
Nutraceutical and Probiotic Approaches to Examine Molecular Interactions of the Amyloid Precursor Protein APP in Drosophila Models of Alzheimer’s Disease


Abstract
:1. Introduction
2. AD Symptoms, Progression and Diagnosis of Neuropathology
3. AD Pathogenesis and Amyloidogenic APP Processing
4. APP Genetic and Molecular Interactions
5. Recent Research on APP: Insights from Drosophila
6. The Use of Nutraceuticals as Promising Treatment Options
7. Concluding Thoughts
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Masters, C.L.; Bateman, R.; Blennow, K.; Rowe, C.C.; Sperling, R.A.; Cummings, J.L. Alzheimer’s disease. Nat. Rev. Dis. Primers. 2015, 1, 15056. [Google Scholar] [CrossRef] [PubMed]
- Avila, J.; Perry, G. A Multilevel View of the Development of Alzheimer’s Disease. Neuroscience 2021, 457, 283–293. [Google Scholar] [CrossRef] [PubMed]
- Livingston, G.; Huntley, J.; Sommerlad, A.; Ames, D.; Ballard, C.; Banerjee, S.; Brayne, C.; Burns, A.; Cohen-Mansfield, J.; Cooper, C.; et al. Dementia prevention, intervention, and care: 2020 report of the Lancet Commission. Lancet 2020, 396, 413–446. [Google Scholar] [CrossRef]
- Fink, H.A.; Linskens, E.J.; MacDonald, R.; Silverman, P.C.; McCarten, J.R.; Talley, K.M.; Forte, M.L.; Desai, P.J.; Nelson, V.A.; Miller, M.A.; et al. Benefits and harms of prescription drugs and supplements for treatment of clinical Alzheimer-type dementia: A systematic review and meta-analysis. Ann. Intern. Med. 2020, 172, 656–668. [Google Scholar] [CrossRef] [PubMed]
- Murray, M.E.; Graff-Radford, N.R.; Ross, O.A.; Petersen, R.C.; Duara, R.; Dickson, D.W. Neuropathologically defined subtypes of Alzheimer’s disease with distinct clinical characteristics: A retrospective study. Lancet Neurol. 2011, 10, 785–796. [Google Scholar] [CrossRef] [Green Version]
- Querfurth, H.; LaFerla, F. Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 1844–1845. [Google Scholar] [CrossRef] [Green Version]
- Glenner, G.G.; Wong, C.W. Alzheimer’s disease: Initial report of the purification and characterization of a novel cerebrovascular amyloid protein. Biochem. Biophys. Res. Commun. 1984, 120, 885–890. [Google Scholar] [CrossRef]
- Brion, J.P.; Passareiro, H.; Nunez, J.; Flament-Durand, J. Mise en évidence immunologique de la protéine tau au niveau des lésions de dégénérescence neurofibrillaire de la maladie d’Alzheimer. Arch. Biol. 1985, 95, 229–235. [Google Scholar]
- Rinne, J.O.; Rummukainen, J.; Paljärvi, L.; Säkö, E.; Mölsä, P.; Rinne, U.K. Neuronal loss in the substantia nigra in patients with Alzheimer’s disease and Parkinson’s disease in relation to extrapyramidal symptoms and dementia. Prog. Clin. Biol. Res. 1989, 317, 325–332. [Google Scholar]
- Bondareff, W.; Mountjoy, C.; Roth, M.; Hauser, D. Neurofibrillary degeneration and neuronal loss in alzheimer’s disease. Neurobiol. Aging 1989, 10, 709–715. [Google Scholar] [CrossRef]
- Braak, H.; Braak, E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol. 1991, 82, 239–259. [Google Scholar] [CrossRef]
- Braak, H.; Alafuzoff, I.; Arzberger, T.; Kretzschmar, H.; Del Tredici, K. Staging of Alzheimer disease-associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006, 112, 389–404. [Google Scholar] [CrossRef] [Green Version]
- Ohm, T.; Müller, H.; Braak, H.; Bohl, J. Close-meshed prevalence rates of different stages as a tool to uncover the rate of Alzheimer’s disease-related neurofibrillary changes. Neuroscience 1995, 64, 209–217. [Google Scholar] [CrossRef]
- Hardy, J. A hundred years of Alzheimer’s disease research. Neuron 2006, 52, 3–13. [Google Scholar] [CrossRef] [Green Version]
- Jelicic, M.; Bonebakker, A.E.; Bonke, B. Implicit memory performance of patients with Alzheimer’s disease: A brief review. Int. Psychogeriatr. 1995, 7, 385–392. [Google Scholar] [CrossRef] [PubMed]
- Förstl, H.; Kurz, A. Clinical features of Alzheimer’s disease. Eur. Arch. Psychiatry Clin. Neurosci. 1999, 249, 288–290. [Google Scholar] [CrossRef] [PubMed]
- Bäckman, L.; Jones, S.; Berger, A.K.; Laukka, E.J.; Small, B.J. Multiple cognitive deficits during the transition to Alzheimer’s disease. J. Intern. Med. 2004, 256, 195–204. [Google Scholar] [CrossRef]
- Gold, D.P.; Reis, M.F.; Markiewicz, D.; Andres, D. When Home Caregiving Ends: A Longitudinal Study of Outcomes for Caregivers of Relatives with Dementia. J. Am. Geriatr. Soc. 1995, 43, 10–16. [Google Scholar] [CrossRef]
- Quertinmount, K.; Rizzo, B.; Swann, C.; Coram, L.; Klein, M.; Detillion, W.N. A Novel Approach to Treating Alzheimer’s Disease. Pharm. Wellness Rev. 2010, 1, 17–18. [Google Scholar]
- Jaul, E.; Meiron, O. Dementia and Pressure Ulcers: Is There a Close Pathophysiological Interrelation? J. Alzheimer’s Dis. 2017, 56, 861–866. [Google Scholar] [CrossRef]
- Dubois, B.; Hampel, H.; Feldman, H.H.; Scheltens, P.; Aisen, P.; Andrieu, S.; Bakardjian, H.; Benali, H.; Bertram, L.; Blennow, K.; et al. Preclinical Alzheimer’s disease: Definition, natural history, and diagnostic criteria. Alzheimer’s Dement. J. Alzheimer’s Assoc. 2016, 12, 292–323. [Google Scholar] [CrossRef] [PubMed]
- Bateman, R.J.; Xiong, C.; Benzinger, T.L.; Fagan, A.M.; Goate, A.; Fox, N.C.; Marcus, D.S.; Cairns, N.J.; Xie, X.; Blazey, T.M.; et al. Clinical and Biomarker Changes in Dominantly Inherited Alzheimer’s Disease. N. Engl. J. Med. 2012, 367, 795–804. [Google Scholar] [CrossRef] [Green Version]
- Fagan, A.M.; Xiong, C.; Jasielec, M.S.; Bateman, R.J.; Goate, A.; Benzinger, T.; Ghetti, B.; Martins, R.; Masters, C.L.; Mayeux, R.; et al. Longitudinal Change in CSF Biomarkers in Autosomal-Dominant Alzheimer’s Disease. Sci. Transl. Med. 2014, 6, 226ra30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reiman, E.M.; Langbaum, J.B.; Fleisher, A.S.; Caselli, R.J.; Chen, K.; Ayutyanont, N.; Quiroz, Y.T.; Kosik, K.S.; Lopera, F.; Tariot, P.N. Alzheimer’s Prevention Initiative: A Plan to Accelerate the Evaluation of Presymptomatic Treatments. J. Alzheimer’s Dis. 2011, 26, 321–329. [Google Scholar] [CrossRef] [PubMed]
- Langbaum, J.B.; Ellison, N.N.; Caputo, A.; Thomas, R.G.; Langlois, C.; Riviere, M.-E.; Graf, A.; Lopez, C.L.; Reiman, E.M.; Tariot, P.N.; et al. The Alzheimer’s Prevention Initiative Composite Cognitive Test: A practical measure for tracking cognitive decline in preclinical Alzheimer’s disease. Alzheimer’s Res. Ther. 2020, 12, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Arendt, T.; Stieler, J.; Ueberham, U. Is sporadic Alzheimer’s disease a developmental disorder? J. Neurochem. 2017, 143, 396–408. [Google Scholar] [CrossRef] [Green Version]
- Penner, G.; Lecocq, S.; Chopin, A.; Vedoya, X.; Lista, S.; Vergallo, A.; Lejeune, F.X.; Hampel, H. Blood-based diagnostics of Alzheimer’s disease. Expert Rev. Mol. Diagn. 2019, 19, 613–616. [Google Scholar] [CrossRef] [PubMed]
- Wenk, G.L. Neuropathologic changes in Alzheimer’s disease. J. Clin. Psychiatry 2003, 64, 7–10. [Google Scholar]
- Schroeter, M.L.; Stein, T.; Maslowski, N.; Neumann, J. Neural correlates of Alzheimer’s disease and mild cognitive impairment: A systematic and quantitative meta-analysis involving 1351 patients. NeuroImage 2009, 47, 1196–1206. [Google Scholar] [CrossRef] [Green Version]
- Yeo, J.M.; Waddell, B.; Khan, Z.; Pal, S. A systematic review and meta-analysis of (18)F-labeled amyloid imaging in Alzheimer’s disease. Alzheimer’s Dement. 2015, 1, 5–13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barthel, H. First Tau PET Tracer Approved: Toward Accurate In Vivo Diagnosis of Alzheimer Disease. J. Nucl. Med. 2020, 61, 1409–1410. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Bennett, D.A.; Blennow, K.; Carrillo, M.C.; Dunn, B.; Haeberlein, S.B.; Holtzman, D.M.; Jagust, W.; Jessen, F.; Karlawish, J.; et al. NIA-AA research framework: Toward a biological definition of Alzheimer’s disease. Alzheimers Dement. 2018, 14, 535–562. [Google Scholar] [CrossRef]
- Zhang, X.; Alshakhshir, N.; Zhao, L. Glycolytic Metabolism, Brain Resilience, and Alzheimer’s Disease. Front. Neurosci. 2021, 15. [Google Scholar] [CrossRef] [PubMed]
- Costantini, L.C.; Barr, L.J.; Vogel, J.L.; Henderson, S.T. Hypometabolism as a therapeutic target in Alzheimer’s disease. BMC Neurosci. 2008, 9, S16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ou, Y.N.; Xu, W.; Li, J.Q.; Guo, Y.; Cui, M.; Chen, K.-L.; Huang, Y.-Y.; Dong, Q.; Tan, L.; Yu, J.-T.; et al. FDG-PET as an independent biomarker for Alzheimer’s biological diagnosis: A longitudinal study. Alz Res Therapy. 2019, 11, 57. [Google Scholar] [CrossRef] [Green Version]
- Sims, N.; Smith, C.; Davison, A.; Bowen, D.; Flack, R.; Snowden, J.; Neary, D. Glucose metabolism and acetylcholine synthesis in relation to neuronal activity in alzheimer’s disease. Lancet 1980, 315, 333–336. [Google Scholar] [CrossRef]
- Iwangoff, P.; Armbruster, R.; Enz, A.; Meier-Ruge, W. Glycolytic enzymes from human autoptic brain cortex: Normal aged and demented cases. Mech. Ageing Dev. 1980, 14, 203–209. [Google Scholar] [CrossRef]
- Hoyer, S. Causes and Consequences of Disturbances of Cerebral Glucose Metabolism in Sporadic Alzheimer Disease: Therapeutic Implications. Adv. Exp. Med. Biol. 2004, 541, 135–152. [Google Scholar] [CrossRef]
- De La Monte, S.M.; Wands, J.R. Alzheimer’s Disease is Type 3 Diabetes—Evidence Reviewed. J. Diabetes Sci. Technol. 2008, 2, 1101–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fyfe, I. Brain organoids shed light on APOE genotype and Alzheimer disease pathology. Nat. Rev. Neurol. 2021, 17, 1. [Google Scholar] [CrossRef]
- Papaspyropoulos, A.; Tsolaki, M.; Foroglou, N.; Pantazaki, A.A. Modeling and Targeting Alzheimer’s Disease With Organoids. Front. Pharmacol. 2020, 11, 396. [Google Scholar] [CrossRef] [PubMed]
- Venkataraman, L.; Fair, S.R.; McElroy, C.A.; Hester, M.E.; Fu, H. Modeling neurodegenerative diseases with cerebral organoids and other three-dimensional culture systems: Focus on Alzheimer’s disease. Stem Cell Rev. Rep. 2020, 1–22. [Google Scholar] [CrossRef] [PubMed]
- Alzheimer, A.; Stelzmann, R.A.; Schnitzlein, H.N.; Murtagh, F.R. An English translatio of Alzheimer’s 1907 paper, “Uber eine eigenartige Erkankung der Hirnrinde”. Clin Anat. 1995, 8, 429–431. [Google Scholar] [PubMed]
- Bloom, G.S. Amyloid-β and tau: The trigger and bullet in Alzheimer disease pathogenesis. JAMA Neurol. 2014, 71, 505–508. [Google Scholar] [CrossRef] [Green Version]
- Goedert, M.; Wischik, C.M.; Crowther, R.A.; Walker, J.; Klug, A. Cloning and sequencing of the cDNA encoding a core protein of the paired helical filament of Alzheimer disease: Identification as the microtubule-associated protein tau. Proc. Natl. Acad. Sci. USA 1988, 85, 4051–4055. [Google Scholar] [CrossRef] [Green Version]
- Lei, P.; Ayton, S.; Finkelstein, D.I.; Adlard, P.A.; Masters, C.L.; Bush, A.I. Tau protein: Relevance to Parkinson’s disease. Int. J. Biochem. Cell Biol. 2010, 42, 1775–1778. [Google Scholar] [CrossRef]
- Jack, C.R., Jr.; Petersen, R.C.; Xu, Y.C.; O’Brien, P.C.; Smith, G.E.; Ivnik, R.J.; Boeve, B.F.; Waring, S.C.; Tangalos, E.G.; Kokmen, E. Prediction of AD with MRI-based hippocampal volume in mild cognitive impairment. Neurology 1999, 52, 1397–1403. [Google Scholar] [CrossRef] [Green Version]
- DeTure, M.A.; Dickson, D.W. The neuropathological diagnosis of Alzheimer’s disease. Mol. Neurodegener. 2019, 14, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Subramanian, J.; Savage, J.C.; Tremblay, M.-E. Synaptic Loss in Alzheimer’s Disease: Mechanistic Insights Provided by Two-Photon in vivo Imaging of Transgenic Mouse Models. Front. Cell. Neurosci. 2020, 14. [Google Scholar] [CrossRef]
- Meyer-Luehmann, M.; Spires-Jones, T.; Prada, C.; Garcia-Alloza, M.; De Calignon, A.; Rozkalne, A.; Koenigsknecht-Talboo, J.; Holtzman, D.M.; Bacskai, B.J.; Hyman, B.T. Rapid appearance and local toxicity of amyloid-β plaques in a mouse model of Alzheimer’s disease. Nat. Cell Biol. 2008, 451, 720–724. [Google Scholar] [CrossRef] [Green Version]
- Blazquez-Llorca, L.; Valero-Freitag, S.; Rodrigues, E.F.; Merchán-Pérez, Á.; Rodriguez, J.-R.; Dorostkar, M.M.; DeFelipe, J.; Herms, J. High plasticity of axonal pathology in Alzheimer’s disease mouse models. Acta Neuropathol. Commun. 2017, 5, 14. [Google Scholar] [CrossRef] [Green Version]
- Peters, F.; Salihoglu, H.; Rodrigues, E.; Herzog, E.; Blume, T.; Filser, S.; Dorostkar, M.; Shimshek, D.R.; Brose, N.; Neumann, U.; et al. BACE1 inhibition more effectively suppresses initiation than progression of β-amyloid pathology. Acta Neuropathol. 2018, 135, 695–710. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Busche, M.A.; Hyman, B.T. Synergy between amyloid-β and tau in Alzheimer’s disease. Nat. Neurosci. 2020, 23, 1183–1193. [Google Scholar] [CrossRef] [PubMed]
- Vilchez, D.; Saez, I.; Dillin, A. The role of protein clearance mechanisms in organismal ageing and age-related diseases. Nat. Commun. 2014, 5, 5659. [Google Scholar] [CrossRef]
- Turner, P.R.; O’Connor, K.; Tate, W.P.; Abraham, W.C. Roles of amyloid precursor protein and its fragments in regulating neural activity, plasticity and memory. Prog. Neurobiol. 2003, 70, 1–32. [Google Scholar] [CrossRef]
- Muller-Hill, B.; Beyreuther, K. Molecular biology ofAlzheimer’s disease. Annu. Rev. Biochem. 1989, 58, 287–307. [Google Scholar] [CrossRef]
- Priller, C.; Bauer, T.; Mitteregger, G.; Krebs, B.; Kretzschmar, H.A.; Herms, J. Synapse Formation and Function Is Modulated by the Amyloid Precursor Protein. J. Neurosci. 2006, 26, 7212–7221. [Google Scholar] [CrossRef]
- Zheng, H.; Koo, E.H. The amyloid precursor protein: Beyond amyloid. Mol. Neurodegener. 2006, 1, 5. [Google Scholar] [CrossRef] [Green Version]
- Dawkins, E.; Small, D.H. Insights into the physiological function of the β-amyloid precursor protein: Beyond Alzheimer’s disease. J. Neurochem. 2014, 129, 756–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li Puma, D.D.; Piacentini, R.; Grassi, C. Does Impairment of Adult Neurogenesis Contribute to Pathophysiology of Alzheimer’s Disease? A Still Open Question. Front. Mol. Neurosci. 2021, 13. [Google Scholar] [CrossRef]
- Porayette, P.; Gallego, M.J.; Kaltcheva, M.M.; Meethal, S.V.; Atwood, C.S. Amyloid-β precursor protein expression and modulation in human embryonic stem cells: A novel role for human chorionic gonadotropin. Biochem. Biophys. Res. Commun. 2007, 364, 522–527. [Google Scholar] [CrossRef]
- Lamb, B.T.; Sisodia, S.S.; Lawler, A.M.; Slunt, H.H.; Kitt, C.A.; Kearns, W.G.; Pearson, P.L.; Price, D.L.; Gearhart, J.D. Introduction and expression of the 400 kilobase precursor amyloid protein gene in transgenic mice. Nat. Genet. 1993, 5, 22–30. [Google Scholar] [CrossRef]
- Nalivaeva, N.N.; Turner, A.J. The amyloid precursor protein: A biochemical enigma in brain development, function and disease. FEBS Lett. 2013, 587, 2046–2054. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akasaka-Manya, K.; Manya, H. The Role of APP O-Glycosylation in Alzheimer’s Disease. Biomolecules 2020, 10, 1569. [Google Scholar] [CrossRef] [PubMed]
- Das, B.; Yan, R. A Close Look at BACE1 Inhibitors for Alzheimer’s Disease Treatment. CNS Drugs 2019, 33, 251–263. [Google Scholar] [CrossRef] [PubMed]
- Zhao, J.; Deng, Y.; Jiang, Z.; Qing, H. G protein-coupled receptors (GPCRs) in Alzheimer’s disease: A focus on BACE1 related GPCRs. Front. Aging Neurosci. 2016, 8, 58. [Google Scholar] [CrossRef] [Green Version]
- Nistor, M.; Don, M.; Parekh, M.; Sarsoza, F.; Goodus, M.; Lopez, G.; Kawas, C.; Leverenz, J.; Doran, E.; Lott, I.; et al. Alpha- and beta-secretase activity as a function of age and beta-amyloid in Down syndrome and normal brain. Neurobiol. Aging 2007, 28, 1493–1506. [Google Scholar] [CrossRef] [Green Version]
- Hartley, D.; Blumenthal, T.; Carrillo, M.; Di Paolo, G.; Esralew, L.; Gardiner, K.; Granholm, A.C.; Iqbal, K.; Krams, M.; Lemere, C.; et al. Down syndrome and Alzheimer’s disease: Common pathways, common goals. Alzheimer’s Dement. 2015, 11, 700–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matsui, T.; Ingelsson, M.; Fukumoto, H.; Ramasamy, K.; Kowa, H.; Frosch, M.P.; Irizarry, M.C.; Hyman, B.T. Expression of APP pathway mRNAs and proteins in Alzheimer’s disease. Brain Res. 2007, 1161, 116–123. [Google Scholar] [CrossRef]
- Haass, C.; Lemere, C.A.; Capell, A.; Citron, M.; Seubert, P.; Schenk, D.; Lannfelt, L.; Selkoe, D.J. The Swedish mutation causes early-onset Alzheimer’s disease by β-secretase cleavage within the secretory pathway. Nat. Med. 1995, 1, 1291–1296. [Google Scholar] [CrossRef]
- Li, N.-M.; Liu, K.-F.; Qiu, Y.-J.; Zhang, H.-H.; Nakanishi, H. Mutations of beta-amyloid precursor protein alter the consequence of Alzheimer’s disease pathogenesis. Neural. Regen. Res. 2019, 14, 658–665. [Google Scholar] [CrossRef] [PubMed]
- Nilsberth, C.; Westlind-Danielsson, A.; Eckman, C.B.; Condron, M.M.; Axelman, K.; Forsell, C.; Stenh, C.; Luthman, J.; Teplow, D.B.; Younkin, S.G.; et al. The ’Arctic’ APP mutation (E693G) causes Alzheimer’s disease by enhanced Aβ protofibril formation. Nat. Neurosci. 2001, 4, 887–893. [Google Scholar] [CrossRef]
- Westerman, M.A.; Cooper-Blacketer, D.; Mariash, A.; Kotilinek, L.; Kawarabayashi, T.; Younkin, L.H.; Carlson, G.A.; Younkin, S.G.; Ashe, K.H. The relationship between Aβ and memory in the Tg2576 mouse model of Alzheimer’s disease. J. Neurosci. 2002, 22, 1858–1867. [Google Scholar] [CrossRef] [Green Version]
- Jonsson, T.; Atwal, J.K.; Steinberg, S.; Snaedal, J.; Jonsson, P.V.; Bjornsson, S.; Stefansson, H.; Sulem, P.; Gudbjartsson, D.F.; Maloney, J.; et al. A mutation in APP protects against Alzheimer’s disease and age-related cognitive decline. Nature 2012, 488, 96–99. [Google Scholar] [CrossRef]
- Kero, M.; Paetau, A.; Polvikoski, T.; Tanskanen, M.; Sulkava, R.; Jansson, L.; Myllykangas, L.; Tienari, P.J. Amyloid precursor protein (APP) A673T mutation in the elderly Finnish population. Neurobiol. Aging 2013, 34, 1518.e1–1518.e3. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.S.; Naj, A.C.; Graham, R.R.; Crane, P.K.; Kunkle, B.W.; Cruchaga, C.; Murcia, J.D.G.; Cannon-Albright, L.; Baldwin, C.T.; Zetterberg, H.; et al. Rarity of the Alzheimer Disease–Protective APP A673T Variant in the United States. JAMA Neurol. 2015, 72, 209–216. [Google Scholar] [CrossRef] [Green Version]
- Maloney, J.A.; Bainbridge, T.; Gustafson, A.; Zhang, S.; Kyauk, R.; Steiner, P.; van der Brug, M.; Liu, Y.; Ernst, J.A.; Watts, R.J.; et al. Molecular Mechanisms of Alzheimer Disease Protection by the A673T Allele of Amyloid Precursor Protein. J. Biol. Chem. 2014, 289, 30990–31000. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kokawa, A.; Ishihara, S.; Fujiwara, H.; Nobuhara, M.; Iwata, M.; Ihara, Y.; Funamoto, S. The A673T mutation in the amyloid precursor protein reduces the production of β-amyloid protein from its β-carboxyl terminal fragment in cells. Acta Neuropathol. Commun. 2015, 3, 1–12. [Google Scholar] [CrossRef] [Green Version]
- Limegrover, C.S.; LeVine, H.; Izzo, N.J.; Yurko, R.; Mozzoni, K.; Rehak, C.; Sadlek, K.; Safferstein, H.; Catalano, S.M. Alzheimer’s protection effect of A673T mutation may be driven by lower Aβ oligomer binding affinity. J. Neurochem. 2021, 157, 1316–1330. [Google Scholar] [CrossRef] [PubMed]
- Tambini, M.D.; Norris, K.; D’Adamio, L. Opposite changes in APP processing and human Aβ levels in rats carrying either a protective or a pathogenic APP mutation. eLife 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Cassar, M.; Kretzschmar, D. Analysis of Amyloid Precursor Protein Function in Drosophila melanogaster. Front. Mol. Neurosci. 2016, 9, 61. [Google Scholar] [CrossRef] [PubMed]
- Copenhaver, P.F.; Kögel, D. Role of APP Interactions with Heterotrimeric G Proteins: Physiological Functions and Pathological Consequences. Front. Mol. Neurosci. 2017, 10, 3. [Google Scholar] [CrossRef] [Green Version]
- Southan, C.; Hancock, J.M. A tale of two drug targets: The evolutionary history of BACE1 and BACE2. Front. Genet. 2013, 4, 293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wong, Y.Y.; Le, P.; Elkhatib, W.; Piekut, T.; Senatore, A. Transcriptome profiling of Trichoplax adhaerens highlights its digestive epithelium and a rich set of genes for fast electrogenic and slow neuromodulatory cellular signaling. Res. Sq. 2019. in preprint research square. [Google Scholar]
- Kaiser, T.; Feng, G. Modeling psychiatric disorders for developing effective treatments. Nat. Med. 2015, 21, 979–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vonhoff, F.; Williams, A.; Ryglewski, S.; Duch, C. Drosophila as a Model for MECP2 Gain of Function in Neurons. PLoS ONE 2012, 7, e31835. [Google Scholar] [CrossRef] [Green Version]
- Ugur, B.; Chen, K.; Bellen, H.J. Drosophila tools and assays for the study of human diseases. Dis. Models Mechanisms 2016, 9, 235–244. [Google Scholar] [CrossRef] [Green Version]
- Bolus, H.; Crocker, K.; Boekhoff-Falk, G.; Chtarbanova, S. Modeling Neurodegenerative Disorders in Drosophila melanogaster. Int. J. Mol. Sci. 2020, 21, 3055. [Google Scholar] [CrossRef]
- Luo, L.Q.; Martin-Morris, L.; White, K. Identification, secretion, and neural expression of APPL, a Drosophila protein similar to human amyloid protein precursor. J. Neurosci. 1990, 10, 3849–3861. [Google Scholar] [CrossRef] [Green Version]
- Guo, Q.; Wang, Z.; Li, H.; Wiese, M.; Zheng, H. APP physiological and pathophysiological functions: Insights from animal models. Cell Res. 2011, 22, 78–89. [Google Scholar] [CrossRef] [Green Version]
- Poeck, B.; Strauss, R.; Kretzschmar, D. Analysis of amyloid precursor protein function in Drosophila melanogaster. Exp. Brain Res. 2011, 217, 413–421. [Google Scholar] [CrossRef] [Green Version]
- Nicolas, M.; Hassan, B.A. Amyloid precursor protein and neural development. Development 2014, 141, 2543–2548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmine-Simmen, K.; Proctor, T.; Tschäpe, J.; Poeck, B.; Triphan, T.; Strauss, R.; Kretzschmar, D. Neurotoxic effects induced by the Drosophila amyloid-β peptide suggest a conserved toxic function. Neurobiol. Dis. 2009, 33, 274–281. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bolkan, B.J.; Triphan, T.; Kretzschmar, D. -Secretase Cleavage of the Fly Amyloid Precursor Protein Is Required for Glial Survival. J. Neurosci. 2012, 32, 16181–16192. [Google Scholar] [CrossRef] [Green Version]
- Luo, L.; Tully, T.; White, K. Human amyloid precursor protein ameliorates behavioral deficit of flies deleted for appl gene. Neuron 1992, 9, 595–605. [Google Scholar] [CrossRef]
- Fernandez-Funez, P.; de Mena, L.; Rincon-Limas, D.E. Modeling the complex pathology of Alzheimer’s disease in Drosophila. Exp. Neurol. 2015, 274, 58–71. [Google Scholar] [CrossRef] [Green Version]
- Prüßing, K.; Voigt, A.; Schulz, J.B. Drosophila melanogaster as a model organism for Alzheimer’s disease. Mol. Neurodegener. 2013, 8, 35. [Google Scholar] [CrossRef] [Green Version]
- Li, M.; Ping, Y. Regulation of Sleep Behavior by Overexpression of Amyloid Precursor Protein in Drosophila Neurons. J. Shanghai Jiaotong Univ. 2021, 26, 63–68. [Google Scholar] [CrossRef]
- Cordone, S.; Scarpelli, S.; Alfonsi, V.; De Gennaro, L.; Gorgoni, M. Sleep-Based Interventions in Alzheimer’s Disease: Promising Approaches from Prevention to Treatment along the Disease Trajectory. Pharmaceuticals. 2021, 14, 383. [Google Scholar] [CrossRef]
- Holth, J.K.; Patel, T.K.; Holtzman, D.M. Sleep in Alzheimer’s disease–beyond amyloid. Neurobiol. Sleep Circadian Rhythm. 2017, 2, 4–14. [Google Scholar] [CrossRef] [Green Version]
- Chakravarti, L.; Moscato, E.; Kayser, M. Unraveling the Neurobiology of Sleep and Sleep Disorders Using Drosophila. Planar Cell Polarity Dur. Dev. 2017, 121, 253–285. [Google Scholar] [CrossRef]
- Kayser, M.S.; Biron, D. Sleep and Development in Genetically Tractable Model Organisms. Genetics 2016, 203, 21–33. [Google Scholar] [CrossRef]
- Donelson, N.C.; Sanyal, S. Use of Drosophila in the investigation of sleep disorders. Exp. Neurol. 2015, 274, 72–79. [Google Scholar] [CrossRef] [PubMed]
- Shukla, A.; Spurrier, J.; Kuzina, I.; Giniger, E. Hyperactive Innate Immunity Causes Degeneration of Dopamine Neurons upon Altering Activity of Cdk5. Cell Rep. 2019, 26, 131–144.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shukla, A.; Giniger, E. Reduced autophagy efficiency induces innate immune activation leading to neurodegeneration. Autophagy 2019, 15, 1117–1119. [Google Scholar] [CrossRef]
- Gjoneska, E.; Pfenning, A.R.; Mathys, H.; Quon, G.; Kundaje, A.; Tsai, L.-H.; Kellis, M. Conserved epigenomic signals in mice and humans reveal immune basis of Alzheimer’s disease. Nat. Cell Biol. 2015, 518, 365–369. [Google Scholar] [CrossRef] [PubMed]
- Zhuang, L.; Peng, F.; Huang, Y.; Li, W.; Huang, J.; Chu, Y.; Ren, P.; Sun, Y.; Zhang, Y.; Xue, E.; et al. CHIP modulates APP-induced autophagy-dependent pathological symptoms in Drosophila. Aging Cell 2019, 19, e13070. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paul, I.; Ghosh, M.K. The E3 ligase CHIP: Insights into its structure and regulation. BioMed Res. Int. 2014, 918183. [Google Scholar] [CrossRef] [Green Version]
- Silva, B.; Niehage, C.; Maglione, M.; Hoflack, B.; Sigrist, S.J.; Wassmer, T.; Pavlowsky, A.; Preat, T. Interactions between amyloid precursor protein-like (APPL) and MAGUK scaffolding proteins contribute to appetitive long-term memory in Drosophila melanogaster. J. Neurogenet. 2020, 34, 92–105. [Google Scholar] [CrossRef]
- Goguel, V.; Belair, A.-L.; Ayaz, D.; Lampin-Saint-Amaux, A.; Scaplehorn, N.; Hassan, B.A.; Preat, T. Drosophila Amyloid Precursor Protein-Like Is Required for Long-Term Memory. J. Neurosci. 2011, 31, 1032–1037. [Google Scholar] [CrossRef]
- Preat, T.; Goguel, V. Role of Drosophila Amyloid Precursor Protein in Memory Formation. Front. Mol. Neurosci. 2016, 9, 142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senechal, Y.; Kelly, P.H.; Dev, K.K. Amyloid precursor protein knockout mice show age-dependent deficits in passive avoidance learning. Behav. Brain Res. 2008, 186, 126–132. [Google Scholar] [CrossRef]
- Soldano, A.; Okray, Z.; Janovská, P.; Tmejová, K.; Reynaud, E.; Claeys, A.; Yan, J.; Atak, Z.K.; De Strooper, B.; Dura, J.-M.; et al. The Drosophila Homologue of the Amyloid Precursor Protein Is a Conserved Modulator of Wnt PCP Signaling. PLoS Biol. 2013, 11, e1001562. [Google Scholar] [CrossRef]
- Marquilly, C.; Busto, G.U.; Leger, B.S.; Boulanger, A.; Giniger, E.; Walker, J.A.; Fradkin, L.G.; Dura, J.-M. Htt is a repressor of Abl activity required for APP-induced axonal growth. PLoS Genet. 2021, 17, e1009287. [Google Scholar] [CrossRef]
- Kessissoglou, I.A.; Langui, D.; Hasan, A.; Maral, M.; Dutta, S.B.; Hiesinger, P.R.; Hassan, B.A. The Drosophila amyloid precursor protein homologue mediates neuronal survival and neuroglial interactions. PLoS Biol. 2020, 18, e3000703. [Google Scholar] [CrossRef]
- Koike, M.A.; Lin, A.J.; Pham, J.; Nguyen, E.; Yeh, J.J.; Rahimian, R.; Tromberg, B.J.; Choi, B.; Green, K.N.; LaFerla, F.M. APP Knockout Mice Experience Acute Mortality as the Result of Ischemia. PLoS ONE 2012, 7, e42665. [Google Scholar] [CrossRef]
- Doody, R.S.; Raman, R.; Farlow, M.; Iwatsubo, T.; Vellas, B.; Joffe, S.; Kieburtz, K.; He, F.; Sun, X.; Thomas, R.G.; et al. A phase 3 trial of semagacestat for treatment of Alzheimer’s disease. N. Engl. J. Med. 2013, 369, 341–350. [Google Scholar] [CrossRef] [PubMed]
- Egan, M.F.; Mukai, Y.; Voss, T.; Kost, J.; Stone, J.; Furtek, C.; Mahoney, E.; Cummings, J.L.; Tariot, P.N.; Aisen, P.S.; et al. Further analyses of the safety of verubecestat in the phase 3 EPOCH trial of mild-to-moderate Alzheimer’s disease. Alzheimer’s Res. Ther. 2019, 11, 1–12. [Google Scholar] [CrossRef]
- Sperling, R.; Henley, D.; Aisen, P.S.; Raman, R.; Donohue, M.C.; Ernstrom, K.; Rafii, M.S.; Streffer, J.; Shi, Y.; Karcher, K.; et al. Findings of efficacy, safety, and biomarker outcomes of atabecestat in preclinical Alzheimer disease: A truncated randomized phase 2b/3 clinical trial. JAMA Neurol. 2021, 78, 293–301. [Google Scholar] [CrossRef]
- Ettcheto, M.; Busquets, O.; Espinosa-Jiménez, T.; Verdaguer, E.; Auladell, C.; Camins, A. A Chronological Review of Potential Disease-Modifying Therapeutic Strategies for Alzheimer’s Disease. Curr. Pharm. Des. 2020, 26, 1286–1299. [Google Scholar] [CrossRef] [PubMed]
- Ettcheto, M.; Busquets, O.; Cano, A.; Sánchez-Lopez, E.; Manzine, P.R.; Espinosa-Jimenez, T.; Verdaguer, E.; Sureda, F.X.; Olloquequi, J.; Castro-Torres, R.D.; et al. Pharmacological Strategies to Improve Dendritic Spines in Alzheimer’s Disease. J. Alzheimer’s Dis. 2021, 82, S91–S107. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Song, C.; Du, Y.; Gaur, U.; Yang, M. Pharmacological Treatment of Alzheimer’s Disease: Insights from Drosophila melanogaster. Int. J. Mol. Sci. 2020, 21, 4621. [Google Scholar] [CrossRef] [PubMed]
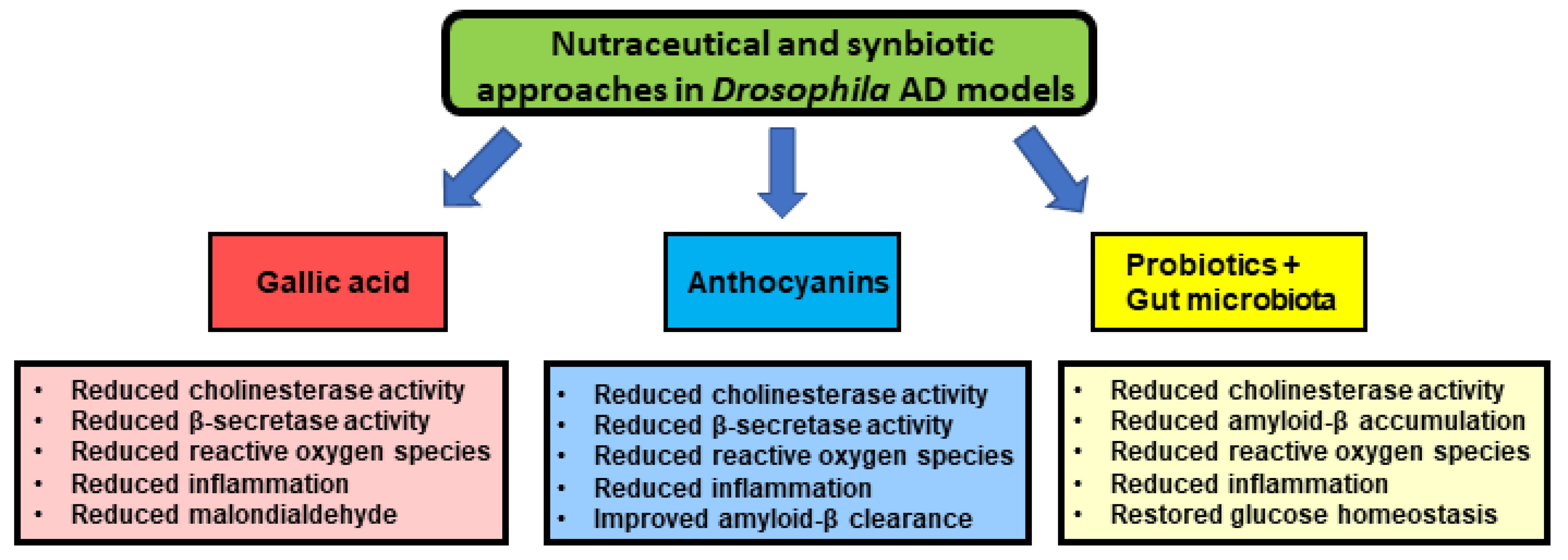
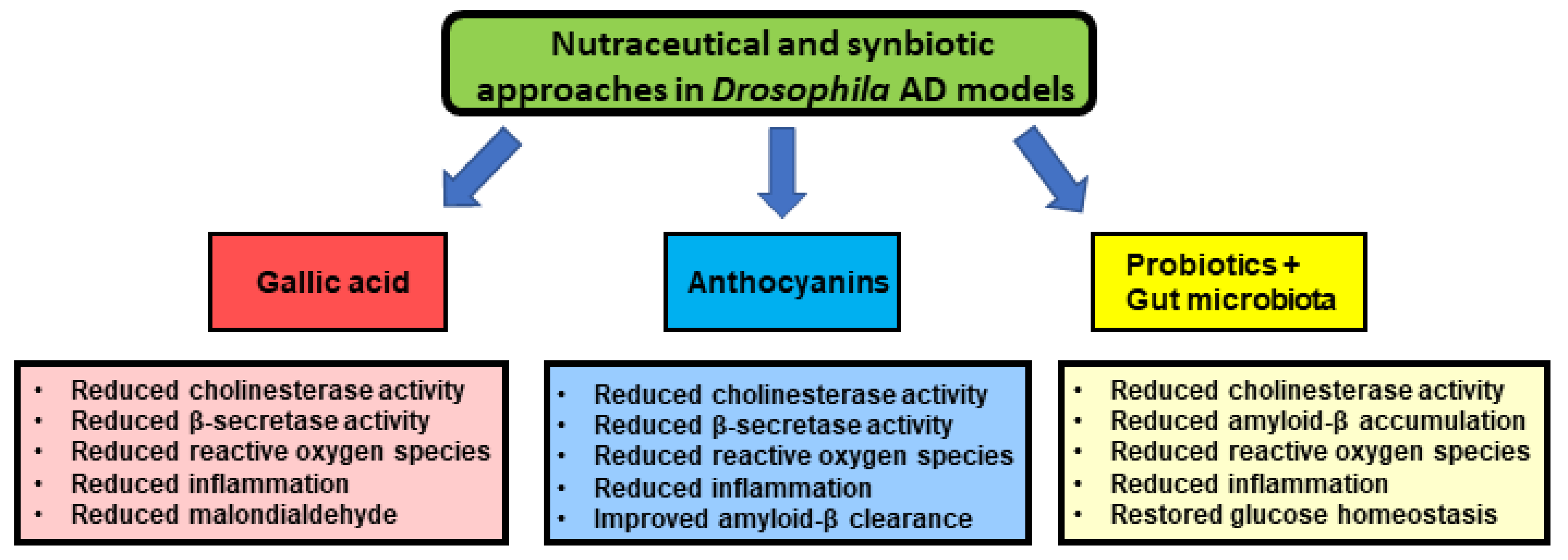
- Ogunsuyi, O.B.; Oboh, G.; Oluokun, O.O.; Ademiluyi, A.O.; Ogunruku, O.O. Gallic acid protects against neurochemical alterations in transgenic Drosophila model of Alzheimer’s disease. Adv. Tradit. Med. 2020, 20, 89–98. [Google Scholar] [CrossRef]
- Hajipour, S.; Sarkaki, A.; Farbood, Y.; Eidi, A.; Mortazavi, P.; Valizadeh, Z. Effect of Gallic Acid on Dementia Type of Alzheimer Disease in Rats: Electrophysiological and Histological Studies. Basic Clin. Neurosci. J. 2016, 7, 97–106. [Google Scholar] [CrossRef] [PubMed]
- Mori, T.; Koyama, N.; Yokoo, T.; Segawa, T.; Maeda, M.; Sawmiller, D.; Tan, J.; Town, T. Gallic acid is a dual α/β-secretase modulator that reverses cognitive impairment and remediates pathology in Alzheimer mice. J. Biol. Chem. 2020, 295, 16251–16266. [Google Scholar] [CrossRef]
- Gao, J.; Hu, J.; Hu, D.; Yang, X. A Role of Gallic Acid in Oxidative Damage Diseases: A Comprehensive Review. Nat. Prod. Commun. 2019, 14, 1934578–19874174. [Google Scholar] [CrossRef] [Green Version]
- Suttisansanee, U.; Charoenkiatkul, S.; Jongruaysup, B.; Tabtimsri, S.; Siriwan, D.; Temviriyanukul, P. Mulberry fruit cultivar ‘Chiang Mai’prevents beta-amyloid toxicity in PC12 neuronal cells and in a Drosophila model of Alzheimer’s disease. Molecules 2020, 25, 1837. [Google Scholar] [CrossRef] [Green Version]
- McDougall, G.J.; Fyffe, S.; Dobson, P.; Stewart, D. Anthocyanins from red cabbage – stability to simulated gastrointestinal digestion. Phytochemistry 2007, 68, 1285–1294. [Google Scholar] [CrossRef]
- Shih, P.-H.; Chan, Y.-C.; Liao, J.-W.; Wang, M.-F.; Yen, G.-C. Antioxidant and cognitive promotion effects of anthocyanin-rich mulberry (Morus atropurpurea L.) on senescence-accelerated mice and prevention of Alzheimer’s disease. J. Nutr. Biochem. 2010, 21, 598–605. [Google Scholar] [CrossRef]
- Roidoung, S.; Dolan, K.D.; Siddiq, M. Gallic acid as a protective antioxidant against anthocyanin degradation and color loss in vitamin-C fortified cranberry juice. Food Chem. 2016, 210, 422–427. [Google Scholar] [CrossRef]
- Fornasaro, S.; Ziberna, L.; Gasperotti, M.; Tramer, F.; Vrhovšek, U.; Mattivi, F.; Passamonti, S. Determination of cyanidin 3-glucoside in rat brain, liver and kidneys by UPLC/MS-MS and its application to a short-term pharmacokinetic study. Sci. Rep. 2016, 6, 22815. [Google Scholar] [CrossRef] [PubMed]
- Pervin, M.; Unno, K.; Nakagawa, A.; Takahashi, Y.; Iguchi, K.; Yamamoto, H.; Hoshino, M.; Hara, A.; Takagaki, A.; Nanjo, F.; et al. Blood brain barrier permeability of (−)-epigallocatechin gallate, its proliferation-enhancing activity of human neuroblastoma SH-SY5Y cells, and its preventive effect on age-related cognitive dysfunction in mice. Biochem. Biophys. Rep. 2017, 9, 180–186. [Google Scholar] [CrossRef]
- Thelen, M.; Brown-Borg, H.M. Does Diet Have a Role in the Treatment of Alzheimer’s Disease? Front. Aging Neurosci. 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Gruendler, R.; Hippe, B.; Jengic, V.S.; Peterlin, B.; Haslberger, A.G. Nutraceutical Approaches of Autophagy and Neuroinflammation in Alzheimer’s Disease: A Systematic Review. Molecules 2020, 25, 6018. [Google Scholar] [CrossRef]
- Frydman-Marom, A.; Levin, A.; Farfara, D.; Benromano, T.; Scherzer-Attali, R.; Peled, S.; Vassar, R.; Segal, D.; Gazit, E.; Frenkel, D.; et al. Orally administrated cinnamon extract reduces β-amyloid oligomerization and corrects cognitive impairment in Alzheimer’s disease animal models. PLoS ONE 2011, 6, e16564. [Google Scholar] [CrossRef] [Green Version]
- Pham, H.M.; Xu, A.; Schriner, S.E.; Sevrioukov, E.A.; Jafari, M. Cinnamaldehyde Improves Lifespan and Healthspan in Drosophila melanogaster Models for Alzheimer’s Disease. BioMed Res. Int. 2018, 2018, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Momtaz, S.; Hassani, S.; Khan, F.; Ziaee, M.; Abdollahi, M. Cinnamon, a promising prospect towards Alzheimer’s disease. Pharmacol. Res. 2018, 130, 241–258. [Google Scholar] [CrossRef] [PubMed]
- García-Viñuales, S.; Ahmed, R.; Sciacca, M.F.M.; Lanza, V.; Giuffrida, M.L.; Zimbone, S.; Romanucci, V.; Zarrelli, A.; Bongiorno, C.; Spinella, N.; et al. Trehalose Conjugates of Silybin as Prodrugs for Targeting Toxic Aβ Aggregates. ACS Chem. Neurosci. 2020, 11, 2566–2576. [Google Scholar] [CrossRef] [PubMed]
- Sciacca, M.F.M.; Romanucci, V.; Zarrelli, A.; Monaco, I.; Lolicato, F.; Spinella, N.; Galati, C.; Grasso, G.; D’Urso, L.; Romeo, M.; et al. Inhibition of Aβ Amyloid Growth and Toxicity by Silybins: The Crucial Role of Stereochemistry. ACS Chem. Neurosci. 2017, 8, 1767–1778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shabbir, U.; Rubab, M.; Tyagi, A.; Oh, D.-H. Curcumin and Its Derivatives as Theranostic Agents in Alzheimer’s Disease: The Implication of Nanotechnology. Int. J. Mol. Sci. 2020, 22, 196. [Google Scholar] [CrossRef]
- Saeedi, M.; Rashidy-Pour, A. Association between chronic stress and Alzheimer’s disease: Therapeutic effects of Saffron. Biomed. Pharmacother. 2021, 133, 110995. [Google Scholar] [CrossRef] [PubMed]
- Kim, J. Pre-Clinical Neuroprotective Evidences and Plausible Mechanisms of Sulforaphane in Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 2929. [Google Scholar] [CrossRef]
- Massie, H.R.; Aiello, V.R.; Williams, T.R. Iron accumulation during development and ageing of Drosophila. Mech. Ageing Dev. 1985, 29, 215–220. [Google Scholar] [CrossRef]
- Lumsden, A.; Rogers, J.; Majd, S.; Newman, M.; Sutherland, G.T.; Verdile, G.; Lardelli, M. Dysregulation of Neuronal Iron Homeostasis as an Alternative Unifying Effect of Mutations Causing Familial Alzheimer’s Disease. Front. Neurosci. 2018, 12, 533. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Kerman, K. Electrochemical biosensors for biometal-protein interactions in neurodegenerative diseases. Biosens. Bioelectron. 2021, 179, 113035. [Google Scholar] [CrossRef] [PubMed]
- Alaraby, M.; Romero, S.; Hernandez, A.; Marcos, R. Toxic and Genotoxic Effects of Silver Nanoparticles in Drosophila. Environ. Mol. Mutagen. 2019, 60, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Greish, K.; Alqahtani, A.A.; Alotaibi, A.F.; Abdulla, A.M.; Bukelly, A.T.; Alsobyani, F.M.; Alharbi, G.H.; Alkiyumi, I.S.; Aldawish, M.M.; Alshahrani, T.F.; et al. The Effect of Silver Nanoparticles on Learning, Memory and Social Interaction in BALB/C Mice. Int. J. Environ. Res. Public Health 2019, 16, 148. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez-Carter, D.A.; Leo, B.F.; Ruenraroengsak, P.; Chen, S.; Goode, A.E.; Theodorou, I.; Chung, K.F.; Carzaniga, R.; Shaffer, M.S.P.; Dexter, D.T.; et al. Silver nanoparticles reduce brain inflammation and related neurotoxicity through induction of H2S-synthesizing enzymes. Sci. Rep. 2017, 7, 42871. [Google Scholar] [CrossRef] [Green Version]
- Carranza-Naval, M.J.; Vargas-Soria, M.; Hierro-Bujalance, C.; Baena-Nieto, G.; Garcia-Alloza, M.; Infante-Garcia, C.; del Marco, A. Alzheimer’s Disease and Diabetes: Role of Diet, Microbiota and Inflammation in Preclinical Models. Biomolecules 2021, 11, 262. [Google Scholar] [CrossRef]
- Shabbir, U.; Arshad, M.; Sameen, A.; Oh, D.-H. Crosstalk between Gut and Brain in Alzheimer’s Disease: The Role of Gut Microbiota Modulation Strategies. Nutrients 2021, 13, 690. [Google Scholar] [CrossRef]
- Shukla, A.K.; Johnson, K.; Giniger, E. Common features of aging fail to occur in Drosophila raised without a bacterial microbiome. iScience 2021. [Google Scholar] [CrossRef]
- Borsom, E.; Lee, K.; Cope, E. Do the Bugs in Your Gut Eat Your Memories? Relationship between Gut Microbiota and Alzheimer’s Disease. Brain Sci. 2020, 10, 814. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.-C.; Cao, Z.-S.; Chang, K.-M.; Juang, J.-L. Intestinal microbial dysbiosis aggravates the progression of Alzheimer’s disease in Drosophila. Nat. Commun. 2017, 8, 1–9. [Google Scholar] [CrossRef]
- Wang, X.; Sun, G.; Feng, T.; Zhang, J.; Huang, X.; Wang, T.; Xie, Z.; Chu, X.; Yang, J.; Wang, H.; et al. Sodium oligomannate therapeutically remodels gut microbiota and suppresses gut bacterial amino acids-shaped neuroinflammation to inhibit Alzheimer’s disease progression. Cell Res. 2019, 29, 787–803. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Chan, P.; Wang, T.; Hong, Z.; Wang, S.; Kuang, W.; He, J.; Pan, X.; Zhou, Y.; Ji, Y.; et al. A 36-week multicenter, randomized, double-blind, placebo-controlled, parallel-group, phase 3 clinical trial of sodium oligomannate for mild-to-moderate Alzheimer’s dementia. Alzheimer’s Res. Ther. 2021, 13, 1–11. [Google Scholar] [CrossRef]
- Vonhoff, F.; Keshishian, H. Activity-Dependent Synaptic Refinement: New Insights from Drosophila. Front. Syst. Neurosci. 2017, 11, 23. [Google Scholar] [CrossRef] [Green Version]
- Westfall, S.; Lomis, N.; Kahouli, I.; Dia, S.Y.; Singh, S.P.; Prakash, S. Microbiome, probiotics and neurodegenerative diseases: Deciphering the gut brain axis. Cell. Mol. Life Sci. 2017, 74, 3769–3787. [Google Scholar] [CrossRef] [PubMed]
- Westfall, S.; Lomis, N.; Prakash, S. A novel synbiotic delays Alzheimer’s disease onset via combinatorial gut-brain-axis signaling in Drosophila melanogaster. PLoS ONE 2019, 14, e0214985. [Google Scholar] [CrossRef]
- Bonfili, L.; Cecarini, V.; Gogoi, O.; Berardi, S.; Scarpona, S.; Angeletti, M.; Rossi, G.; Eleuteri, A.M. Gut microbiota manipulation through probiotics oral administration restores glucose homeostasis in a mouse model of Alzheimer’s disease. Neurobiol. Aging 2020, 87, 35–43. [Google Scholar] [CrossRef]
- Nagpal, R.; Neth, B.J.; Wang, S.; Craft, S.; Yadav, H. Modified Mediterranean-ketogenic diet modulates gut microbiome and short-chain fatty acids in association with Alzheimer’s disease markers in subjects with mild cognitive impairment. EBioMedicine 2019, 47, 529–542. [Google Scholar] [CrossRef] [Green Version]
- Rosenthal, S.L.; Kamboh, M.I. Late-Onset Alzheimer’s Disease Genes and the Potentially Implicated Pathways. Curr. Genet. Med. Rep. 2014, 2, 85–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shukla, S.; Tekwani, B.L. Histone Deacetylases Inhibitors in Neurodegenerative Diseases, Neuroprotection and Neuronal Differentiation. Front. Pharmacol. 2020, 11, 537. [Google Scholar] [CrossRef]
- Lee, S.-H.; Gomes, S.M.; Ghalayini, J.; Iliadi, K.G.; Boulianne, G.L. Angiotensin Converting Enzyme Inhibitors and Angiotensin Receptor Blockers Rescue Memory Defects in Drosophila-Expressing Alzheimer’s Disease-Related Transgenes Independently of the Canonical Renin Angiotensin System. eNeuro 2020, 7. [Google Scholar] [CrossRef]
- Gabrawy, M.M.; Campbell, S.; Carbone, M.A.; Morozova, T.V.; Arya, G.H.; Turlapati, L.B.; Walston, J.D.; Starz-Gaiano, M.; Everett, L.; Mackay, T.F.C.; et al. Lisinopril Preserves Physical Resilience and Extends Life Span in a Genotype-Specific Manner in Drosophila melanogaster. J. Gerontol. Ser. A Boil. Sci. Med Sci. 2019, 74, 1844–1852. [Google Scholar] [CrossRef]
- Ederer, K.A.; Jin, K.; Bouslog, S.; Wang, L.; Gorman, G.S.; Rowe, G.C.; Abadir, P.; Raftery, D.; Moellering, D.; Promislow, D.; et al. Age- and Genotype-Specific Effects of the Angiotensin-Converting Enzyme Inhibitor Lisinopril on Mitochondrial and Metabolic Parameters in Drosophila melanogaster. Int. J. Mol. Sci. 2018, 19, 3351. [Google Scholar] [CrossRef] [Green Version]
- Ancidoni, A.; Bacigalupo, I.; Remoli, G.; Lacorte, E.; Piscopo, P.; Sarti, G.; Corbo, M.; Vanacore, N.; Canevelli, M. Anticancer drugs repurposed for Alzheimer’s disease: A systematic review. Alzheimer’s Res. Ther. 2021, 13, 1–15. [Google Scholar] [CrossRef]
- Ozlu, C.; Bailey, R.M.; Sinnett, S.; Goodspeed, K.D. Gene Transfer Therapy for Neurodevelopmental Disorders. Dev. Neurosci. 2021, 1–11. [Google Scholar] [CrossRef]
- Khan, I.; Preeti, K.; Fernandes, V.; Khatri, D.K.; Singh, S.B. Role of MicroRNAs, Aptamers in Neuroinflammation and Neurodegenerative Disorders. Cell. Mol. Neurobiol. 2021, 1–21. [Google Scholar] [CrossRef]
- Barman, N.C.; Khan, N.M.; Islam, M.; Nain, Z.; Roy, R.K.; Haque, A.; Barman, S.K. CRISPR-Cas9: A Promising Genome Editing Therapeutic Tool for Alzheimer’s Disease—A Narrative Review. Neurol. Ther. 2020, 9, 419–434. [Google Scholar] [CrossRef] [PubMed]
- Douglas, A.E. Drosophila and its gut microbes: A model for drug-microbiome interactions. Drug Discov. Today Dis. Model. 2018, 28, 43–49. [Google Scholar] [CrossRef]
- Ludington, W.B.; Ja, W.W. Drosophila as a model for the gut microbiome. PLoS Pathog. 2020, 16, e1008398. [Google Scholar] [CrossRef] [PubMed]
- Bothwell, M.; Giniger, E. Alzheimer’s Disease: Neurodevelopment Converges with Neurodegeneration. Cell 2000, 102, 271–273. [Google Scholar] [CrossRef] [Green Version]
- Karisetty, B.C.; Bhatnagar, A.; Armour, E.M.; Beaver, M.; Zhang, H.; Elefant, F. Amyloid-β Peptide Impact on Synaptic Function and Neuroepigenetic Gene Control Reveal New Therapeutic Strategies for Alzheimer’s Disease. Front. Mol. Neurosci. 2020, 13. [Google Scholar] [CrossRef] [PubMed]
- Esposito, M.; Sherr, G.L. Epigenetic Modifications in Alzheimer’s Neuropathology and Therapeutics. Front. Neurosci. 2019, 13, 476. [Google Scholar] [CrossRef] [PubMed] [Green Version]

{kind=link}
| Compound | Preferred IUPAC name | Chemical Formula | Type of Molecule | Occurrence | References (AD Models) |
|---|---|---|---|---|---|
| Gallic acid | 3,4,5-Trihydroxybenzoic acid | 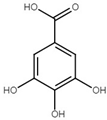 | Phenolic acid | Sumac (Rhus), tea leaves, strawberry, grape | [123,124,125] |
| Cyanidin | 2-(3,4-Dihydroxyphenyl)-3,5,7-trihydroxy-1λ4-benzopyran-1-ylium | 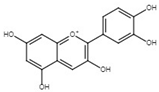 | Pigment | Mulberry (Morus nigra) Blueberry (Vaccinium) | [127,129,130] |
| Cinnamaldehyde | (2E)-3-Phenylprop-2-enal |  | Phenylpropanoid | Cinnamon | [135,136,137] |
| Curcumin | (1E,6E)-1,7-Bis(4-hydroxy-3-methoxyphenyl)hepta-1,6-diene-3,5-dione |  | Diarylheptanoid | Turmeric (Curcuma longa) | [140] |
| Silybin | (2R,3R)-3,5,7-trihydroxy-2-[(2R*,3R*)-3-(4-hydroxy-3-methoxyphenyl)-2-(hydroxymethyl)-2,3-dihydrobenzo[b][1,4]dioxin-6-yl]chroman-4-one |  | Flavonolignan | Milk thistle (Silibum marianum) | [138,139] |
| Crocin | Bis[(2S,3R,4S,5S,6R)-3,4,5-trihydroxy-6-({[(2R,3R,4S,5S,6R)-3,4,5-trihydroxy-6-(hydroxymethyl)oxan-2-yl]oxy}methyl)oxan-2-yl] (2E,4E,6E,8E,10E,12E,14E)-2,6,11,15-tetramethylhexadeca-2,4,6,8,10,12,14-heptaenedioate | 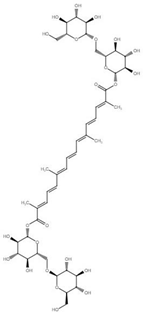 | Carotenoid | Saffron | [141] |
| Sulforaphane | 1-Isothiocyanato-4-(methanesulfinyl) butane |  | Isothiocyanate | Broccoli Brussel sprouts Cabbage | [142] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jalali, D.; Guevarra, J.A.; Martinez, L.; Hung, L.; Vonhoff, F.J. Nutraceutical and Probiotic Approaches to Examine Molecular Interactions of the Amyloid Precursor Protein APP in Drosophila Models of Alzheimer’s Disease. Int. J. Mol. Sci. 2021, 22, 7022. https://doi.org/10.3390/ijms22137022
Jalali D, Guevarra JA, Martinez L, Hung L, Vonhoff FJ. Nutraceutical and Probiotic Approaches to Examine Molecular Interactions of the Amyloid Precursor Protein APP in Drosophila Models of Alzheimer’s Disease. International Journal of Molecular Sciences. 2021; 22(13):7022. https://doi.org/10.3390/ijms22137022
Chicago/Turabian StyleJalali, David, Justine Anne Guevarra, Luz Martinez, Lily Hung, and Fernando J Vonhoff. 2021. "Nutraceutical and Probiotic Approaches to Examine Molecular Interactions of the Amyloid Precursor Protein APP in Drosophila Models of Alzheimer’s Disease" International Journal of Molecular Sciences 22, no. 13: 7022. https://doi.org/10.3390/ijms22137022