
Oral Treatment with RD2RD2 Impedes Development of Motoric Phenotype and Delays Symptom Onset in SOD1G93A Transgenic Mice

 , , , , , , , and
, , , , , , , and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
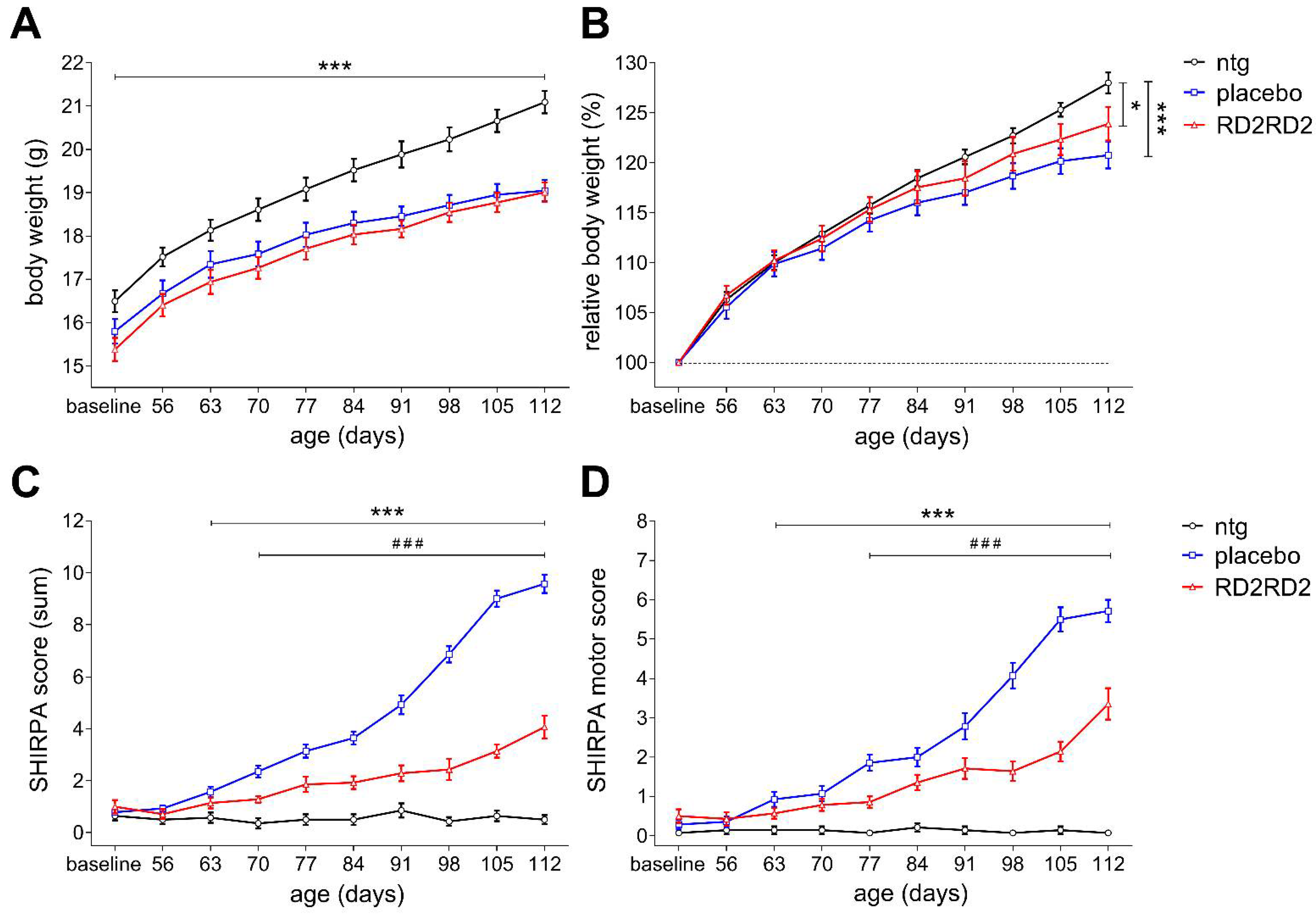
2.1. Oral Treatment with RD2RD2 Led to an Amelioration of the SOD1G93A Phenotype
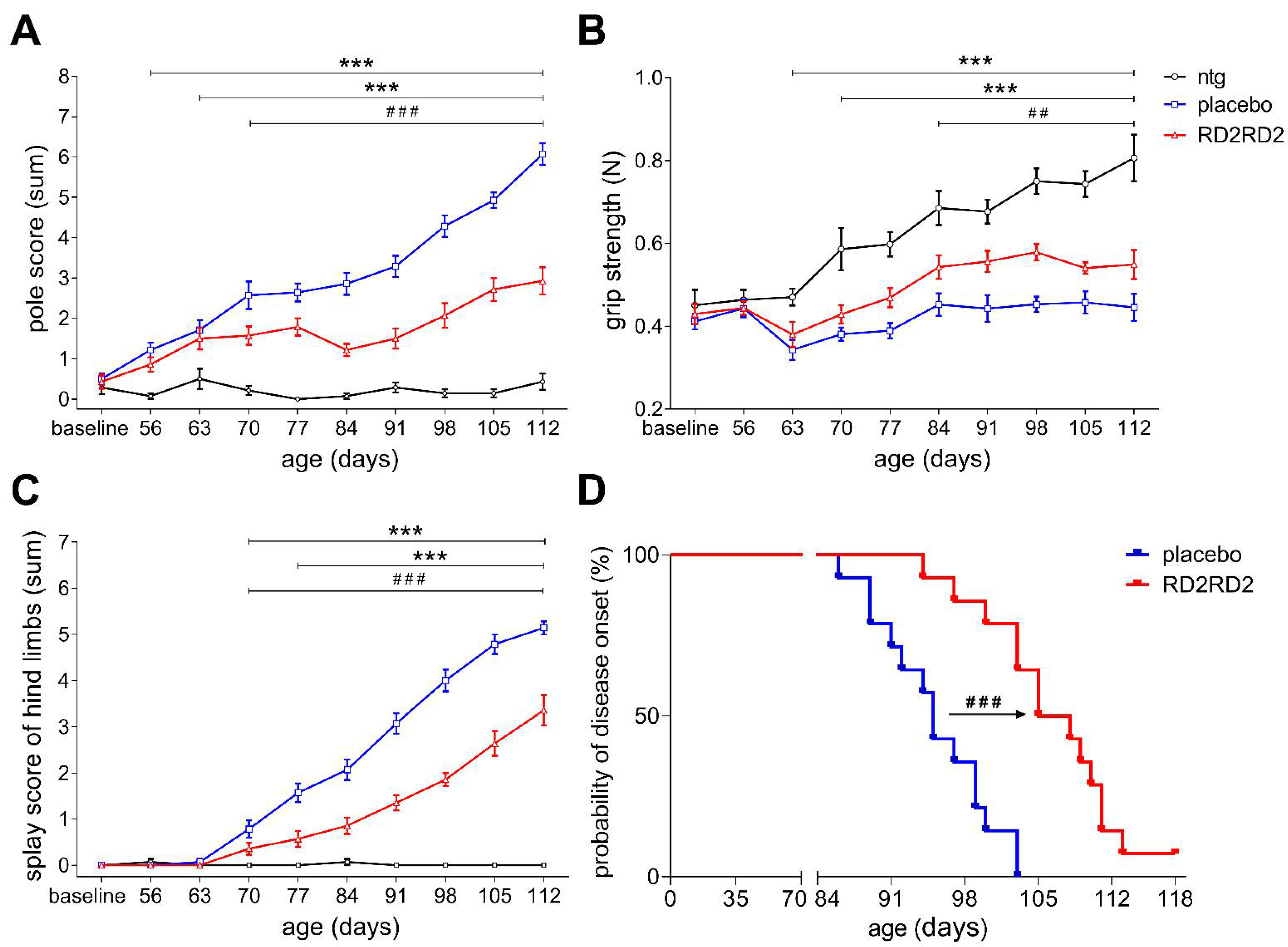
2.2. RD2RD2 Treatment Improved Motor Performance and Led to Delay of the Disease Onset in SOD1G93A Mice
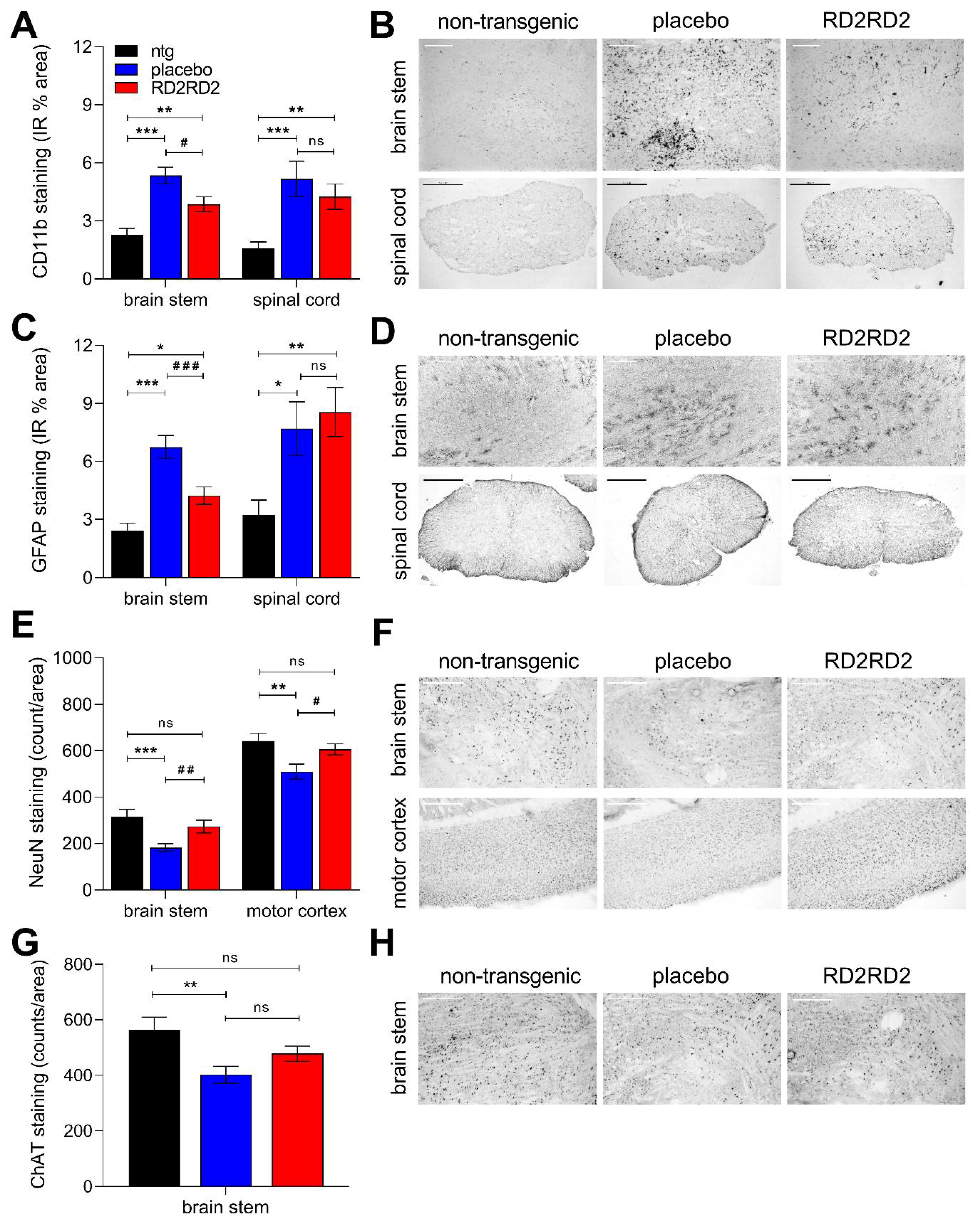
2.3. Reduced Gliosis and Neurodegeneration in RD2RD2-Treated SOD1G93A Mice
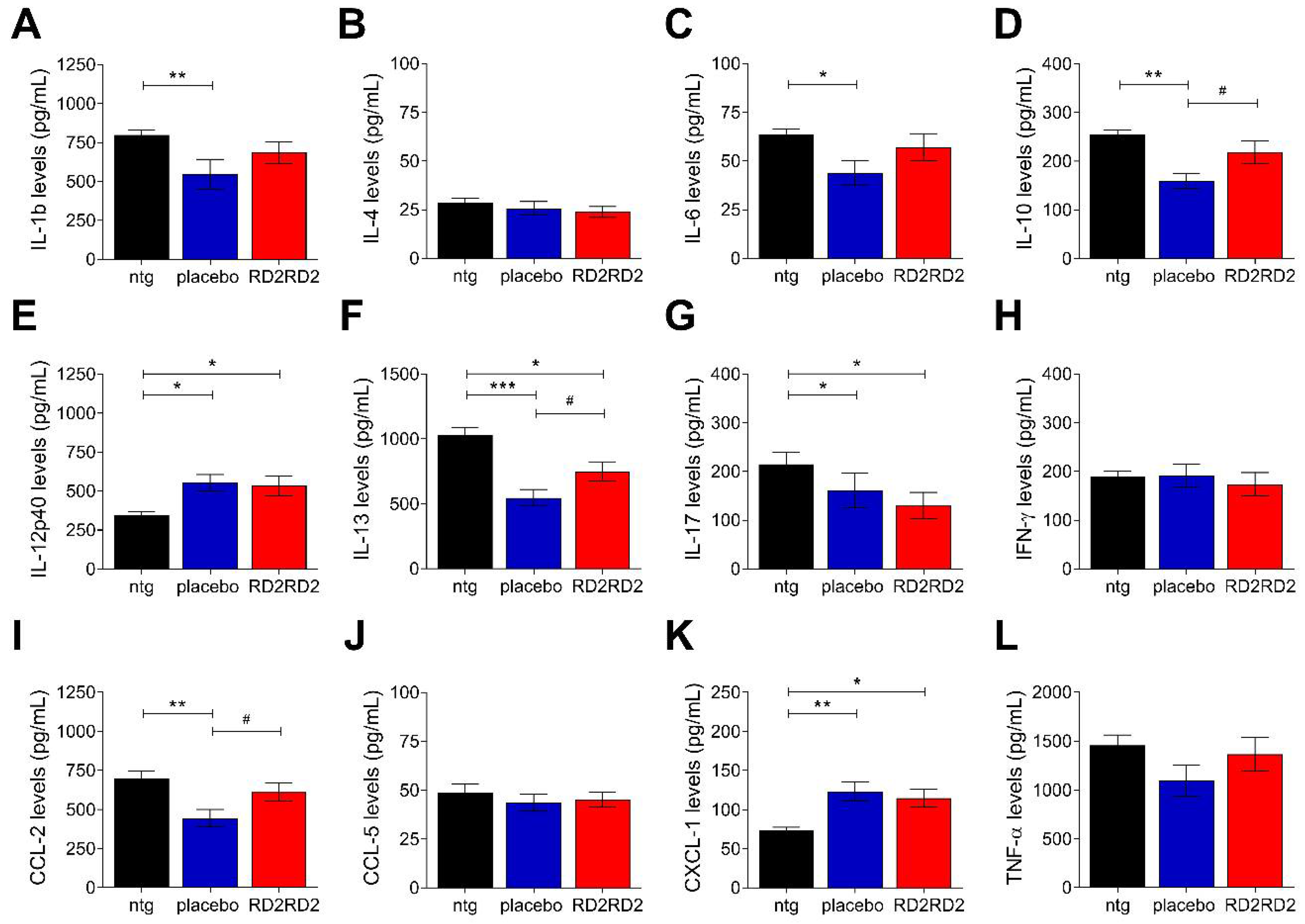
2.4. RD2RD2 Administration Suppressed the Activation of Inflammatory Plasma Markers in SOD1G93A Mice
3. Discussion
4. Conclusions
5. Methods
5.1. Animals
5.2. Drug Candidate
5.3. Study Design
5.4. Treatment
5.5. Body Weight of SOD1G93A
5.6. SHIRPA Phenotype Assessment
5.7. Modified Pole Test
5.8. Grip Strength Test
5.9. Splay Reflex Test of Hind Limbs
5.10. Disease Onset Analysis
5.11. Plasma and Tissue Collection
5.12. Immunohistochemistry
5.13. Quantification
5.14. Cytokine Assay
5.15. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Magnus, T.; Beck, M.; Giess, R.; Puls, I.; Naumann, M.; Toyka, K. Disease progression in amyotrophic lateral sclerosis: Predictors of survival. Muscle Nerve 2002, 25, 709–714. [Google Scholar] [CrossRef] [PubMed]
- McDermott, C.; Shaw, P. Diagnosis and management of motor neurone disease. BMJ 2008, 336, 658–662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niedermeyer, S.; Murn, M.; Choi, P.J. Respiratory Failure in Amyotrophic Lateral Sclerosis. Chest 2019, 155, 401–408. [Google Scholar] [CrossRef]
- Renton, A.E.; Chio, A.; Traynor, B.J. State of play in amyotrophic lateral sclerosis genetics. Nat. Neurosci. 2014, 17, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Taylor, J.P.; Brown, R.H.; Cleveland, D.W. Decoding ALS: From genes to mechanism. Nat. Cell Biol. 2016, 539, 197–206. [Google Scholar] [CrossRef] [Green Version]
- Andersen, P.M.; Sims, K.B.; Xin, W.W.; Kiely, R.; O’Neill, G.; Ravits, J.; Pioro, E.; Harati, Y.; Brower, R.D.; Levine, J.S.; et al. Sixteen novel mutations in the Cu/Zn superoxide dismutase gene in amyotrophic lateral sclerosis: A decade of discoveries, defects and disputes. Amyotroph. Lateral Scler. Other Mot. Neuron Disord. 2003, 4, 62–73. [Google Scholar] [CrossRef]
- Andersen, P.M. Amyotrophic lateral sclerosis associated with mutations in the CuZn superoxide dismutase gene. Curr. Neurol. Neurosci. Rep. 2006, 6, 37–46. [Google Scholar] [CrossRef]
- Rosen, D.R.; Siddique, T.; Patterson, D.; Figlewicz, D.A.; Sapp, P.C.; Hentati, A.; Donaldson, D.H.; Goto, J.; O’Regan, J.P.; Deng, H.-X.; et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature 1993, 362, 59–62. [Google Scholar] [CrossRef]
- Cudkowicz, M.E.; McKenna-Yasek, D.; Sapp, P.E.; Chin, W.; Geller, B.; Hayden, D.L.; Schoenfeld, D.A.; Hosler, B.A.; Horvitz, H.R.; Brown, R.H. Epidemiology of mutations in superoxide dismutase in amyotrophic lateal sclerosis. Ann. Neurol. 1997, 41, 210–221. [Google Scholar] [CrossRef]
- Corrado, L.; D’Alfonso, S.; Bergamaschi, L.; Testa, L.; Leone, M.; Nasuelli, N.; Momigliano-Richiardi, P.; Mazzini, L. SOD1 gene mutations in Italian patients with Sporadic Amyotrophic Lateral Sclerosis (ALS). Neuromuscul. Disord. 2006, 16, 800–804. [Google Scholar] [CrossRef]
- Kwon, M.-J.; Baek, W.; Ki, C.-S.; Kim, H.Y.; Koh, S.-H.; Kim, J.-W.; Kim, S.H. Screening of the SOD1, FUS, TARDBP, ANG, and OPTN mutations in Korean patients with familial and sporadic ALS. Neurobiol. Aging 2012, 33, 1017.e17–1017.e23. [Google Scholar] [CrossRef]
- McGoldrick, P.; Joyce, P.I.; Fisher, E.M.; Greensmith, L. Rodent models of amyotrophic lateral sclerosis. Biochim. Biophys. Acta (BBA)-Mol. Basis Dis. 2013, 1832, 1421–1436. [Google Scholar] [CrossRef] [Green Version]
- Philips, T.; Rothstein, J.D. Rodent Models of Amyotrophic Lateral Sclerosis. Curr. Protoc. Pharmacol. 2015, 69, 5.67.1–5.67.21. [Google Scholar] [CrossRef] [Green Version]
- Picher-Martel, V.; Valdmanis, P.N.; Gould, P.V.; Julien, J.-P.; Dupré, N. From animal models to human disease: A genetic approach for personalized medicine in ALS. Acta Neuropathol. Commun. 2016, 4, 70. [Google Scholar] [CrossRef]
- Lutz, C. Mouse models of ALS: Past, present and future. Brain Res. 2018, 1693, 1–10. [Google Scholar] [CrossRef]
- Gurney, M.E.; Pu, H.; Chiu, A.Y.; Dal Canto, M.C.; Polchow, C.Y.; Alexander, D.D.; Caliendo, J.; Hentati, A.; Kwon, Y.W.; Deng, H.X.; et al. Motor neuron degeneration in mice that express a human Cu,Zn superoxide dismutase mutation. Science 1994, 264, 1772–1775. [Google Scholar] [CrossRef]
- Chiu, A.Y.; Zhai, P.; Canto, M.C.D.; Peters, T.M.; Kwon, Y.W.; Prattis, S.M.; Gurney, M.E. Age-Dependent Penetrance of Disease in a Transgenic Mouse Model of Familial Amyotrophic Lateral Sclerosis. Mol. Cell. Neurosci. 1995, 6, 349–362. [Google Scholar] [CrossRef]
- Alexander, G.M.; Erwin, K.L.; Byers, N.; Deitch, J.S.; Augelli, B.J.; Blankenhorn, E.P.; Heiman-Patterson, T.D. Effect of transgene copy number on survival in the G93A SOD1 transgenic mouse model of ALS. Mol. Brain Res. 2004, 130, 7–15. [Google Scholar] [CrossRef]
- Marcuzzo, S.; Zucca, I.; Mastropietro, A.; de Rosbo, N.K.; Cavalcante, P.; Tartari, S.; Bonanno, S.; Preite, L.; Mantegazza, R.; Bernasconi, P. Hind limb muscle atrophy precedes cerebral neuronal degeneration in G93A-SOD1 mouse model of amyotrophic lateral sclerosis: A longitudinal MRI study. Exp. Neurol. 2011, 231, 30–37. [Google Scholar] [CrossRef]
- Ferrante, R.J.; Browne, S.E.; Shinobu, L.A.; Bowling, A.C.; Baik, M.J.; MacGarvey, U.; Kowall, N.W.; Brown, R.H., Jr.; Beal, M.F. Evidence of Increased Oxidative Damage in Both Sporadic and Familial Amyotrophic Lateral Sclerosis. J. Neurochem. 1997, 69, 2064–2074. [Google Scholar] [CrossRef]
- Yoshihara, T.; Ishigaki, S.; Yamamoto, M.; Liang, Y.; Jun Niwa, I.; Takeuchi, H.; Doyu, M.; Sobue, G. Differential expression of inflammation-and apoptosis-related genes in spinal cords of a mutant SOD1 transgenic mouse model of familial amyotrophic lateral sclerosis. J. Neurochem. 2002, 80, 158–167. [Google Scholar] [CrossRef]
- Philips, T.; Robberecht, W. Neuroinflammation in amyotrophic lateral sclerosis: Role of glial activation in motor neuron disease. Lancet Neurol. 2011, 10, 253–263. [Google Scholar] [CrossRef]
- Beers, D.R.; Zhao, W.; Liao, B.; Kano, O.; Wang, J.; Huang, A.; Appel, S.H.; Henkel, J.S. Neuroinflammation modulates distinct regional and temporal clinical responses in ALS mice. Brain Behav. Immun. 2011, 25, 1025–1035. [Google Scholar] [CrossRef] [Green Version]
- McCombe, P.A.; Henderson, R.D. The Role of Immune and Inflammatory Mechanisms in ALS. Curr. Mol. Med. 2011, 11, 246–254. [Google Scholar] [CrossRef]
- Evans, M.; Couch, Y.; Sibson, N.; Turner, M. Inflammation and neurovascular changes in amyotrophic lateral sclerosis. Mol. Cell. Neurosci. 2013, 53, 34–41. [Google Scholar] [CrossRef]
- Hall, E.D.; Oostveen, J.A.; Gurney, M.E. Relationship of microglial and astrocytic activation to disease onset and progression in a transgenic model of familial ALS. Glia 1998, 23, 249–256. [Google Scholar] [CrossRef]
- Haidet-Phillips, A.M.; Hester, M.E.; Miranda, C.; Meyer, K.; Braun, L.; Frakes, A.; Song, S.; Likhite, S.; Murtha, M.J.; Foust, K.D.; et al. Astrocytes from familial and sporadic ALS patients are toxic to motor neurons. Nat. Biotechnol. 2011, 29, 824–828. [Google Scholar] [CrossRef] [Green Version]
- Geloso, M.C.; Corvino, V.; Marchese, E.; Serrano, A.; Michetti, F.; D’Ambrosi, N. The Dual Role of Microglia in ALS: Mechanisms and Therapeutic Approaches. Front. Aging Neurosci. 2017, 9, 242. [Google Scholar] [CrossRef] [Green Version]
- Elliott, J.L. Cytokine upregulation in a murine model of familial amyotrophic lateral sclerosis. Mol. Brain Res. 2001, 95, 172–178. [Google Scholar] [CrossRef]
- Jeyachandran, A.; Mertens, B.; McKissick, E.A.; Mitchell, C.S. Type I vs. Type II cytokine levels as a function of SOD1 G93A mouse amyotrophic lateral sclerosis disease progression. Front. Cell. Neurosci. 2015, 9, 462. [Google Scholar] [CrossRef] [Green Version]
- Miller, R.G.; Appel, S.H. Introduction to supplement: The current status of treatment for ALS. Amyotroph. Lateral Scler. Front. Degener. 2017, 18, 1–4. [Google Scholar] [CrossRef]
- Mathis, S.; Couratier, P.; Julian, A.; Corcia, P.; Le Masson, G. Current view and perspectives in amyotrophic lateral sclerosis. Neural Regen. Res. 2017, 12, 181–184. [Google Scholar] [CrossRef]
- Gordon, P.H. Amyotrophic lateral sclerosis. CNS Drugs 2011, 25, 1–15. [Google Scholar] [CrossRef]
- Dorst, J.; Ludolph, A.C.; Huebers, A. Disease-modifying and symptomatic treatment of amyotrophic lateral sclerosis. Ther. Adv. Neurol. Disord. 2017, 11. [Google Scholar] [CrossRef] [PubMed]
- Leithold, L.H.; Jiang, N.; Post, J.; Niemietz, N.; Schartmann, E.; Ziehm, T.; Kutzsche, J.; Shah, N.J.; Breitkreutz, J.; Langen, K.-J.; et al. Pharmacokinetic properties of tandem d-peptides designed for treatment of Alzheimer’s disease. Eur. J. Pharm. Sci. 2016, 89, 31–38. [Google Scholar] [CrossRef]
- Schartmann, E.; Schemmert, S.; Niemietz, N.; Honold, D.; Ziehm, T.; Tusche, M.; Elfgen, A.; Gering, I.; Brener, O.; Shah, N.J.; et al. In Vitro Potency and Preclinical Pharmacokinetic Comparison of All-D-Enantiomeric Peptides Developed for the Treatment of Alzheimer’s Disease. J. Alzheimer’s Dis. 2018, 64, 859–873. [Google Scholar] [CrossRef] [Green Version]
- Elfgen, A.; Hupert, M.; Bochinsky, K.; Tusche, M.; Martin, E.G.D.S.R.; Gering, I.; Sacchi, S.; Pollegioni, L.; Huesgen, P.F.; Hartmann, R.; et al. Metabolic resistance of the D-peptide RD2 developed for direct elimination of amyloid-β oligomers. Sci. Rep. 2019, 9, 5715. [Google Scholar] [CrossRef] [Green Version]
- Kutzsche, J.; Jürgens, D.; Willuweit, A.; Adermann, K.; Fuchs, C.; Simons, S.; Windisch, M.; Hümpel, M.; Rossberg, W.; Wolzt, M.; et al. Safety and pharmacokinetics of the orally available antiprionic compound PRI-002: A single and multiple ascending dose phase I study. Alzheimer’s Dementia Transl. Res. Clin. Interv. 2020, 6, e12001. [Google Scholar] [CrossRef] [PubMed]
- Van Groen, T.; Schemmert, S.; Brener, O.; Gremer, L.; Ziehm, T.; Tusche, M.; Nagel-Steger, L.; Kadish, I.; Schartmann, E.; Elfgen, A.; et al. The Aβ oligomer eliminating D-enantiomeric peptide RD2 improves cognition without changing plaque pathology. Sci. Rep. 2017, 7, 16275. [Google Scholar] [CrossRef] [Green Version]
- Schemmert, S.; Schartmann, E.; Zafiu, C.; Kass, B.; Hartwig, S.; Lehr, S.; Bannach, O.; Langen, K.-J.; Shah, N.J.; Kutzsche, J.; et al. Aβ Oligomer Elimination Restores Cognition in Transgenic Alzheimer’s Mice with Full-blown Pathology. Mol. Neurobiol. 2019, 56, 2211–2223. [Google Scholar] [CrossRef] [Green Version]
- Schemmert, S.; Schartmann, E.; Honold, D.; Zafiu, C.; Ziehm, T.; Langen, K.-J.; Shah, N.J.; Kutzsche, J.; Willuweit, A.; Willbold, D. Deceleration of the neurodegenerative phenotype in pyroglutamate-Aβ accumulating transgenic mice by oral treatment with the Aβ oligomer eliminating compound RD2. Neurobiol. Dis. 2019, 124, 36–45. [Google Scholar] [CrossRef]
- Zhang, T.; Loschwitz, J.; Strodel, B.; Nagel-Steger, L.; Willbold, D. Interference with Amyloid-β Nucleation by Transient Ligand Interaction. Molecules 2019, 24, 2129. [Google Scholar] [CrossRef] [Green Version]
- Kutzsche, J.; Schemmert, S.; Tusche, M.; Neddens, J.; Rabl, R.; Jürgens, D.; Brener, O.; Willuweit, A.; Hutter-Paier, B.; Willbold, D. Large-Scale Oral Treatment Study with the Four Most Promising D3-Derivatives for the Treatment of Alzheimer’s Disease. Molecules 2017, 22, 1693. [Google Scholar] [CrossRef] [Green Version]
- Post, J.; Kogel, V.; Schaffrath, A.; Lohmann, P.; Shah, N.J.; Langen, K.J.; Willbold, D.; Willuweit, A.; Kutzsche, J. A Novel Anti-Inflammatory d-Peptide Inhibits Disease Phenotype Progression in an ALS Mouse Model. Molecules 2021, 26, 1590. [Google Scholar] [CrossRef]
- Mead, R.J.; Bennett, E.J.; Kennerley, A.J.; Sharp, P.; Sunyach, C.; Kasher, P.; Berwick, J.; Pettmann, B.; Battaglia, G.; Azzouz, M.; et al. Optimised and Rapid Pre-clinical Screening in the SOD1G93A Transgenic Mouse Model of Amyotrophic Lateral Sclerosis (ALS). PLoS ONE 2011, 6, e23244. [Google Scholar] [CrossRef] [PubMed]
- Ferrucci, M.; Spalloni, A.; Bartalucci, A.; Cantafora, E.; Fulceri, F.; Nutini, M.; Longone, P.; Paparelli, A.; Fornai, F. A systematic study of brainstem motor nuclei in a mouse model of ALS, the effects of lithium. Neurobiol. Dis. 2010, 37, 370–383. [Google Scholar] [CrossRef] [PubMed]
- Dibaj, P.; Zschüntzsch, J.; Steffens, H.; Scheffel, J.; Göricke, B.; Weishaupt, J.H.; Le Meur, K.; Kirchhoff, F.; Hanisch, U.-K.; Schomburg, E.D.; et al. Influence of Methylene Blue on Microglia-Induced Inflammation and Motor Neuron Degeneration in the SOD1G93A Model for ALS. PLoS ONE 2012, 7, e43963. [Google Scholar] [CrossRef]
- Rudnick, N.D.; Griffey, C.J.; Guarnieri, P.; Gerbino, V.; Wang, X.; Piersaint, J.A.; Tapia, J.C.; Rich, M.M.; Maniatis, T. Distinct roles for motor neuron autophagy early and late in the SOD1G93A mouse model of ALS. Proc. Natl. Acad. Sci. USA 2017, 114, E8294–E8303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jara, J.H.; Genç, B.; Stanford, M.J.; Pytel, P.; Roos, R.P.; Weintraub, S.; Mesulam, M.M.; Bigio, E.H.; Miller, R.J.; Özdinler, P.H. Evidence for an early innate immune response in the motor cortex of ALS. J. Neuroinflamm. 2017, 14, 129. [Google Scholar] [CrossRef] [Green Version]
- Cleveland, D.W.; Rothstein, J.D. From charcot to lou gehrig: Deciphering selective motor neuron death in als. Nat. Rev. Neurosci. 2001, 2, 806–819. [Google Scholar] [CrossRef]
- Arthur, K.C.; Calvo, A.; Price, T.R.; Geiger, J.T.; Chiò, A.C.A.; Traynor, K.C.A.B.J. Projected increase in amyotrophic lateral sclerosis from 2015 to 2040. Nat. Commun. 2016, 7, 12408. [Google Scholar] [CrossRef] [Green Version]
- McCauley, M.E.; Baloh, R.H. Inflammation in ALS/FTD pathogenesis. Acta Neuropathol. 2019, 137, 715–730. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beers, D.R.; Appel, S.H. Immune dysregulation in amyotrophic lateral sclerosis: Mechanisms and emerging therapies. Lancet Neurol. 2019, 18, 211–220. [Google Scholar] [CrossRef]
- Boillée, S.; Yamanaka, K.; Lobsiger, C.S.; Copeland, N.G.; Jenkins, N.A.; Kassiotis, G.; Kollias, G.; Cleveland, D.W. Onset and Progression in Inherited ALS Determined by Motor Neurons and Microglia. Science 2006, 312, 1389–1392. [Google Scholar] [CrossRef] [Green Version]
- Hensley, K.; Abdel-Moaty, H.; Hunter, J.; Mhatre, M.; Mou, S.; Nguyen, K.; Potapova, T.; Pye, Q.N.; Qi, M.; Rice, H.; et al. Primary glia expressing the G93A-SOD1 mutation present a neuroinflammatory phenotype and provide a cellular system for studies of glial inflammation. J. Neuroinflamm. 2006, 3, 2. [Google Scholar] [CrossRef] [Green Version]
- Hooten, K.G.; Beers, D.R.; Zhao, W.; Appel, S.H. Protective and Toxic Neuroinflammation in Amyotrophic Lateral Sclerosis. Neurotherapeutics 2015, 12, 364–375. [Google Scholar] [CrossRef] [Green Version]
- Becher, B.; Spath, B.B.S.; Goverman, J. Cytokine networks in neuroinflammation. Nat. Rev. Immunol. 2017, 17, 49–59. [Google Scholar] [CrossRef]
- Mammana, S.; Fagone, P.; Cavalli, E.; Basile, M.S.; Petralia, M.C.; Nicoletti, F.; Bramanti, P.; Mazzon, E. The Role of Macrophages in Neuroinflammatory and Neurodegenerative Pathways of Alzheimer’s Disease, Amyotrophic Lateral Sclerosis, and Multiple Sclerosis: Pathogenetic Cellular Effectors and Potential Therapeutic Targets. Int. J. Mol. Sci. 2018, 19, 831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Henkel, J.S.; Beers, D.R.; Zhao, W.; Appel, S.H. Microglia in ALS: The Good, The Bad, and The Resting. J. Neuroimmune Pharmacol. 2009, 4, 389–398. [Google Scholar] [CrossRef]
- Zhao, W.; Beers, D.R.; Appel, S.H. Immune-mediated Mechanisms in the Pathoprogression of Amyotrophic Lateral Sclerosis. J. Neuroimmune Pharmacol. 2013, 8, 888–899. [Google Scholar] [CrossRef]
- Keller, A.F.; Gravel, M.; Kriz, J. Treatment with minocycline after disease onset alters astrocyte reactivity and increases microgliosis in SOD1 mutant mice. Exp. Neurol. 2011, 228, 69–79. [Google Scholar] [CrossRef]
- Patel, P.; Julien, J.-P.; Kriz, J. Early-Stage Treatment with Withaferin A Reduces Levels of Misfolded Superoxide Dismutase 1 and Extends Lifespan in a Mouse Model of Amyotrophic Lateral Sclerosis. Neurotherapeutics 2015, 12, 217–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Apolloni, S.; Fabbrizio, P.; Amadio, S.; Volonté, C. Actions of the antihistaminergic clemastine on presymptomatic SOD1-G93A mice ameliorate ALS disease progression. J. Neuroinflamm. 2016, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, K.E.; Rasmussen, A.L.; Bennett, W.; King, A.; West, A.K.; Chung, R.S.; Chuah, M.I. Microglia and motor neurons during disease progression in the SOD1G93A mouse model of amyotrophic lateral sclerosis: Changes in arginase1 and inducible nitric oxide synthase. J. Neuroinflamm. 2014, 11, 55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beers, D.R.; Henkel, J.S.; Zhao, W.; Wang, J.; Huang, A.; Wen, S.; Liao, B.; Appel, S.H. Endogenous regulatory T lymphocytes ameliorate amyotrophic lateral sclerosis in mice and correlate with disease progression in patients with amyotrophic lateral sclerosis. Brain 2011, 134, 1293–1314. [Google Scholar] [CrossRef] [PubMed]
- Prado, L.D.G.R.; Rocha, N.P.; De Souza, L.C.; Bicalho, I.C.S.; Gomez, R.S.; Vidigal-Lopes, M.; Braz, N.F.T.; Vieira, É.L.M.; Teixeira, A.L. Longitudinal assessment of clinical and inflammatory markers in patients with amyotrophic lateral sclerosis. J. Neurol. Sci. 2018, 394, 69–74. [Google Scholar] [CrossRef] [PubMed]
- McCombe, P.A.; Henderson, R.D. Effects of gender in amyotrophic lateral sclerosis. Gend. Med. 2010, 7, 557–570. [Google Scholar] [CrossRef]
- Veldink, J.; Bär, P.; Joosten, E.; Otten, M.; Wokke, J.; Berg, L.V.D. Sexual differences in onset of disease and response to exercise in a transgenic model of ALS. Neuromuscul. Disord. 2003, 13, 737–743. [Google Scholar] [CrossRef]
- Jankowsky, J.L.; Younkin, L.H.; Gonzales, V.; Fadale, D.J.; Slunt, H.H.; Lester, H.A.; Younkin, S.G.; Borchelt, D.R. Rodent Aβ Modulates the Solubility and Distribution of Amyloid Deposits in Transgenic Mice. J. Biol. Chem. 2007, 282, 22707–22720. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, Z.; Cheng, X.; Zhong, S.; Zhang, X.; Liu, C.; Liu, F.; Zhao, C. Peripheral and Central Nervous System Immune Response Crosstalk in Amyotrophic Lateral Sclerosis. Front. Neurosci. 2020, 14, 575. [Google Scholar] [CrossRef]
- Ohgomori, T.; Yamasaki, R.; Takeuchi, H.; Kadomatsu, K.; Jinno, S.; Kira, J.-I. Differential activation of neuronal and glial STAT3 in the spinal cord of theSOD1G93Amouse model of amyotrophic lateral sclerosis. Eur. J. Neurosci. 2017, 46, 2001–2014. [Google Scholar] [CrossRef]
- Dunkelmann, T.; Schemmert, S.; Honold, D.; Teichmann, K.; Butzküven, E.; DeMuth, H.-U.; Shah, N.J.; Langen, K.-J.; Kutzsche, J.; Willbold, D.; et al. Comprehensive Characterization of the Pyroglutamate Amyloid-β Induced Motor Neurodegenerative Phenotype of TBA2.1 Mice. J. Alzheimer’s Dis. 2018, 63, 115–130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rogers, D.C.; Fisher, E.M.C.; Brown, S.D.M.; Peters, J.; Hunter, A.J.; Martin, J.E. Behavioral and functional analysis of mouse phenotype: SHIRPA, a proposed protocol for comprehensive phenotype assessment. Mamm. Genome 1997, 8, 711–713. [Google Scholar] [CrossRef]
- Rogers, D.C.; Peters, J.; Martin, J.E.; Ball, S.; Nicholson, S.J.; Witherden, A.S.; Hafezparast, M.; Latcham, J.; Robinson, T.L.; Quilter, C.A.; et al. SHIRPA, a protocol for behavioral assessment: Validation for longitudinal study of neurological dysfunction in mice. Neurosci. Lett. 2001, 306, 89–92. [Google Scholar] [CrossRef]
- Ogawa, N.; Hirose, Y.; Ohara, S.; Ono, T.; Watanabe, Y. A simple quantitative bradykinesia test in MPTP-treated mice. Res. Commun. Chem. Pathol. Pharmacol. 1985, 50, 435–441. [Google Scholar]
- Abramow-Newerly, W.; Lipina, T.; Abramow-Newerly, M.; Kim, D.; Bechard, A.R.; Xie, G.; Clapcote, S.J.; Roder, J.C. Methods to rapidly and accurately screen a large number of ENU mutagenized mice for abnormal motor phenotypes. Amyotroph. Lateral Scler. 2006, 7, 112–118. [Google Scholar] [CrossRef]
- Tadros, M.A.; Harris, B.M.; Anderson, W.B.; Brichta, A.M.; Graham, B.A.; Callister, R.J. Are all spinal segments equal: Intrinsic membrane properties of superficial dorsal horn neurons in the developing and mature mouse spinal cord. J. Physiol. 2012, 590, 2409–2425. [Google Scholar] [CrossRef] [PubMed]
- Hugnot, J.-P. Isolate and culture neural stem cells from the mouse adult spinal cord. In Neural Progenitor Cells. Methods in Molecular Biology (Methods and Protocols); Reynolds, B., Deleyrolle, L., Eds.; Humana Press: Totowa, NJ, USA, 2013; Volume 1059, pp. 53–63. [Google Scholar]
- Paxinos, G.; Franklin, K.B. Paxinos and Franklin’s the Mouse Brain in Stereotaxic Coordinates; Academic Press: Cambridge, MA, USA, 2019. [Google Scholar]
- McQuin, C.; Goodman, A.; Chernyshev, V.; Kamentsky, L.; Cimini, B.A.; Karhohs, K.W.; Doan, M.; Ding, L.; Rafelski, S.M.; Thirstrup, D.; et al. CellProfiler 3.0: Next-generation image processing for biology. PLoS Biol. 2018, 16, e2005970. [Google Scholar] [CrossRef] [Green Version]
- Clark, R.A.; Shoaib, M.; Hewitt, K.N.; Stanford, S.C.; Bate, S.T. A comparison of InVivoStat with other statistical software packages for analysis of data generated from animal experiments. J. Psychopharmacol. 2012, 26, 1136–1142. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Post, J.; Schaffrath, A.; Gering, I.; Hartwig, S.; Lehr, S.; Shah, N.J.; Langen, K.-J.; Willbold, D.; Kutzsche, J.; Willuweit, A. Oral Treatment with RD2RD2 Impedes Development of Motoric Phenotype and Delays Symptom Onset in SOD1G93A Transgenic Mice. Int. J. Mol. Sci. 2021, 22, 7066. https://doi.org/10.3390/ijms22137066
Post J, Schaffrath A, Gering I, Hartwig S, Lehr S, Shah NJ, Langen K-J, Willbold D, Kutzsche J, Willuweit A. Oral Treatment with RD2RD2 Impedes Development of Motoric Phenotype and Delays Symptom Onset in SOD1G93A Transgenic Mice. International Journal of Molecular Sciences. 2021; 22(13):7066. https://doi.org/10.3390/ijms22137066
Chicago/Turabian StylePost, Julia, Anja Schaffrath, Ian Gering, Sonja Hartwig, Stefan Lehr, N. Jon Shah, Karl-Josef Langen, Dieter Willbold, Janine Kutzsche, and Antje Willuweit. 2021. "Oral Treatment with RD2RD2 Impedes Development of Motoric Phenotype and Delays Symptom Onset in SOD1G93A Transgenic Mice" International Journal of Molecular Sciences 22, no. 13: 7066. https://doi.org/10.3390/ijms22137066