
Comparative Transcriptome Analysis Reveals Regulatory Networks during the Maize Ear Shank Elongation Process


Abstract
:1. Introduction
2. Results
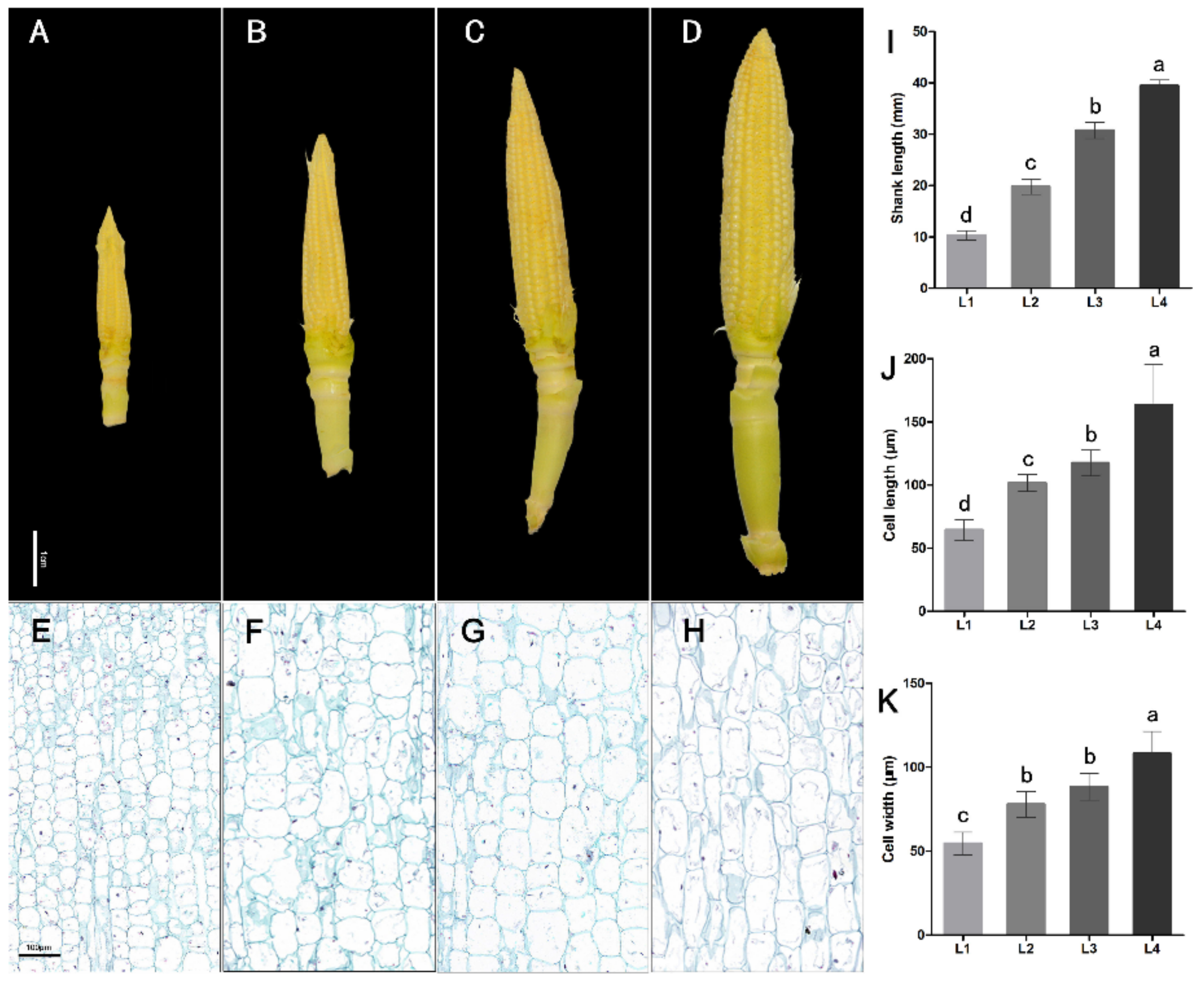
2.1. Phenotype Analysis of Maize Ear Shanks at Different Developing Stages
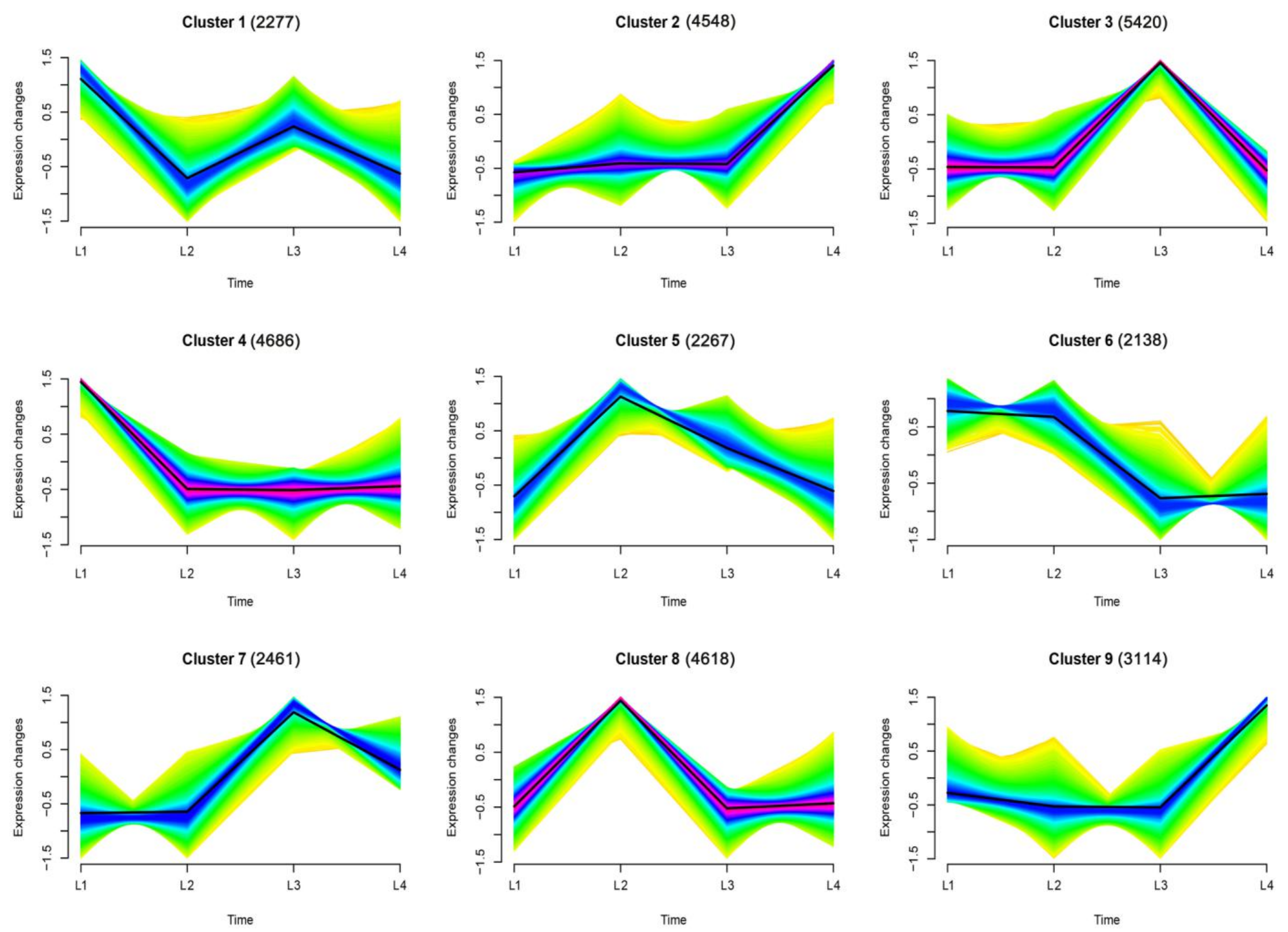
2.2. Temporal Expression Patterns of Genes
2.3. Functional Classification of DEGs
2.4. DEGs Involved in the Plant Hormone Signal Transduction Pathway
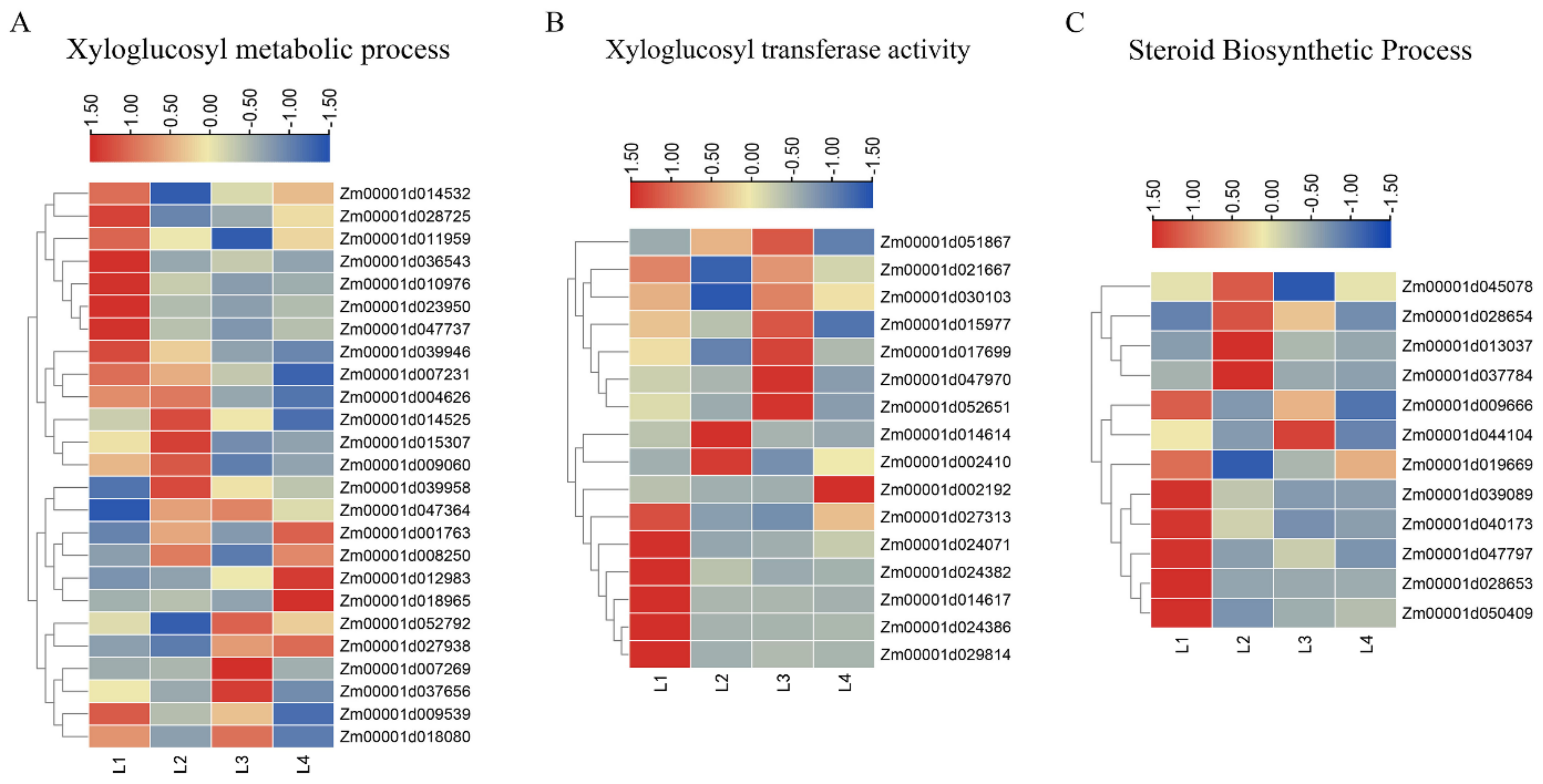
2.5. DEGs Involved in the Xyloglucan Metabolic Process and Xyloglucan Xyloglucosyl Transferase Activity
2.6. DEGs Involved in Steroid Biosynthetic Process
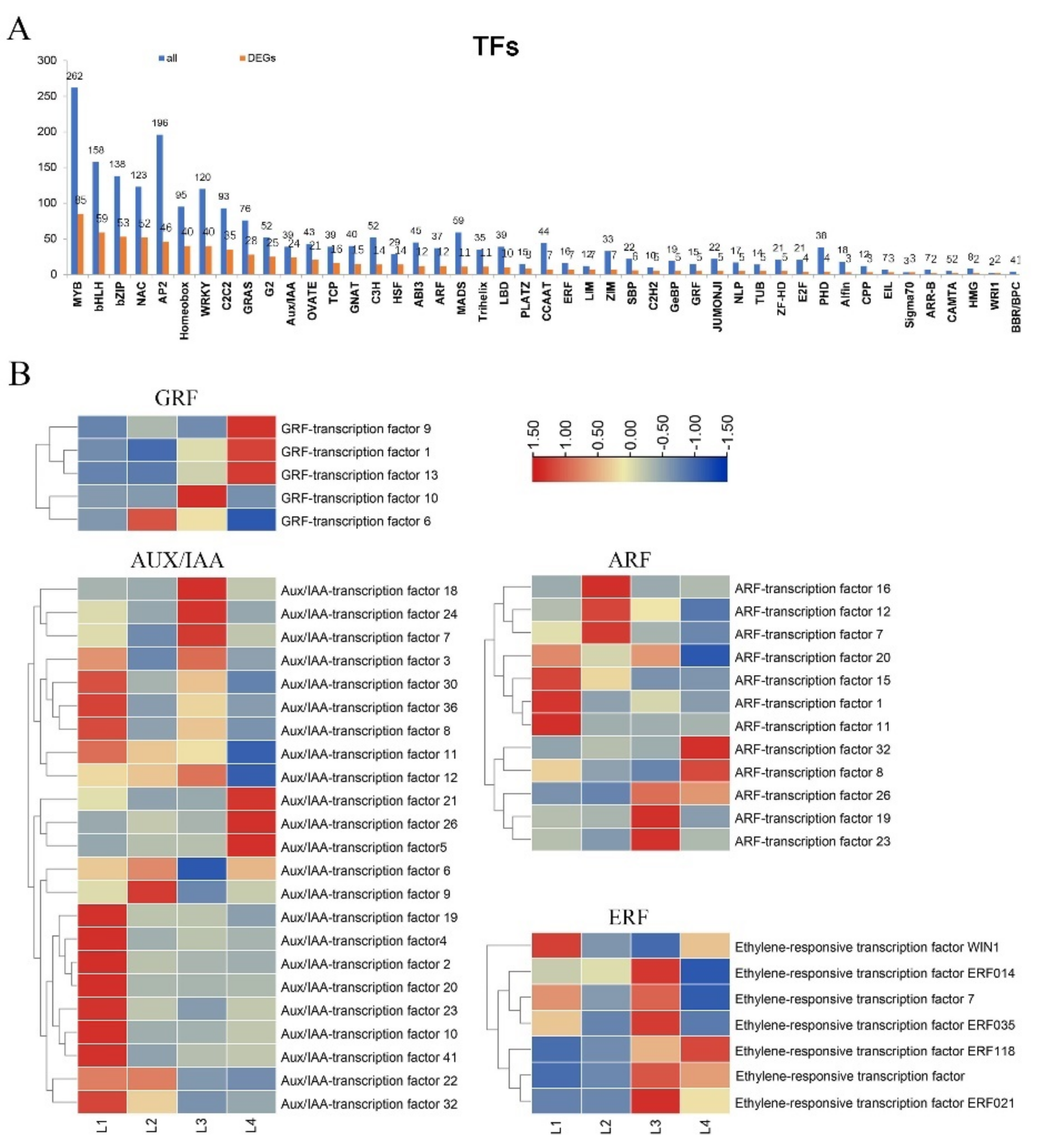
2.7. Expression of TFs Involved in the Phytohormone Response Factor
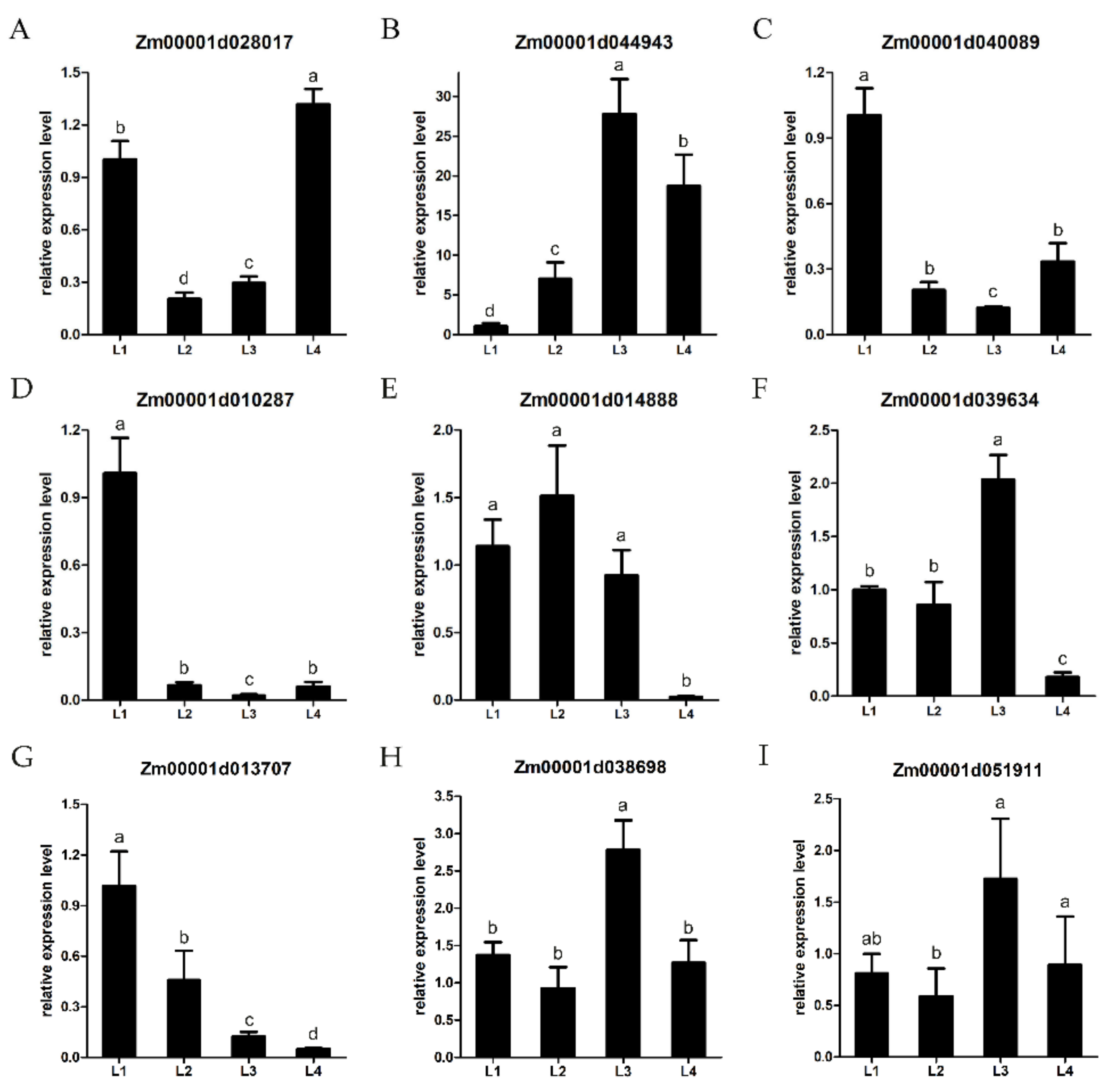
2.8. Validation of DEGs by qRT-PCR
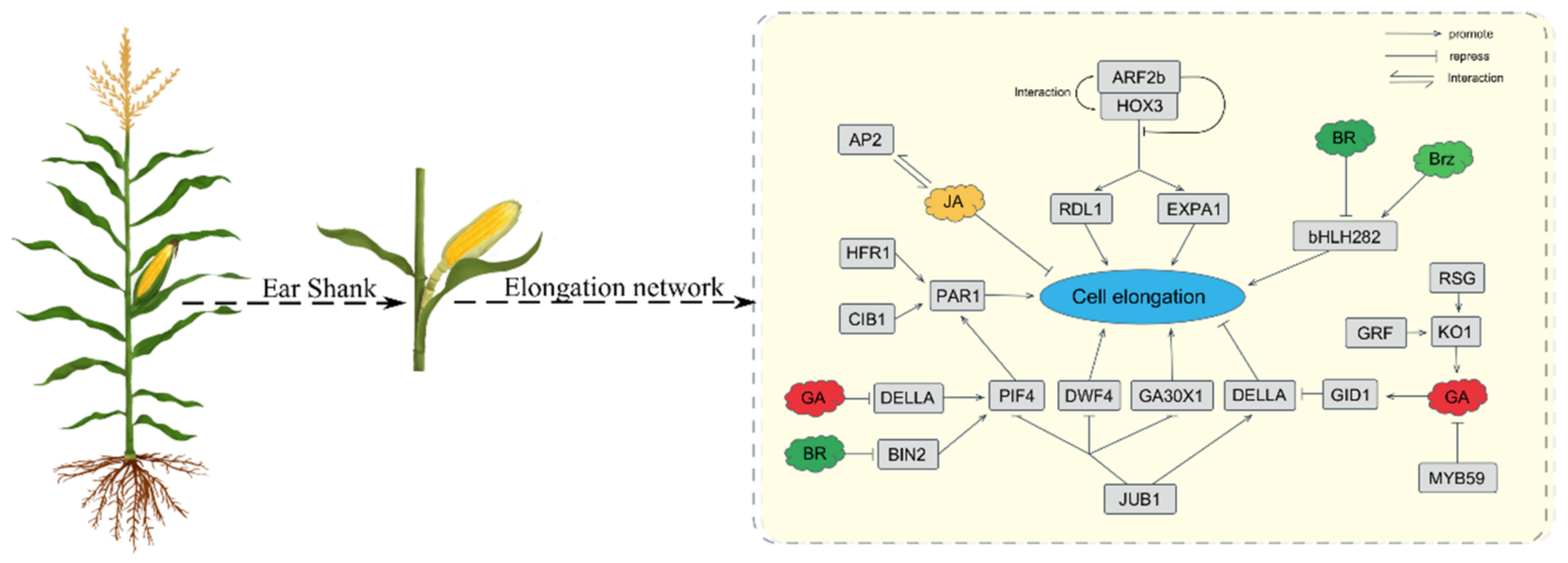
3. Discussion
4. Materials and Method
4.1. Plant Materials
4.2. RNA Extraction and Library Preparation
4.3. Preprocessing of RNA-Seq Data
4.4. Identification of DEGs and Functional Classification
4.5. Real-Time PCR Analysis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Conflicts of Interest
Abbreviations
| ABA | Abscisic acid |
| ARF | Auxin response factor |
| BR | Brassinosteroids |
| CTK | Cytokinin |
| DEGs | Differentially expressed genes |
| ERF | Ethylene responsive factor |
| ETH | Ethylene |
| FDR | False discovery rate |
| FPKM | Fragments per kilobase of transcript per million reads |
| GO | Gene ontology |
| GER | Gibberellin ear rot |
| GRF | Gibberellin response factor |
| IAA | Auxin |
| JA | Jasmonic acid |
| KEGG | Kyoto Encyclopedia of Genes and Genomes |
| KO | Ent-kaurene oxidase |
| qRT-PCR | Quantitative real-time polymerase chain reaction |
| QTL | Quantitative trait locus |
| SA | Salicylic acid |
| TFs | Transcription factors |
References
- Shiferaw, B.; Prasanna, B.M.; Hellin, J.; Banziger, M. Crops that feed the world 6. Past successes and future challenges to the role played by maize in global food security. Food Secur. 2011, 3, 307–327. [Google Scholar] [CrossRef] [Green Version]
- Ren, H.; Jiang, Y.; Zhao, M.; Qi, H.; Li, C. Nitrogen supply regulates vascular bundle structure and matter transport characteristics of spring maize under high plant density. Front. Plant Sci. 2020, 11, 602739. [Google Scholar] [CrossRef] [PubMed]
- Suwa, R.; Hakata, H.; Hara, H.; El-Shemy, H.A.; Adu-Gyamfi, J.J.; Nguyen, N.T.; Kanai, S.; Lightfoot, D.A.; Mohapatra, P.K.; Fujita, K. High temperature effects on photosynthate partitioning and sugar metabolism during ear expansion in maize (Zea mays L.) genotypes. Plant Physiol. Biochem. 2010, 48, 124–130. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; He, W.; Zhang, A.; Zhang, L.; Sun, D.; Gao, Y.; Ni, P.; Ma, X.; Cui, Z.; Ruan, Y. Genetic analysis of maize shank length by QTL mapping in three recombinant inbred line populations. Plant Sci. 2021, 303, 110767. [Google Scholar] [CrossRef]
- Troyer, A.F.; Ambrose, W.B. Plant characteristics affecting field drying rate of ear corn1. Crop Sci. 1971, 11, 529–531. [Google Scholar] [CrossRef]
- Sun, C.; Yang, B.; Gao, Z.; Zheng, Z.; Ni, Z. Field investigation of ear picking in corn mechanical harvesting. J. Chin. Agric. Mech. 2014, 35, 15–18. [Google Scholar] [CrossRef]
- Kang, M.S.; Zuber, M.S. Combining ability for grain moisture, husk moisture, and maturity in maize with yellow and white endosperms. Crop Sci. 1989, 29, 689–692. [Google Scholar] [CrossRef]
- Li, Y.; Tao, H.; Zhang, B.; Huang, S.; Wang, P. Timing of water deficit limits maize kernel setting in association with changes in the Source-Flow-Sink Relationship. Front. Plant Sci. 2018, 9, 1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, W.; Song, Q.; Zuo, B.; Han, Q.; Jia, Z. Effects of gibberellin (GA4+7) in grain filling, hormonal behavior, and antioxidants in high-density maize (Zea mays L.). Plants 2020, 9, 978. [Google Scholar] [CrossRef]
- Travaglia, C.; Balboa, G.; Espósito, G.; Reinoso, H. ABA action on the production and redistribution of field-grown maize carbohydrates in semiarid regions. Plant Growth Regul. 2012, 67, 27–34. [Google Scholar] [CrossRef]
- Hansen, L.A. The Inheritance of Ten Quantitative Characteristics in Sweet Corn (Zea mays L.). Master’s Thesis, Oregon State University, Corvallis, OR, USA, 1976. [Google Scholar]
- Yan, Y.; Christensen, S.; Isakeit, T.; Engelberth, J.; Meeley, R.; Hayward, A.; Emery, R.J.; Kolomiets, M.V. Disruption of OPR7 and OPR8 reveals the versatile functions of jasmonic acid in maize development and defense. Plant Cell 2012, 24, 1420–1436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Q.; Kong, F.; Zhang, H.; Jiang, Y.; Heng, S.; Liang, R.; Ma, L.; Liu, J.; Lu, X.; Li, P.; et al. Molecular mechanisms governing shade responses in maize. Biochem. Biophys. Res. Commun. 2019, 516, 112–119. [Google Scholar] [CrossRef]
- Ma, N.; Dong, L.; Lü, W.; Lü, J.; Meng, Q.; Liu, P. Transcriptome analysis of maize seedling roots in response to nitrogen-, phosphorus-, and potassium deficiency. Plant Soil 2020, 447, 637–658. [Google Scholar] [CrossRef]
- Kebede, A.Z.; Johnston, A.; Schneiderman, D.; Bosnich, W.; Harris, L.J. Transcriptome profiling of two maize inbreds with distinct responses to Gibberella ear rot disease to identify candidate resistance genes. BMC Genom. 2018, 19, 131. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheller, H.V.; Ulvskov, P. Hemicelluloses. Annu. Rev. Plant Biol. 2010, 61, 263–289. [Google Scholar] [CrossRef]
- Stratilova, B.; Kozmon, S.; Stratilova, E.; Hrmova, M. Plant xyloglucan xyloglucosyl transferases and the cell wall structure: Subtle but significant. Molecules 2020, 25, 5619. [Google Scholar] [CrossRef] [PubMed]
- Ferreira-Guerra, M.; Marques-Bueno, M.; Mora-Garcia, S.; Cano-Delgado, A.I. Delving into the evolutionary origin of steroid sensing in plants. Curr. Opin. Plant Biol. 2020, 57, 87–95. [Google Scholar] [CrossRef]
- Zhang, H.; Jin, J.; Tang, L.; Zhao, Y.; Gu, X.; Gao, G.; Luo, J. PlantTFDB 2.0: Update and improvement of the comprehensive plant transcription factor database. Nucleic Acids Res. 2011, 39, D1114–D1117. [Google Scholar] [CrossRef] [Green Version]
- Adiconis, X.; Borges-Rivera, D.; Satija, R.; DeLuca, D.S.; Busby, M.A.; Berlin, A.M.; Sivachenko, A.; Thompson, D.A.; Wysoker, A.; Fennell, T.; et al. Comparative analysis of RNA sequencing methods for degraded or low-input samples. Nat. Methods 2013, 10, 623–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbez, E.; Dunser, K.; Gaidora, A.; Lendl, T.; Busch, W. Auxin steers root cell expansion via apoplastic pH regulation in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2017, 114, E4884–E4893. [Google Scholar] [CrossRef] [Green Version]
- Yang, S.; Zhang, K.; Zhu, H.; Zhang, X.; Yan, W.; Xu, N.; Liu, D.; Hu, J.; Wu, Y.; Weng, Y.; et al. Melon short internode (CmSi) encodes an ERECTA-like receptor kinase regulating stem elongation through auxin signaling. Hortic. Res. 2020, 7, 202. [Google Scholar] [CrossRef] [PubMed]
- Yue, J.-H.; Zhang, D.; Ren, L.; Shen, X.-H. Gibberellin and auxin signals control scape cell elongation and proliferation in Agapanthus praecox ssp. orientalis. J. Plant Biol. 2016, 59, 358–368. [Google Scholar] [CrossRef]
- Ayano, M.; Kani, T.; Kojima, M.; Sakakibara, H.; Kitaoka, T.; Kuroha, T.; Angeles-Shim, R.B.; Kitano, H.; Nagai, K.; Ashikari, M. Gibberellin biosynthesis and signal transduction is essential for internode elongation in deepwater rice. Plant Cell Environ. 2014, 37, 2313–2324. [Google Scholar] [CrossRef] [PubMed]
- Bai, M.Y.; Shang, J.X.; Oh, E.; Fan, M.; Bai, Y.; Zentella, R.; Sun, T.P.; Wang, Z.Y. Brassinosteroid, gibberellin and phytochrome impinge on a common transcription module in Arabidopsis. Nat. Cell Biol. 2012, 14, 810–817. [Google Scholar] [CrossRef] [Green Version]
- Band, L.R.; Ubeda-Tomas, S.; Dyson, R.J.; Middleton, A.M.; Hodgman, T.C.; Owen, M.R.; Jensen, O.E.; Bennett, M.J.; King, J.R. Growth-induced hormone dilution can explain the dynamics of plant root cell elongation. Proc. Natl. Acad. Sci. USA 2012, 109, 7577–7582. [Google Scholar] [CrossRef] [Green Version]
- Tong, H.; Xiao, Y.; Liu, D.; Gao, S.; Liu, L.; Yin, Y.; Jin, Y.; Qian, Q.; Chu, C. Brassinosteroid regulates cell elongation by modulating gibberellin metabolism in rice. Plant Cell 2014, 26, 4376–4393. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Xu, P.; Wang, W.; Wang, S.; Caruana, J.C.; Yang, H.Q.; Lian, H. Arabidopsis G-Protein beta subunit AGB1 interacts with BES1 to regulate brassinosteroid signaling and cell elongation. Front. Plant Sci. 2017, 8, 2225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oh, E.; Zhu, J.Y.; Bai, M.Y.; Arenhart, R.A.; Sun, Y.; Wang, Z.Y. Cell elongation is regulated through a central circuit of interacting transcription factors in the Arabidopsis hypocotyl. Elife 2014, 3. [Google Scholar] [CrossRef]
- Dai, X.; Lu, Q.; Wang, J.; Wang, L.; Xiang, F.; Liu, Z. MiR160 and its target genes ARF10, ARF16 and ARF17 modulate hypocotyl elongation in a light, BRZ, or PAC-dependent manner in Arabidopsis: miR160 promotes hypocotyl elongation. Plant Sci. 2021, 303, 110686. [Google Scholar] [CrossRef]
- Majda, M.; Robert, S. The role of auxin in cell wall expansion. Int. J. Mol. Sci. 2018, 19, 951. [Google Scholar] [CrossRef] [Green Version]
- Christiaens, A.; Pauwels, E.; Gobin, B.; Van Labeke, M.C. Flower differentiation of azalea depends on genotype and not on the use of plant growth regulators. Plant Growth Regul. 2015, 75, 245–252. [Google Scholar] [CrossRef]
- Dockter, C.; Gruszka, D.; Braumann, I.; Druka, A.; Druka, I.; Franckowiak, J.; Gough, S.P.; Janeczko, A.; Kurowska, M.; Lundqvist, J.; et al. Induced variations in brassinosteroid genes define barley height and sturdiness, and expand the green revolution genetic toolkit. Plant Physiol. 2014, 166, 1912–1927. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ross, J.J.; O’Neill, D.P.; Smith, J.J.; Kerckhoffs, L.; Elliott, R.C. Evidence that auxin promotes gibberellin A1 biosynthesis in pea. Plant J. 2001, 21, 547–552. [Google Scholar] [CrossRef] [PubMed]
- Goda, H.; Sawa, S.; Asami, T.; Fujioka, S.; Shimada, Y.; Yoshida, S. Comprehensive comparison of auxin-regulated and brassinosteroid-regulated genes in Arabidopsis. Plant Physiol. 2004, 134, 1555–1573. [Google Scholar] [CrossRef] [Green Version]
- Patil, V.; McDermott, H.I.; McAllister, T.; Cummins, M.; Silva, J.C.; Mollison, E.; Meikle, R.; Morris, J.; Hedley, P.E.; Waugh, R.; et al. APETALA2 control of barley internode elongation. Development 2019, 146. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Wang, Y.; Ye, D.; Xing, J.; Duan, L.; Li, Z.; Zhang, M. Ethephon-regulated maize internode elongation associated with modulating auxin and gibberellin signal to alter cell wall biosynthesis and modification. Plant Sci. 2020, 290, 110196. [Google Scholar] [CrossRef]
- Liu, C.; Bi, J.; Kang, L.; Zhou, J.; Liu, X.; Liu, Z.; Yuan, S. The molecular mechanism of stipe cell wall extension for mushroom stipe elongation growth. Fungal Biol. Rev. 2021, 35, 14–26. [Google Scholar] [CrossRef]
- Cosgrove, D.J. Growth of the plant cell wall. Nat. Rev. Mol. Cell Biol. 2005, 6, 850–861. [Google Scholar] [CrossRef]
- Hrmova, M.; Farkas, V.; Lahnstein, J.; Fincher, G.B. A Barley xyloglucan xyloglucosyl transferase covalently links xyloglucan, cellulosic substrates, and (1,3;1,4)-beta-D-glucans. J. Biol. Chem. 2007, 282, 12951–12962. [Google Scholar] [CrossRef] [Green Version]
- Behar, H.; Graham, S.W.; Brumer, H. Comprehensive cross-genome survey and phylogeny of glycoside hydrolase family 16 members reveals the evolutionary origin of EG16 and XTH proteins in plant lineages. Plant J. 2018, 95, 1114–1128. [Google Scholar] [CrossRef] [Green Version]
- Vaaje-Kolstad, G.; Farkas, V.; Hrmova, M.; Fincher, G.B. Xyloglucan xyloglucosyl transferases from barley (Hordeum vulgare L.) bind oligomeric and polymeric xyloglucan molecules in their acceptor binding sites. Biochim. Biophys. Acta 2010, 1800, 674–684. [Google Scholar] [CrossRef]
- Takeda, T.; Furuta, Y.; Awano, T.; Mizuno, K.; Mitsuishi, Y.; Hayashi, T. Suppression and acceleration of cell elongation by integration of xyloglucans in pea stem segments. Proc. Natl. Acad. Sci. USA 2002, 99, 9055–9060. [Google Scholar] [CrossRef] [Green Version]
- Zhai, K.; Zhao, G.; Jiang, H.; Sun, C.; Ren, J. Overexpression of maize ZmMYB59 gene plays a negative regulatory role in seed germination in Nicotiana tabacum and Oryza sativa. Front. Plant Sci. 2020, 11, 564665. [Google Scholar] [CrossRef]
- Lu, R.; Zhang, J.; Liu, D.; Wei, Y.L.; Wang, Y.; Li, X.B. Characterization of bHLH/HLH genes that are involved in brassinosteroid (BR) signaling in fiber development of cotton (Gossypium hirsutum). BMC Plant Biol. 2018, 18, 304. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, Y.; Zhao, J.; Huang, J.; Shi, Y.; Deng, D. Unveiling gibberellin-responsive coding and long noncoding RNAs in maize. Plant Mol. Biol. 2018, 98, 427–438. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.; Lu, Y.; Yang, H.; He, J.; Hu, Z.; Hu, X.; Luan, M.; Zhang, L.; Fan, Y.; Wang, L. ZmGRF, a GA regulatory factor from maize, promotes flowering and plant growth in Arabidopsis. Plant Mol. Biol. 2015, 87, 157–167. [Google Scholar] [CrossRef]
- Shahnejat-Bushehri, S.; Allu, A.D.; Mehterov, N.; Thirumalaikumar, V.P.; Alseekh, S.; Fernie, A.R.; Mueller-Roeber, B.; Balazadeh, S. Arabidopsis NAC transcription factor JUNGBRUNNEN1 exerts conserved control over gibberellin and brassinosteroid Metabolism and signaling genes in tomato. Front. Plant Sci. 2017, 8, 214. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Zhao, Y.; Chu, H.; Wang, L.; Fu, Y.; Liu, P.; Upadhyaya, N.; Chen, C.; Mou, T.; Feng, Y.; et al. SHOEBOX modulates root meristem size in rice through dose-dependent effects of gibberellins on cell elongation and proliferation. PLoS Genet. 2015, 11, e1005464. [Google Scholar] [CrossRef]
- Jiao, Y.; Peluso, P.; Shi, J.; Liang, T.; Stitzer, M.C.; Wang, B.; Campbell, M.S.; Stein, J.C.; Wei, X.; Chin, C.S.; et al. Improved maize reference genome with single-molecule technologies. Nature 2017, 546, 524–527. [Google Scholar] [CrossRef]
- Pertea, M.; Kim, D.; Pertea, G.M.; Leek, J.T.; Salzberg, S.L. Transcript-level expression analysis of RNA-seq experiments with HISAT, StringTie and Ballgown. Nat. Protoc. 2016, 11, 1650–1667. [Google Scholar] [CrossRef]
- Trapnell, C.; Williams, B.A.; Pertea, G.; Mortazavi, A.; Kwan, G.; van Baren, M.J.; Salzberg, S.L.; Wold, B.J.; Pachter, L. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat. Biotechnol. 2010, 28, 511–515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, L.; M, E.F. Mfuzz: A software package for soft clustering of microarray data. Bioinformation 2007, 2, 5–7. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Qian, J.; Fang, Z.; Tang, J. Transcriptomic and physiological analyses of rice seedlings under different nitrogen supplies provide insight into the regulation involved in axillary bud outgrowth. BMC Plant. Biol. 2020, 20, 197. [Google Scholar] [CrossRef]
- Chen, C.; Chen, H.; Zhang, Y.; Thomas, H.R.; Frank, M.H.; He, Y.; Xia, R. TBtools: An integrative toolkit developed for interactive analyses of big biological data. Mol. Plant 2020, 13, 1194–1202. [Google Scholar] [CrossRef] [PubMed]
- Ashburner, M.; Ball, C.; Blake, J.; Botstein, D.; Butler, H.; Cherry, J.; Davis, A.; Dolinski, K.; Dwight, S.; Eppig, T.; et al. Gene Ontology: Tool for the unification of biology. Nat. Genet. 2000, 25, 25–29. [Google Scholar] [CrossRef] [Green Version]
- Kanehisa, M.; Goto, S. KEGG: Kyoto Encyclopedia of Genes and Genomes. Nucleic Acids Res. 2000, 28, 27–30. [Google Scholar] [CrossRef]
- Livak, K.J.; Schmittgen, T.J.M. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| GO ID | GO Term | Gene Number | p-Value | FDR |
|---|---|---|---|---|
| GO:0016798 | hydrolase activity, acting on glycosyl bonds | 157 | 6.00 × 10−10 | 3.95 × 10−7 |
| GO:0004672 | protein kinase activity | 436 | 1.10 × 10−9 | 3.95 × 10−7 |
| GO:0006468 | protein phosphorylation | 441 | 8.92 × 10−9 | 1.13 × 10−5 |
| GO:0004553 | hydrolase activity, hydrolyzing O-glycosyl compounds | 143 | 9.34 × 10−9 | 2.23 × 10−6 |
| GO:0015979 | photosynthesis | 57 | 1.99 × 10−8 | 1.26 × 10−5 |
| GO:0008017 | microtubule binding | 50 | 4.64 × 10−8 | 8.32 × 10−6 |
| GO:0015631 | tubulin binding | 50 | 6.95 × 10−8 | 9.64 × 10−6 |
| GO:0016773 | phosphotransferase activity, alcohol group as acceptor | 463 | 8.06 × 10−8 | 9.64 × 10−6 |
| GO:0007017 | microtubule-based process | 78 | 1.54 × 10−7 | 6.46 × 10−5 |
| GO:0015630 | microtubule cytoskeleton | 65 | 2.36 × 10−7 | 2.05 × 10−5 |
| GO:0009579 | protein complex binding | 50 | 3.19 × 10−7 | 3.27 × 10−5 |
| GO:0044436 | thylakoid part | 35 | 3.20 × 10−7 | 2.05 × 10−5 |
| GO:0032403 | thylakoid | 35 | 3.20 × 10−7 | 2.05 × 10−5 |
| GO:0005871 | kinesin complex | 40 | 4.11 × 10−7 | 2.05 × 10−5 |
| GO:0034357 | photosynthetic membrane | 34 | 4.41 × 10−7 | 2.05 × 10−5 |
| GO:0009521 | photosystem | 33 | 6.06 × 10−7 | 2.35 × 10−5 |
| GO:0005875 | microtubule associated complex | 44 | 1.25 × 10−6 | 4.15 × 10−5 |
| GO:0016301 | kinase activity | 461 | 1.29 × 10−6 | 1.16 × 10−4 |
| GO:0016310 | phosphorylation | 459 | 1.52 × 10−6 | 4.80 × 10−4 |
| GO:0043228 | intracellular non-membrane-bounded organelle | 257 | 2.34 × 10−6 | 6.07 × 10−5 |
| Pathway ID | Pathway | Out (1257) | All (4604) | p-Value |
|---|---|---|---|---|
| ko03010 | Ribosome | 151 | 375 | 9.38 × 10−9 |
| ko01110 | Biosynthesis of secondary metabolites | 341 | 1023 | 7.80 × 10−7 |
| ko00062 | Fatty acid elongation | 25 | 39 | 1.56 × 10−6 |
| ko01100 | Metabolic pathways | 565 | 1819 | 2.38 × 10−6 |
| ko00061 | Fatty acid biosynthesis | 23 | 39 | 3.01 × 10−5 |
| ko00195 | Photosynthesis | 37 | 76 | 5.09 × 10−5 |
| ko00196 | Photosynthesis—antenna proteins | 16 | 24 | 6.30 × 10−5 |
| ko01212 | Fatty acid metabolism | 31 | 61 | 7.37 × 10−5 |
| ko04626 | Plant–pathogen interaction | 126 | 348 | 9.81 × 10−5 |
| ko00520 | Amino sugar and nucleotide sugar metabolism | 49 | 116 | 3.14 × 10−4 |
| ko04075 | Plant hormone signal transduction | 96 | 263 | 4.93 × 10−4 |
| ko00940 | Phenylpropanoid biosynthesis | 72 | 190 | 7.54 × 10−4 |
| ko00360 | Phenylalanine metabolism | 20 | 39 | 1.21 × 10−3 |
| ko00941 | Flavonoid biosynthesis | 16 | 29 | 1.36 × 10−3 |
| ko00130 | Ubiquinone and other terpenoid-quinone biosynthesis | 19 | 37 | 1.56 × 10−3 |
| ko00591 | Linoleic acid metabolism | 9 | 13 | 1.94 × 10−3 |
| ko03410 | Base excision repair | 16 | 34 | 1.06 × 10−2 |
| ko00250 | Alanine, aspartate and glutamate metabolism | 19 | 45 | 2.14 × 10−2 |
| ko00780 | Biotin metabolism | 7 | 13 | 3.85 × 10−2 |
| ko00760 | Nicotinate and nicotinamide metabolism | 8 | 16 | 4.44 × 10−2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xiong, C.-Y.; Gong, Q.-Y.; Pei, H.; Liao, C.-J.; Yang, R.-C.; Li, G.-K.; Huang, J. Comparative Transcriptome Analysis Reveals Regulatory Networks during the Maize Ear Shank Elongation Process. Int. J. Mol. Sci. 2021, 22, 7029. https://doi.org/10.3390/ijms22137029
Xiong C-Y, Gong Q-Y, Pei H, Liao C-J, Yang R-C, Li G-K, Huang J. Comparative Transcriptome Analysis Reveals Regulatory Networks during the Maize Ear Shank Elongation Process. International Journal of Molecular Sciences. 2021; 22(13):7029. https://doi.org/10.3390/ijms22137029
Chicago/Turabian StyleXiong, Cai-Yun, Qing-You Gong, Hu Pei, Chang-Jian Liao, Rui-Chun Yang, Gao-Ke Li, and Jun Huang. 2021. "Comparative Transcriptome Analysis Reveals Regulatory Networks during the Maize Ear Shank Elongation Process" International Journal of Molecular Sciences 22, no. 13: 7029. https://doi.org/10.3390/ijms22137029