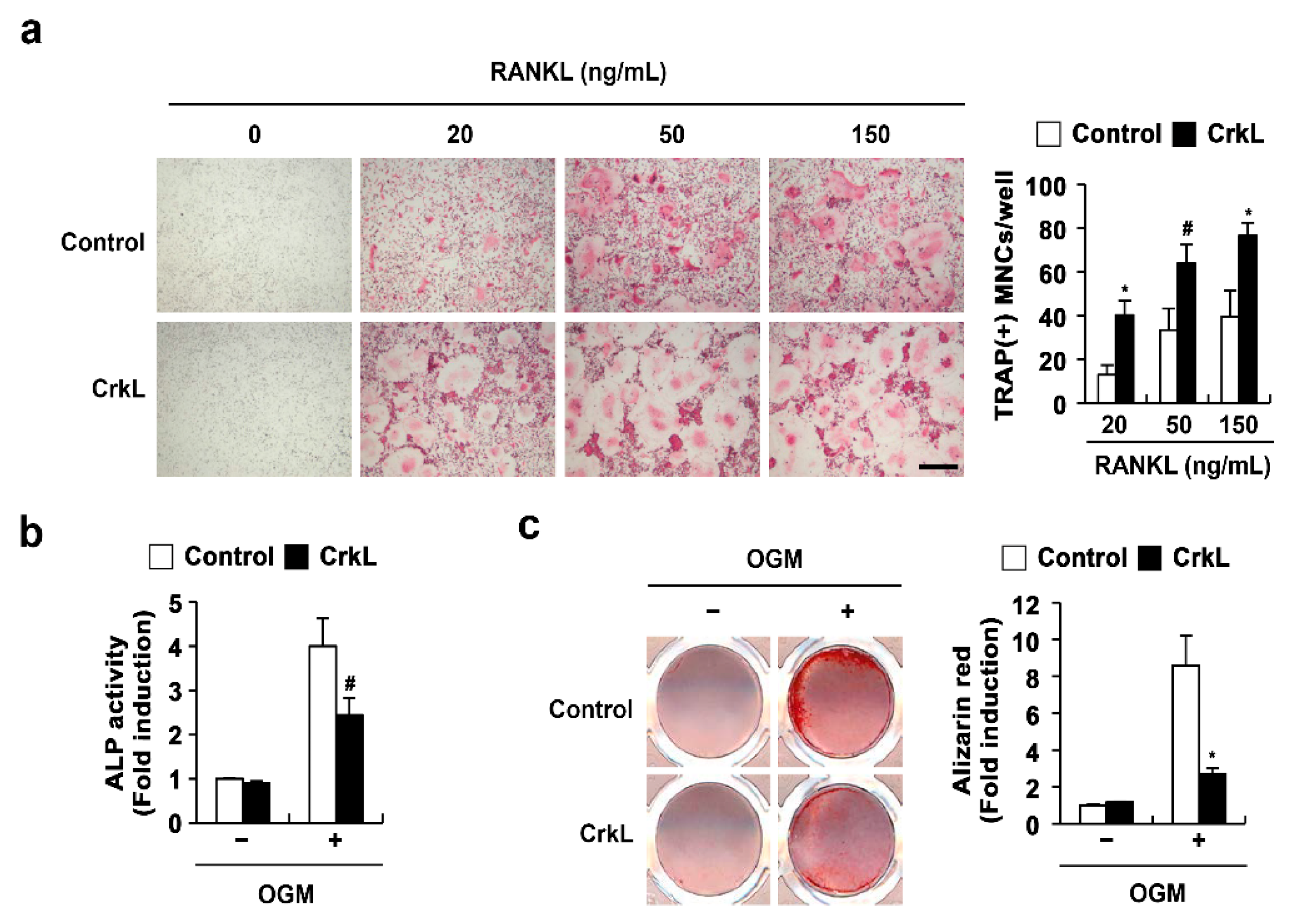
2.1. CrkL Has a Positive and Negative Effect on Osteoclast and Osteoblast Differentiation, Respectively
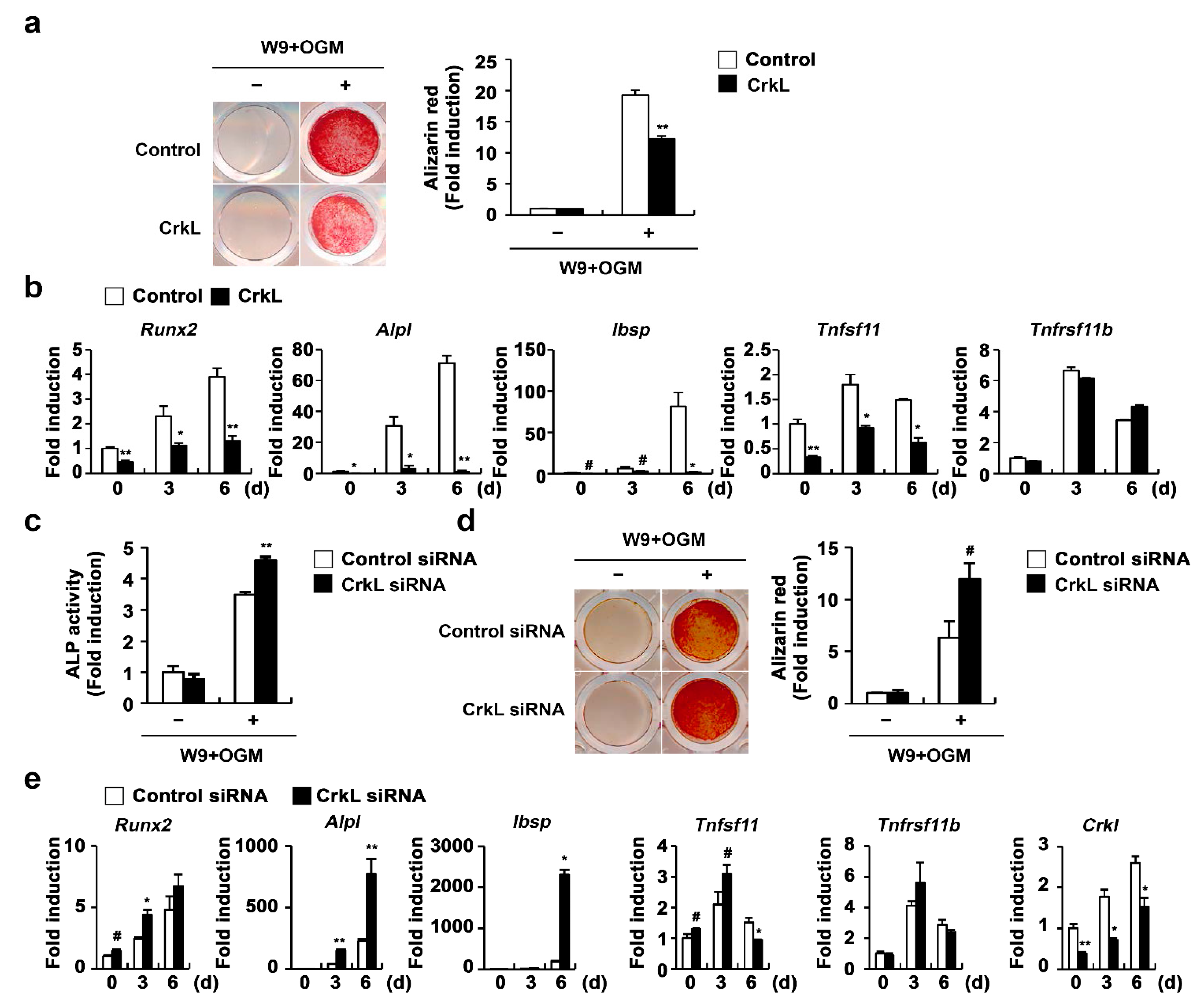
2.2. CrkL Indirectly Inhibits Osteoclast Differentiation by Regulating RANKL (Tnfsf11) Expression
2.3. CrkL Inhibits RANKL-Mediated Osteoblast Differentiation
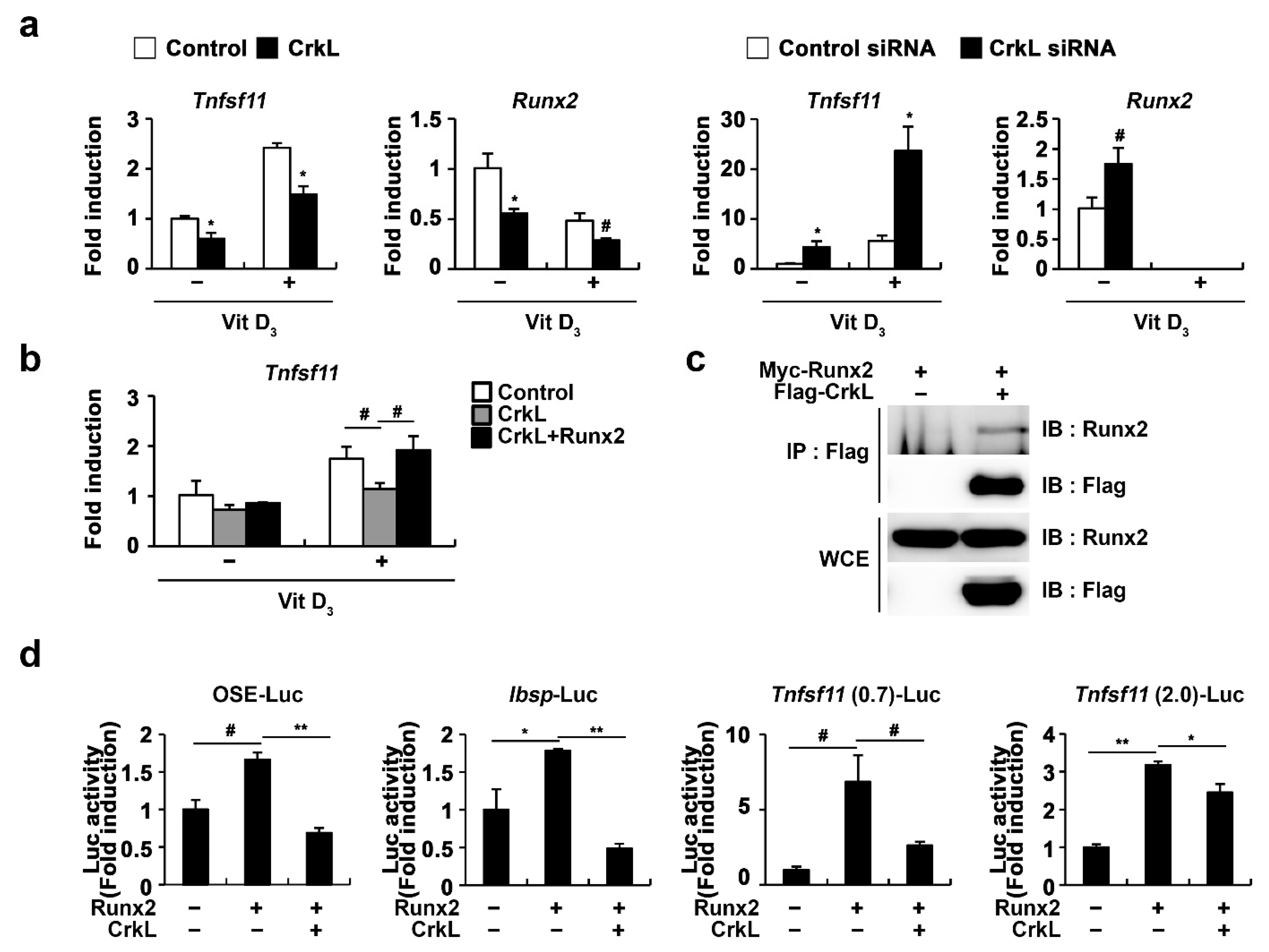
2.4. CrkL Inhibits RANKL Expression via Interaction with Runx2
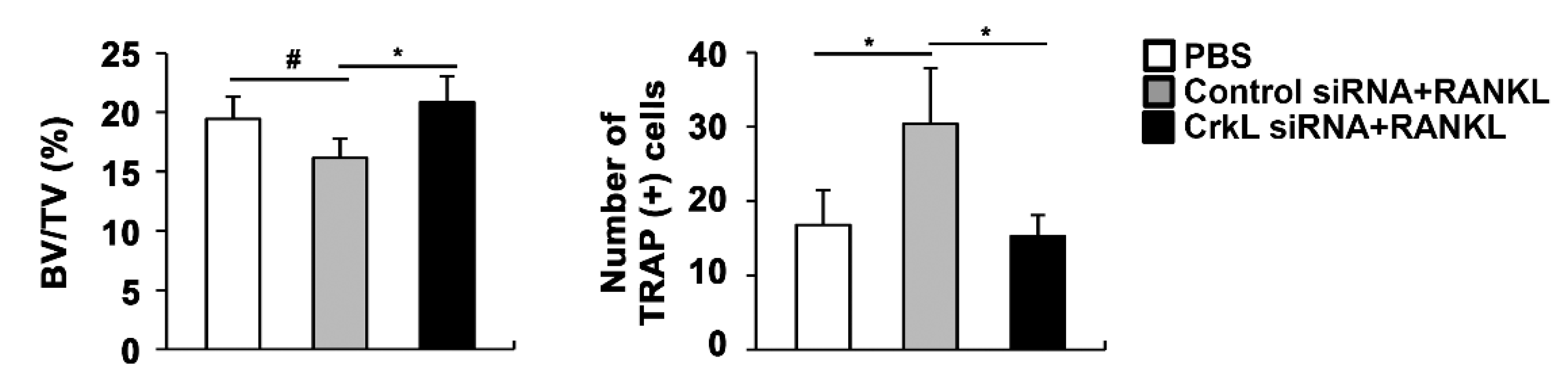
2.5. Downregulation of CrkL Can Protect RANKL-Induced Bone Loss In Vivo
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Osteoclast Differentiation
4.3. Osteoblast Differentiation
4.4. Luciferase Assay
4.5. Quantitative Real-Time PCR Analysis
4.6. Retroviral Infection
4.7. siRNA Transfection
4.8. In Vivo Experiments
4.9. Immunoprecipitation and Immunoblotting
4.10. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| ALP | alkaline phosphatase |
| BMMs | bone marrow-derived monocyte/macrophages lineage cells |
| BMP2 | bone morphogenic protein 2 |
| BSP | bone sialoprotein |
| CrkL | v-crk avian sarcoma virus CT10 oncogene homolog-like |
| M-CSF | macrophage colony-stimulating factor |
| OGM | osteogenic media |
| OPG | osteoprotegerin |
| PGE2 | prostaglandin E2 |
| RANK | receptor activator of nuclear factor kappa-B |
| RANKL | receptor activator of nuclear factor kappa-B ligand |
| Runx2 | runt-related transcription factor 2 |
| TRAP | tartrate-resistant acid phosphatase |
| Vit D3 | 1,25 (OH)2 vitamin D3 |
References
- Walsh, M.C.; Kim, N.; Kadono, Y.; Rho, J.; Lee, S.Y.; Lorenzo, J.; Choi, Y. Osteoimmunology: Interplay between the immune system and bone metabolism. Annu. Rev. Immunol. 2006, 24, 33–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cappariello, A.; Maurizi, A.; Veeriah, V.; Teti, A. The Great Beauty of the osteoclast. Arch. Biochem. Biophys. 2014, 558, 70–78. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, N. Signaling Pathways in Osteoclast Differentiation. Chonnam Med. J. 2016, 52, 12–17. [Google Scholar] [CrossRef] [Green Version]
- Kim, B.J.; Koh, J.M. Coupling factors involved in preserving bone balance. Cell. Mol. Life Sci. 2019, 76, 1243–1253. [Google Scholar] [CrossRef]
- Rodan, G.A.; Martin, T.J. Role of osteoblasts in hormonal control of bone resorption—A hypothesis. Calcif. Tissue Int. 1981, 33, 349–351. [Google Scholar] [CrossRef] [PubMed]
- Tamma, R.; Zallone, A. Osteoblast and osteoclast crosstalks: From OAF to Ephrin. Inflamm. Allergy Drug Targets 2012, 11, 196–200. [Google Scholar] [CrossRef]
- Mundy, G.R.; Eleftesriou, F. Boning up on ephrin signaling. Cell 2006, 126, 441–443. [Google Scholar] [CrossRef] [Green Version]
- Sambandam, Y.; Blanchard, J.J.; Daughtridge, G.; Kolb, R.J.; Shanmugarajan, S.; Pandruvada, S.N.; Bateman, T.A.; Reddy, S.V. Microarray profile of gene expression during osteoclast differentiation in modelled microgravity. J. Cell. Biochem. 2010, 111, 1179–1187. [Google Scholar] [CrossRef]
- Anderson, D.M.; Maraskovsky, E.; Billingsley, W.L.; Dougall, W.C.; Tometsko, M.E.; Roux, E.R.; Teepe, M.C.; DuBose, R.F.; Cosman, D.; Galibert, L. A homologue of the TNF receptor and its ligand enhance T-cell growth and dendritic-cell function. Nature 1997, 390, 175–179. [Google Scholar] [CrossRef] [PubMed]
- Wong, B.R.; Rho, J.; Arron, J.; Robinson, E.; Orlinick, J.; Chao, M.; Kalachikov, S.; Cayani, E.; Bartlett, F.S., 3rd; Frankel, W.N.; et al. TRANCE is a novel ligand of the tumor necrosis factor receptor family that activates c-Jun N-terminal kinase in T cells. J. Biol. Chem. 1997, 272, 25190–25194. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Liu, S.; Zhao, Y.; Liu, D.; Liu, Y.; Chen, C.; Karray, S.; Shi, S.; Jin, Y. Osteoblast-induced osteoclast apoptosis by fas ligand/FAS pathway is required for maintenance of bone mass. Cell Death Differ. 2015, 22, 1654–1664. [Google Scholar] [CrossRef]
- Matsuoka, K.; Park, K.A.; Ito, M.; Ikeda, K.; Takeshita, S. Osteoclast-derived complement component 3a stimulates osteoblast differentiation. J. Bone Miner. Res. 2014, 29, 1522–1530. [Google Scholar] [CrossRef]
- Negishi-Koga, T.; Shinohara, M.; Komatsu, N.; Bito, H.; Kodama, T.; Friedel, R.H.; Takayanagi, H. Suppression of bone formation by osteoclastic expression of semaphorin 4D. Nat. Med. 2011, 17, 1473–1480. [Google Scholar] [CrossRef]
- Zhang, Y.; Wei, L.; Miron, R.J.; Shi, B.; Bian, Z. Anabolic bone formation via a site-specific bone-targeting delivery system by interfering with semaphorin 4D expression. J. Bone Miner. Res. 2015, 30, 286–296. [Google Scholar] [CrossRef]
- Chen, X.; Wang, Z.; Duan, N.; Zhu, G.; Schwarz, E.M.; Xie, C. Osteoblast-osteoclast interactions. Connect. Tissue Res. 2018, 59, 99–107. [Google Scholar] [CrossRef] [PubMed]
- Yuan, F.L.; Wu, Q.Y.; Miao, Z.N.; Xu, M.H.; Xu, R.S.; Jiang, D.L.; Ye, J.X.; Chen, F.H.; Zhao, M.D.; Wang, H.J.; et al. Osteoclast-Derived Extracellular Vesicles: Novel Regulators of Osteoclastogenesis and Osteoclast-Osteoblasts Communication in Bone Remodeling. Front. Physiol. 2018, 9, 628. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sims, N.A.; Martin, T.J. Coupling Signals between the Osteoclast and Osteoblast: How are Messages Transmitted between These Temporary Visitors to the Bone Surface? Front. Endocrinol. 2015, 6, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, B.; Quan, Q.; Li, Y.; Qiu, H.; Peng, J.; Gu, Y. Treatment of Osteoporosis, with a Focus on 2 Monoclonal Antibodies. Med Sci. Monit. 2018, 24, 8758–8766. [Google Scholar] [CrossRef] [PubMed]
- Qaseem, A.; Forciea, M.A.; McLean, R.M.; Denberg, T.D. Treatment of Low Bone Density or Osteoporosis to Prevent Fractures in Men and Women: A Clinical Practice Guideline Update From the American College of Physicians. Ann. Intern. Med. 2017, 166, 818–839. [Google Scholar] [CrossRef] [PubMed]
- Cotts, K.G.; Cifu, A.S. Treatment of Osteoporosis. JAMA 2018, 319, 1040–1041. [Google Scholar] [CrossRef]
- Rachner, T.D.; Khosla, S.; Hofbauer, L.C. Osteoporosis: Now and the future. Lancet 2011, 377, 1276–1287. [Google Scholar] [CrossRef] [Green Version]
- Cosman, F.; de Beur, S.J.; LeBoff, M.S.; Lewiecki, E.M.; Tanner, B.; Randall, S.; Lindsay, R. Clinician’s Guide to Prevention and Treatment of Osteoporosis. Osteoporos Int. 2014, 25, 2359–2381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shigeno-Nakazawa, Y.; Kasai, T.; Ki, S.; Kostyanovskaya, E.; Pawlak, J.; Yamagishi, J.; Okimoto, N.; Taiji, M.; Okada, M.; Westbrook, J.; et al. A pre-metazoan origin of the CRK gene family and co-opted signaling network. Sci. Rep. 2016, 6, 34349. [Google Scholar] [CrossRef] [Green Version]
- Roy, N.H.; Mammadli, M.; Burkhardt, J.K.; Karimi, M. CrkL is required for donor T cell migration to GvHD target organs. Oncotarget 2020, 11, 1505–1514. [Google Scholar] [CrossRef]
- Song, Q.; Yi, F.; Zhang, Y.; Li, D.K.J.; Wei, Y.; Yu, H.; Zhang, Y. CRKL regulates alternative splicing of cancer-related genes in cervical cancer samples and HeLa cell. BMC Cancer 2019, 19, 499. [Google Scholar] [CrossRef]
- Birge, R.B.; Kalodimos, C.; Inagaki, F.; Tanaka, S. Crk and CrkL adaptor proteins: Networks for physiological and pathological signaling. Cell Commun. Signal. 2009, 7, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ren, R.; Ye, Z.S.; Baltimore, D. Abl protein-tyrosine kinase selects the Crk adapter as a substrate using SH3-binding sites. Genes Dev. 1994, 8, 783–795. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakai, R.; Iwamatsu, A.; Hirano, N.; Ogawa, S.; Tanaka, T.; Mano, H.; Yazaki, Y.; Hirai, H. A novel signaling molecule, p130, forms stable complexes in vivo with v-Crk and v-Src in a tyrosine phosphorylation-dependent manner. EMBO J. 1994, 13, 3748–3756. [Google Scholar] [CrossRef] [PubMed]
- De Jong, R.; ten Hoeve, J.; Heisterkamp, N.; Groffen, J. Crkl is complexed with tyrosine-phosphorylated Cbl in Ph-positive leukemia. J. Biol. Chem. 1995, 270, 21468–21471. [Google Scholar] [CrossRef] [Green Version]
- Salgia, R.; Uemura, N.; Okuda, K.; Li, J.L.; Pisick, E.; Sattler, M.; de Jong, R.; Druker, B.; Heisterkamp, N.; Chen, L.B.; et al. CRKL links p210BCR/ABL with paxillin in chronic myelogenous leukemia cells. J. Biol. Chem. 1995, 270, 29145–29150. [Google Scholar] [CrossRef] [Green Version]
- Salgia, R.; Pisick, E.; Sattler, M.; Li, J.L.; Uemura, N.; Wong, W.K.; Burky, S.A.; Hirai, H.; Chen, L.B.; Griffin, J.D. p130CAS forms a signaling complex with the adapter protein CRKL in hematopoietic cells transformed by the BCR/ABL oncogene. J. Biol. Chem. 1996, 271, 25198–25203. [Google Scholar] [CrossRef] [Green Version]
- Ribon, V.; Hubbell, S.; Herrera, R.; Saltiel, A.R. The product of the cbl oncogene forms stable complexes in vivo with endogenous Crk in a tyrosine phosphorylation-dependent manner. Mol. Cell. Biol. 1996, 16, 45–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beitner-Johnson, D.; Blakesley, V.A.; Shen-Orr, Z.; Jimenez, M.; Stannard, B.; Wang, L.M.; Pierce, J.; LeRoith, D. The proto-oncogene product c-Crk associates with insulin receptor substrate-1 and 4PS. Modulation by insulin growth factor-I (IGF) and enhanced IGF-I signaling. J. Biol. Chem. 1996, 271, 9287–9290. [Google Scholar] [CrossRef] [Green Version]
- Sattler, M.; Salgia, R.; Okuda, K.; Uemura, N.; Durstin, M.A.; Pisick, E.; Xu, G.; Li, J.L.; Prasad, K.V.; Griffin, J.D. The proto-oncogene product p120CBL and the adaptor proteins CRKL and c-CRK link c-ABL, p190BCR/ABL and p210BCR/ABL to the phosphatidylinositol-3’ kinase pathway. Oncogene 1996, 12, 839–846. [Google Scholar] [PubMed]
- Akagi, T.; Shishido, T.; Murata, K.; Hanafusa, H. v-Crk activates the phosphoinositide 3-kinase/AKT pathway in transformation. Proc. Natl. Acad. Sci. USA 2000, 97, 7290–7295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sattler, M.; Salgia, R.; Shrikhande, G.; Verma, S.; Uemura, N.; Law, S.F.; Golemis, E.A.; Griffin, J.D. Differential signaling after beta1 integrin ligation is mediated through binding of CRKL to p120(CBL) and p110(HEF1). J. Biol. Chem. 1997, 272, 14320–14326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sattler, M.; Salgia, R.; Shrikhande, G.; Verma, S.; Pisick, E.; Prasad, K.V.; Griffin, J.D. Steel factor induces tyrosine phosphorylation of CRKL and binding of CRKL to a complex containing c-kit, phosphatidylinositol 3-kinase, and p120(CBL). J. Biol. Chem. 1997, 272, 10248–10253. [Google Scholar] [CrossRef] [Green Version]
- Gesbert, F.; Garbay, C.; Bertoglio, J. Interleukin-2 stimulation induces tyrosine phosphorylation of p120-Cbl and CrkL and formation of multimolecular signaling complexes in T lymphocytes and natural killer cells. J. Biol. Chem. 1998, 273, 3986–3993. [Google Scholar] [CrossRef] [Green Version]
- Koval, A.P.; Karas, M.; Zick, Y.; LeRoith, D. Interplay of the proto-oncogene proteins CrkL and CrkII in insulin-like growth factor-I receptor-mediated signal transduction. J. Biol. Chem. 1998, 273, 14780–14787. [Google Scholar] [CrossRef] [Green Version]
- Fish, E.N.; Uddin, S.; Korkmaz, M.; Majchrzak, B.; Druker, B.J.; Platanias, L.C. Activation of a CrkL-stat5 signaling complex by type I interferons. J. Biol. Chem. 1999, 274, 571–573. [Google Scholar] [CrossRef] [Green Version]
- Kim, J.H.; Kim, K.; Kim, I.; Seong, S.; Nam, K.I.; Kim, K.K.; Kim, N. Adaptor protein CrkII negatively regulates osteoblast differentiation and function through JNK phosphorylation. Exp. Mol. Med. 2019, 51, 1–10. [Google Scholar] [CrossRef]
- Kim, J.H.; Kim, K.; Kim, I.; Seong, S.; Nam, K.I.; Lee, S.H.; Kim, K.K.; Kim, N. Role of CrkII Signaling in RANKL-Induced Osteoclast Differentiation and Function. J. Immunol. 2016, 196, 1123–1131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guris, D.L.; Fantes, J.; Tara, D.; Druker, B.J.; Imamoto, A. Mice lacking the homologue of the human 22q11.2 gene CRKL phenocopy neurocristopathies of DiGeorge syndrome. Nat. Genet. 2001, 27, 293–298. [Google Scholar] [CrossRef] [PubMed]
- Park, T.J.; Boyd, K.; Curran, T. Cardiovascular and craniofacial defects in Crk-null mice. Mol. Cell Biol. 2006, 26, 6272–6282. [Google Scholar] [CrossRef] [Green Version]
- Ikebuchi, Y.; Aoki, S.; Honma, M.; Hayashi, M.; Sugamori, Y.; Khan, M.; Kariya, Y.; Kato, G.; Tabata, Y.; Penninger, J.M.; et al. Coupling of bone resorption and formation by RANKL reverse signalling. Nature 2018, 561, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Ozaki, Y.; Koide, M.; Furuya, Y.; Ninomiya, T.; Yasuda, H.; Nakamura, M.; Kobayashi, Y.; Takahashi, N.; Yoshinari, N.; Udagawa, N. Treatment of OPG-deficient mice with WP9QY, a RANKL-binding peptide, recovers alveolar bone loss by suppressing osteoclastogenesis and enhancing osteoblastogenesis. PLoS ONE 2017, 12, e0184904. [Google Scholar] [CrossRef] [Green Version]
- Sawa, M.; Wakitani, S.; Kamei, N.; Kotaka, S.; Adachi, N.; Ochi, M. Local administration of WP9QY (W9) peptide promotes bone formation in a rat femur delayed-union model. J. Bone Miner. Metab. 2018, 36, 383–391. [Google Scholar] [CrossRef]
- Otsuki, Y.; Ii, M.; Moriwaki, K.; Okada, M.; Ueda, K.; Asahi, M. W9 peptide enhanced osteogenic differentiation of human adipose-derived stem cells. Biochem. Biophys. Res. Commun. 2018, 495, 904–910. [Google Scholar] [CrossRef]
- Geoffroy, V.; Kneissel, M.; Fournier, B.; Boyde, A.; Matthias, P. High bone resorption in adult aging transgenic mice overexpressing cbfa1/runx2 in cells of the osteoblastic lineage. Mol. Cell. Biol. 2002, 22, 6222–6233. [Google Scholar] [CrossRef] [Green Version]
- Enomoto, H.; Shiojiri, S.; Hoshi, K.; Furuichi, T.; Fukuyama, R.; Yoshida, C.A.; Kanatani, N.; Nakamura, R.; Mizuno, A.; Zanma, A.; et al. Induction of osteoclast differentiation by Runx2 through receptor activator of nuclear factor-kappa B ligand (RANKL) and osteoprotegerin regulation and partial rescue of osteoclastogenesis in Runx2−/− mice by RANKL transgene. J. Biol. Chem. 2003, 278, 23971–23977. [Google Scholar] [CrossRef] [Green Version]
- Yahiro, Y.; Maeda, S.; Morikawa, M.; Koinuma, D.; Jokoji, G.; Ijuin, T.; Komiya, S.; Kageyama, R.; Miyazono, K.; Taniguchi, N. BMP-induced Atoh8 attenuates osteoclastogenesis by suppressing Runx2 transcriptional activity and reducing the Rankl/Opg expression ratio in osteoblasts. Bone Res. 2020, 8, 32. [Google Scholar] [CrossRef] [PubMed]
- Byon, C.H.; Sun, Y.; Chen, J.; Yuan, K.; Mao, X.; Heath, J.M.; Anderson, P.G.; Tintut, Y.; Demer, L.L.; Wang, D.; et al. Runx2-upregulated receptor activator of nuclear factor κB ligand in calcifying smooth muscle cells promotes migration and osteoclastic differentiation of macrophages. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 1387–1396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Komori, T.; Yagi, H.; Nomura, S.; Yamaguchi, A.; Sasaki, K.; Deguchi, K.; Shimizu, Y.; Bronson, R.T.; Gao, Y.H.; Inada, M.; et al. Targeted disruption of Cbfa1 results in a complete lack of bone formation owing to maturational arrest of osteoblasts. Cell 1997, 89, 755–764. [Google Scholar] [CrossRef] [Green Version]
- Fakhry, M.; Hamade, E.; Badran, B.; Buchet, R.; Magne, D. Molecular mechanisms of mesenchymal stem cell differentiation towards osteoblasts. World J. Stem Cells 2013, 5, 136–148. [Google Scholar] [CrossRef]
- Mori, K.; Kitazawa, R.; Kondo, T.; Maeda, S.; Yamaguchi, A.; Kitazawa, S. Modulation of mouse RANKL gene expression by Runx2 and PKA pathway. J. Cell. Biochem. 2006, 98, 1629–1644. [Google Scholar] [CrossRef] [PubMed]
- O’Brien, C.A.; Kern, B.; Gubrij, I.; Karsenty, G.; Manolagas, S.C. Cbfa1 does not regulate RANKL gene activity in stromal/osteoblastic cells. Bone 2002, 30, 453–462. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.H.; Kim, K.; Kim, I.; Seong, S.; Kook, H.; Kim, K.K.; Koh, J.-T.; Kim, N. Bifunctional Role of CrkL during Bone Remodeling. Int. J. Mol. Sci. 2021, 22, 7007. https://doi.org/10.3390/ijms22137007
Kim JH, Kim K, Kim I, Seong S, Kook H, Kim KK, Koh J-T, Kim N. Bifunctional Role of CrkL during Bone Remodeling. International Journal of Molecular Sciences. 2021; 22(13):7007. https://doi.org/10.3390/ijms22137007
Chicago/Turabian StyleKim, Jung Ha, Kabsun Kim, Inyoung Kim, Semun Seong, Hyun Kook, Kyung Keun Kim, Jeong-Tae Koh, and Nacksung Kim. 2021. "Bifunctional Role of CrkL during Bone Remodeling" International Journal of Molecular Sciences 22, no. 13: 7007. https://doi.org/10.3390/ijms22137007