
Ineffective Erythropoiesis in ?-Thalassaemia: Key Steps and Therapeutic Options by Drugs


Abstract
: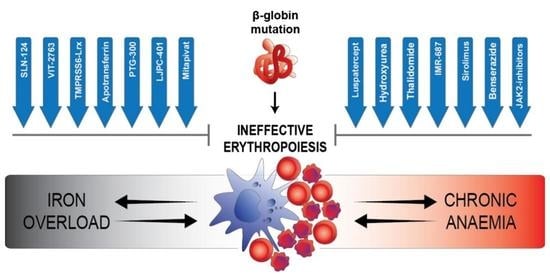
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Erythropoiesis
2.1. Steady-State and Stress Erythropoiesis in Physiological Conditions
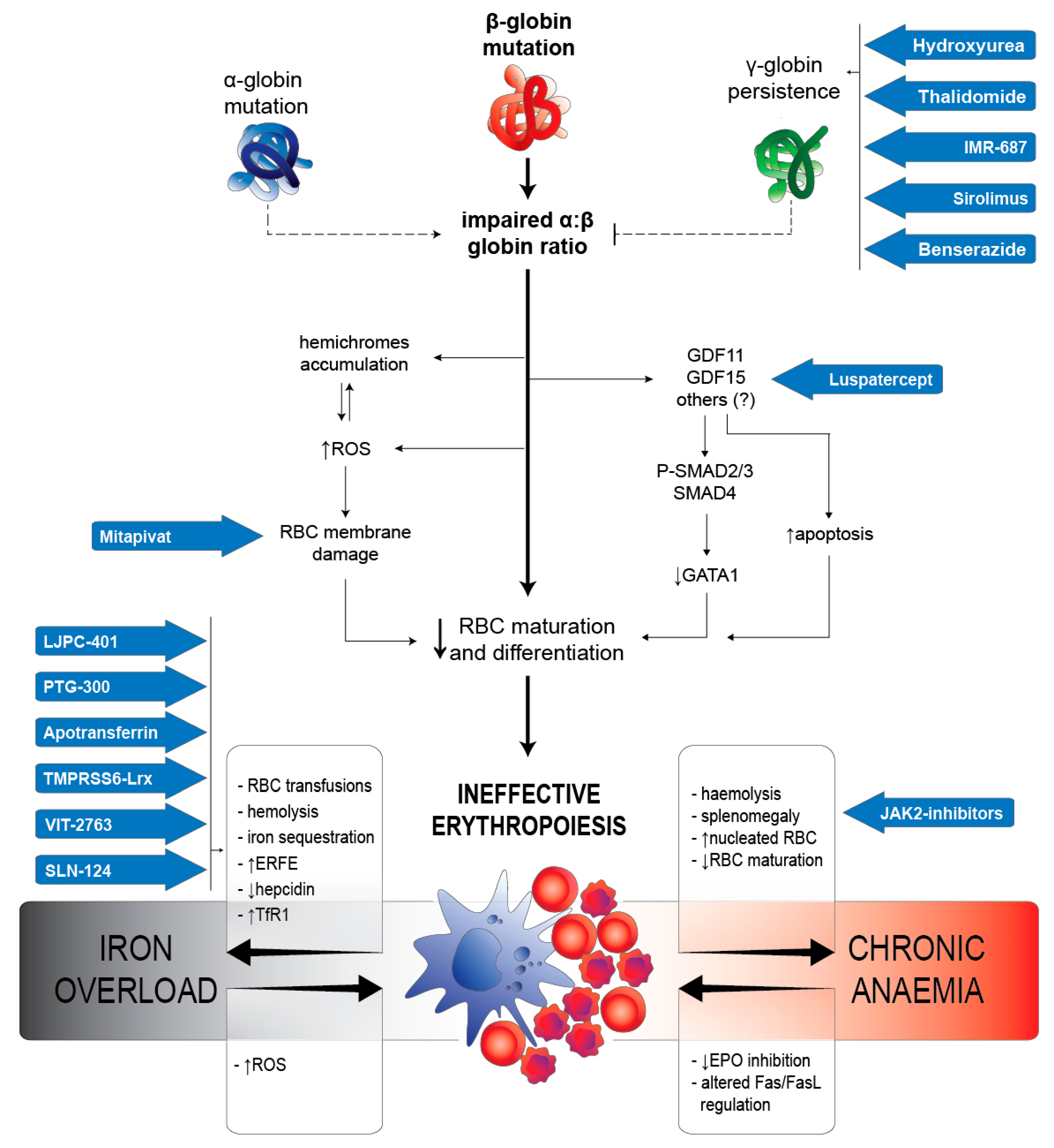
2.2. IE in β-Thalassaemia
2.2.1. RBC Alterations Caused by α:β Chains Impairment
2.2.2. Iron Metabolism Dysregulation
2.2.3. Arrest of Maturation Mediated by TGF-β Superfamily
2.2.4. Regulating Proliferation/Maturation Balance by GATA-1
3. Therapeutic Options by Drugs
3.1. Luspatercept (ACE-536)
3.2. Mitapivat (AG-348)
3.3. Modifiers of Iron Metabolism
3.3.1. Hepcidin Analogues
3.3.2. Apotransferrin
3.3.3. Inhibitors of TMPRSS6
3.3.4. Inhibition of Fpn by VIT-2763
3.4. Multiple Approaches to HbF Induction
3.4.1. Hydroxyurea
3.4.2. Thalidomide
3.4.3. IMR-687
3.4.4. Sirolimus
3.4.5. Benserazide
3.5. EPEG
4. Future Perspectives
Funding
Conflicts of Interest
References
- Tusi, B.K.; Wolock, S.L.; Weinreb, C.; Hwang, Y.; Hidalgo, D.; Zilionis, R.; Waisman, A.; Huh, J.R.; Klein, A.M.; Socolovsky, M. Population snapshots predict early haematopoietic and erythroid hierarchies. Nature 2018, 555, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Chen, K.; Liu, J.; Heck, S.; Chasis, J.A.; An, X.; Mohandas, N. Resolving the distinct stages in erythroid differentiation based on dynamic changes in membrane protein expression during erythropoiesis. Proc. Natl. Acad. Sci. USA 2009, 106, 17413–17418. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dulmovits, B.M.; Hom, J.; Narla, A.; Mohandas, N.; Blanc, L. Characterization, regulation, and targeting of erythroid progenitors in normal and disordered human erythropoiesis. Curr. Opin. Hematol. 2017, 24, 159–166. [Google Scholar] [CrossRef] [PubMed]
- An, X.; Schulz, V.P.; Li, J.; Wu, K.; Liu, J.; Xue, F.; Hu, J.; Mohandas, N.; Gallagher, P.G. Global transcriptome analyses of human and murine terminal erythroid differentiation. Blood 2014, 123, 3466–3477. [Google Scholar] [CrossRef] [Green Version]
- Chasis, J.A.; Mohandas, N. Erythroblastic islands: Niches for erythropoiesis. Blood 2008, 112, 470–478. [Google Scholar] [CrossRef] [Green Version]
- Koulnis, M.; Liu, Y.; Hallstrom, K.; Socolovsky, M. Negative autoregulation by Fas stabilizes adult erythropoiesis and accelerates its stress response. PLoS ONE 2011, 6, e21192. [Google Scholar] [CrossRef] [PubMed]
- Parisi, S.; Finelli, C.; Fazio, A.; De Stefano, A.; Mongiorgi, S.; Ratti, S.; Cappellini, A.; Billi, A.M.; Cocco, L.; Follo, M.Y.; et al. Clinical and Molecular Insights in Erythropoiesis Regulation of Signal Transduction Pathways in Myelodysplastic Syndromes and β-Thalassemia. Int. J. Mol. Sci. 2021, 22, 827. [Google Scholar] [CrossRef] [PubMed]
- Kim, A.; Nemeth, E. New insights into iron regulation and erythropoiesis. Curr. Opin. Hematol. 2015, 22, 199–205. [Google Scholar] [CrossRef]
- Paulson, R.F.; Shi, L.; Wu, D.C. Stress erythropoiesis: New signals and new stress progenitor cells. Curr. Opin. Hematol. 2011, 18, 139–145. [Google Scholar] [CrossRef] [Green Version]
- Paulson, R.F.; Hariharan, S.; Little, J.A. Stress erythropoiesis: Definitions and models for its study. Exp. Hematol. 2020, 89, 43–54. [Google Scholar] [CrossRef]
- Crielaard, B.J.; Rivella, S. β-Thalassemia and Polycythemia vera: Targeting chronic stress erythropoiesis. Int. J. Biochem. Cell Biol. 2014, 51, 89–92. [Google Scholar] [CrossRef] [Green Version]
- Centis, F.; Tabellini, L.; Lucarelli, G.; Buffi, O.; Tonucci, P.; Persini, B.; Annibali, M.; Emiliani, R.; Iliescu, A.; Rapa, S.; et al. The importance of erythroid expansion in determining the extent of apoptosis in erythroid precursors in patients with β-thalassemia major. Blood 2000, 96, 3624–3629. [Google Scholar] [CrossRef] [PubMed]
- Oikonomidou, P.R.; Rivella, S. What can we learn from ineffective erythropoiesis in thalassemia? Blood Rev. 2018, 32, 130–143. [Google Scholar] [CrossRef] [PubMed]
- Libani, I.V.; Guy, E.C.; Melchiori, L.; Schiro, R.; Ramos, P.; Breda, L.; Scholzen, T.; Chadburn, A.; Liu, Y.; Kernbach, M.; et al. Decreased differentiation of erythroid cells exacerbates ineffective erythropoiesis in β-thalassemia. Blood 2008, 112, 875–885. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Talawy, T.S.; Altoum, A.E.A.; Babker, A.M. The Role of Erythroferrone Hormone as Erythroid Regulator of Hepcidin and Iron Metabolism during Thalassemia and in Iron Deficiency Anemia—A Short Review. J. Pharm. Res. Int. 2020, 32, 55–59. [Google Scholar] [CrossRef]
- Cao, A.; Gossens, M.; Pirastu, M. β thalassaemia mutations in Mediterranean populations. Br. J. Haematol. 1989, 71, 309–312. [Google Scholar] [CrossRef] [PubMed]
- Thein, S.L. Dominant β thalassaemia: Molecular basis and pathophysiology. Br. J. Haematol. 1992, 80, 273–277. [Google Scholar] [CrossRef]
- Weatherall, D.J. Phenotype-genotype relationships in monogenic disease: Lessons from the thalassaemias. Nat. Rev. Genet. 2001, 2, 245–255. [Google Scholar] [CrossRef]
- Yuan, J.; Kannan, R.; Shinar, E.; Rachmilewitz, E.A.; Low, P.S. Isolation, characterization, and immunoprecipitation studies of immune complexes from membranes of beta-thalassemic erythrocytes. Blood 1992, 79, 3007–3013. [Google Scholar] [CrossRef]
- Welbourn, E.M.; Wilson, M.T.; Yusof, A.; Metodiev, M.V.; Cooper, C.E. The mechanism of formation, structure and physiological relevance of covalent hemoglobin attachment to the erythrocyte membrane. Free Radic. Biol. Med. 2017, 103, 95–106. [Google Scholar] [CrossRef] [Green Version]
- Mohanty, J.G.; Nagababu, E.; Rifkind, J.M. Red blood cell oxidative stress impairs oxygen delivery and induces red blood cell aging. Front. Physiol. 2014, 5, 84. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zahedpanah, M.; Azarkeivan, A.; Aghaieepour, M.; Nikogoftar, M.; Ahmadinegad, M.; Hajibeigi, B.; Tabatabaiee, M.R.; Maghsudlu, M. Erythrocytic phosphatidylserine exposure and hemostatic alterations in β-thalassemia intermediate patients. Hematology 2014, 19, 472–476. [Google Scholar] [CrossRef] [PubMed]
- Shinar, E.; Shalev, O.; Rachmilewitz, E.A.; Schrier, S.L. Erythrocyte membrane skeleton abnormalities in severe β-thalassemia. Blood 1987, 70, 158–164. [Google Scholar] [CrossRef] [Green Version]
- De Franceschi, L.; Bertoldi, M.; Matte, A.; Santos Franco, S.; Pantaleo, A.; Ferru, E.; Turrini, F. Oxidative stress and β-thalassemic erythroid cells behind the molecular defect. Oxid. Med. Cell. Longev. 2013, 2013, 985210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taher, A.T.; Saliba, A.N. Iron overload in thalassemia: Different organs at different rates. Hematology 2017, 2017, 265–271. [Google Scholar] [CrossRef] [Green Version]
- Razak, S.A.A.; Murad, N.A.A.; Masra, F.; Chong, D.L.S.; Abdullah, N.; Jalil, N.; Alauddin, H.; Sabudin, R.Z.A.R.; Ithnin, A.; Khai, L.C.; et al. Genetic Modifiers of Fetal Haemoglobin (HbF) and Phenotypic Severity in β-Thalassemia Patients. Curr. Mol. Med. 2018, 18, 295–305. [Google Scholar] [CrossRef]
- Gong, Y.; Zhang, X.; Zhang, Q.; Zhang, Y.; Ye, Y.; Yu, W.; Shao, C.; Yan, T.; Huang, J.; Zhong, J.; et al. A natural DNMT1 mutation elevates the fetal hemoglobin level via epigenetic derepression of the γ-globin gene in β-thalassemia. Blood 2021, 137, 1652–1657. [Google Scholar] [CrossRef]
- Beguin, Y.; Stray, S.M.; Cazzola, M.; Huebers, H.A.; Finch, C.A. Ferrokinetic measurement of erythropoiesis. Acta Haematol. 1988, 79, 121–126. [Google Scholar] [CrossRef]
- Kautz, L.; Jung, G.; Du, X.; Gabayan, V.; Chapman, J.; Nasoff, M.; Nemeth, E.; Ganz, T. Erythroferrone contributes to hepcidin suppression and iron overload in a mouse model of β-thalassemia. Blood 2015, 126, 2031–2037. [Google Scholar] [CrossRef] [Green Version]
- Kautz, L.; Jung, G.; Valore, E.V.; Rivella, S.; Nemeth, E.; Ganz, T. Identification of erythroferrone as an erythroid regulator of iron metabolism. Nat. Genet. 2014, 46, 678–684. [Google Scholar] [CrossRef] [Green Version]
- Hentze, M.W.; Muckenthaler, M.U.; Galy, B.; Camaschella, C. Two to tango: Regulation of Mammalian iron metabolism. Cell 2010, 142, 24–38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srole, D.N.; Ganz, T. Erythroferrone structure, function, and physiology: Iron homeostasis and beyond. J. Cell. Physiol. 2021, 236, 4888–4901. [Google Scholar] [CrossRef] [PubMed]
- Nai, A.; Pagani, A.; Mandelli, G.; Lidonnici, M.R.; Silvestri, L.; Ferrari, G.; Camaschella, C. Deletion of TMPRSS6 attenuates the phenotype in a mouse model of β-thalassemia. Blood 2012, 119, 5021–5029. [Google Scholar] [CrossRef] [PubMed]
- Stagg, D.B.; Whittlesey, R.L.; Li, X.; Lozovatsky, L.; Gardenghi, S.; Rivella, S.; Finberg, K.E. Genetic loss of Tmprss6 alters terminal erythroid differentiation in a mouse model of β-thalassemia intermedia. Haematologica 2019, 104, e442–e446. [Google Scholar] [CrossRef] [PubMed]
- Vadolas, J.; Ng, G.Z.; Kysenius, K.; Crouch, P.J.; Dames, S.; Eisermann, M.; Nualkaew, T.; Vilcassim, S.; Schaeper, U.; Grigoriadis, G. SLN124, a GalNac-siRNA targeting transmembrane serine protease 6, in combination with deferiprone therapy reduces ineffective erythropoiesis and hepatic iron-overload in a mouse model of β-thalassaemia. Br. J. Haematol. 2021, 194, 200–210. [Google Scholar] [CrossRef] [PubMed]
- Comporti, M.; Signorini, C.; Buonocore, G.; Ciccoli, L. Iron release, oxidative stress and erythrocyte ageing. Free Radic. Biol. Med. 2002, 32, 568–576. [Google Scholar] [CrossRef]
- Bozza, M.T.; Jeney, V. Pro-inflammatory Actions of Heme and Other Hemoglobin-Derived DAMPs. Front. Immunol. 2020, 11, 1–13. [Google Scholar] [CrossRef]
- Porter, J.; Cappellini, M.D.; Coates, T.; Hermine, O.; Viprakasit, V.; Voskaridou, E.; Liew, H.K.; Perrotta, S.; Khelif, A.; Kattamis, A.; et al. Effects of Luspatercept on Iron Overload and Impact on Responders to Luspatercept: Results from the BELIEVE Trial. Blood 2019, 134, 2245. [Google Scholar] [CrossRef]
- Camaschella, C.; Pagani, A.; Nai, A.; Silvestri, L. The mutual control of iron and erythropoiesis. Int. J. Lab. Hematol. 2016, 38 (Suppl. S1), 20–26. [Google Scholar] [CrossRef]
- Guimarães, J.S.; Cominal, J.G.; Silva-Pinto, A.C.; Olbina, G.; Ginzburg, Y.Z.; Nandi, V.; Westerman, M.; Rivella, S.; de Souza, A.M. Altered erythropoiesis and iron metabolism in carriers of thalassemia. Eur. J. Haematol. 2015, 94, 511–518. [Google Scholar] [CrossRef]
- Li, H.; Choesang, T.; Bao, W.; Chen, H.; Feola, M.; Garcia-Santos, D.; Li, J.; Sun, S.; Follenzi, A.; Pham, P.; et al. Decreasing TfR1 expression reverses anemia and hepcidin suppression in β-thalassemic mice. Blood 2017, 129, 1514–1526. [Google Scholar] [CrossRef] [Green Version]
- Forejtnikovà, H.; Vieillevoye, M.; Zermati, Y.; Lambert, M.; Pellegrino, R.M.; Guihard, S.; Gaudry, M.; Camaschella, C.; Lacombe, C.; Roetto, A.; et al. Transferrin receptor 2 is a component of the erythropoietin receptor complex and is required for efficient erythropoiesis. Blood 2010, 116, 5357–5367. [Google Scholar] [CrossRef] [Green Version]
- Artuso, I.; Lidonnici, M.R.; Altamura, S.; Mandelli, G.; Pettinato, M.; Muckenthaler, M.U.; Silvestri, L.; Ferrari, G.; Camaschella, C.; Nai, A. Transferrin receptor 2 is a potential novel therapeutic target for β-thalassemia: Evidence from a murine model. Blood 2018, 132, 2286–2297. [Google Scholar] [CrossRef] [Green Version]
- Rivella, S. Iron metabolism under conditions of ineffective erythropoiesis in β-Thalassemia. Blood 2019, 133, 51–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mueller, T.D. Mechanisms of BMP-Receptor Interaction and Activation. Vitam. Horm. 2015, 99, 1–61. [Google Scholar] [CrossRef] [PubMed]
- Derynck, R.; Zhang, Y.E. Smad-dependent and Smad-independent pathways in TGF-β family signalling. Nature 2003, 425, 577–584. [Google Scholar] [CrossRef]
- Suragani, R.N.V.S.; Cadena, S.M.; Cawley, S.M.; Sako, D.; Mitchell, D.; Li, R.; Davies, M.V.; Alexander, M.J.; Devine, M.; Loveday, K.S.; et al. Transforming growth factor-β superfamily ligand trap ACE-536 corrects anemia by promoting late-stage erythropoiesis. Nat. Med. 2014, 20, 408–414. [Google Scholar] [CrossRef] [PubMed]
- Martinez, P.A.; Li, R.; Ramanathan, H.N.; Bhasin, M.; Pearsall, R.S.; Kumar, R.; Suragani, R.N.V.S. Smad2/3-pathway ligand trap luspatercept enhances erythroid differentiation in murine β-thalassaemia by increasing GATA-1 availability. J. Cell. Mol. Med. 2020, 24, 6162–6177. [Google Scholar] [CrossRef]
- Guerra, A.; Oikonomidou, P.R.; Sinha, S.; Zhang, J.; Lo Presti, V.; Hamilton, C.R.; Breda, L.; Casu, C.; La, P.; Martins, A.C.; et al. Lack of Gdf11 does not improve anemia or prevent the activity of RAP-536 in a mouse model of b-thalassemia. Blood 2019, 134, 568–572. [Google Scholar] [CrossRef] [PubMed]
- Fang, Z.; Zhu, Z.; Zhang, H.; Peng, Y.; Liu, J.; Lu, H.; Li, J.; Liang, L.; Xia, S.; Wang, Q.; et al. GDF11 contributes to hepatic hepcidin (HAMP) inhibition through SMURF1-mediated BMP-SMAD signalling suppression. Br. J. Haematol. 2020, 188, 321–331. [Google Scholar] [CrossRef]
- Tanno, T.; Noel, P.; Miller, J.L. Growth differentiation factor 15 in erythroid health and disease. Curr. Opin. Hematol. 2010, 17, 184–190. [Google Scholar] [CrossRef]
- Musallam, K.M.; Taher, A.T.; Duca, L.; Cesaretti, C.; Halawi, R.; Cappellini, M.D. Levels of growth differentiation factor-15 are high and correlate with clinical severity in transfusion-independent patients with β thalassemia intermedia. Blood Cells. Mol. Dis. 2011, 47, 232–234. [Google Scholar] [CrossRef]
- Salussoglia, I.; Volpe, G.; Fracchia, S.; Roggero, S.; Longo, F.; Piga, A. Growth Differentiation Factor 15 (GDF15) and Erythropoietin (EPO) Levels in Beta Talassemia Major Patients. Blood 2008, 112, 1881. [Google Scholar] [CrossRef]
- Ranjbaran, R.; Abbasi, M.; Rahimian, E.; Dehbidi, G.R.; Seyyedi, N.; Zare, F.; Behzad-Behbahani, A. GDF-15 negatively regulates excess erythropoiesis and its overexpression is involved in erythroid hyperplasia. Exp. Cell Res. 2020, 397, 112346. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, R.; Ohneda, K.; Yamamoto, M.; Philipsen, S. GATA1 function, a paradigm for transcription factors in hematopoiesis. Mol. Cell. Biol. 2005, 25, 1215–1227. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutiérrez, L.; Caballero, N.; Fernández-Calleja, L.; Karkoulia, E.; Strouboulis, J. Regulation of GATA1 levels in erythropoiesis. IUBMB Life 2020, 72, 89–105. [Google Scholar] [CrossRef]
- Arlet, J.-B.; Ribeil, J.-A.; Guillem, F.; Negre, O.; Hazoume, A.; Marcion, G.; Beuzard, Y.; Dussiot, M.; Moura, I.C.; Demarest, S.; et al. HSP70 sequestration by free α-globin promotes ineffective erythropoiesis in β-thalassaemia. Nature 2014, 514, 242–246. [Google Scholar] [CrossRef] [PubMed]
- De Maria, R.; Zeuner, A.; Eramo, A.; Domenichelli, C.; Bonci, D.; Grignani, F.; Srinivasula, S.M.; Alnemri, E.S.; Testa, U.; Peschle, C. Negative regulation of erythropoiesis by caspase-mediated cleavage of GATA-1. Nature 1999, 401, 489–493. [Google Scholar] [CrossRef] [PubMed]
- Ribeil, J.-A.; Zermati, Y.; Vandekerckhove, J.; Cathelin, S.; Kersual, J.; Dussiot, M.; Coulon, S.; Moura, I.C.; Zeuner, A.; Kirkegaard-Sørensen, T.; et al. Hsp70 regulates erythropoiesis by preventing caspase-3-mediated cleavage of GATA-1. Nature 2007, 445, 102–105. [Google Scholar] [CrossRef]
- Chen, J.-J. Regulation of protein synthesis by the heme-regulated eIF2alpha kinase: Relevance to anemias. Blood 2007, 109, 2693–2699. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.-J.; Zhang, S. Heme-regulated eIF2α kinase in erythropoiesis and hemoglobinopathies. Blood 2019, 134, 1697–1707. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.A.; Walters, M.C.; Kwiatkowski, J.; Rasko, J.E.J.; Ribeil, J.-A.; Hongeng, S.; Magrin, E.; Schiller, G.J.; Payen, E.; Semeraro, M.; et al. Gene Therapy in Patients with Transfusion-Dependent β-Thalassemia. N. Engl. J. Med. 2018, 378, 1479–1493. [Google Scholar] [CrossRef] [PubMed]
- Frangoul, H.; Altshuler, D.; Cappellini, M.D.; Chen, Y.-S.; Domm, J.; Eustace, B.K.; Foell, J.; de la Fuente, J.; Grupp, S.; Handgretinger, R.; et al. CRISPR-Cas9 Gene Editing for Sickle Cell Disease and β-Thalassemia. N. Engl. J. Med. 2020. [Google Scholar] [CrossRef]
- Porter, J. Beyond transfusion therapy: New therapies in thalassemia including drugs, alternate donor transplant, and gene therapy. Hematol. 2014 Am. Soc. Hematol. Educ. Program Book 2018, 2018, 361–370. [Google Scholar] [CrossRef] [Green Version]
- Piga, A.; Perrotta, S.; Gamberini, M.R.; Voskaridou, E.; Melpignano, A.; Filosa, A.; Caruso, V.; Pietrangelo, A.; Longo, F.; Tartaglione, I.; et al. Luspatercept improves hemoglobin levels and blood transfusion requirements in a study of patients with b-thalassemia. Blood 2019, 133, 1279–1289. [Google Scholar] [CrossRef] [Green Version]
- Cappellini, M.D.; Viprakasit, V.; Taher, A.T.; Georgiev, P.; Kuo, K.H.M.; Coates, T.; Voskaridou, E.; Liew, H.-K.; Pazgal-Kobrowski, I.; Forni, G.L.; et al. A Phase 3 Trial of Luspatercept in Patients with Transfusion-Dependent β-Thalassemia. N. Engl. J. Med. 2020, 382, 1219–1231. [Google Scholar] [CrossRef]
- Chen, N.; Kassir, N.; Laadem, A.; Giuseppi, A.C.; Shetty, J.; Maxwell, S.E.; Sriraman, P.; Ritland, S.; Linde, P.G.; Budda, B.; et al. Population Pharmacokinetics and Exposure-Response Relationship of Luspatercept, an Erythroid Maturation Agent, in Anemic Patients With β-Thalassemia. J. Clin. Pharmacol. 2021, 61, 52–63. [Google Scholar] [CrossRef]
- Taher, A.T.; Cappellini, M.D.; Kattamis, A.; Voskaridou, E.; Perrotta, S.; Piga, A.; Filosa, A.; Porter, J.B.; Coates, T.D.; Forni, G.L.; et al. The beyond study: Results of a phase 2, double-blind, randomized, placebo-controlled multi center study of luspatercept in adult patients with non-transfusion dependent β-thalassemia. In Proceedings of the 26th Congress of the European Hematology Association, Hague, The Netherlands, 9–17 June 2021; p. S101. Available online: https://eha2021.ehaweb.org/program/eha/eha2021/en-US?filter=abstract (accessed on 30 May 2021).
- Kung, C.; Hixon, J.; Kosinski, P.A.; Cianchetta, G.; Histen, G.; Chen, Y.; Hill, C.; Gross, S.; Si, Y.; Johnson, K.; et al. AG-348 enhances pyruvate kinase activity in red blood cells from patients with pyruvate kinase deficiency. Blood 2017, 130, 1347–1356. [Google Scholar] [CrossRef]
- Grace, R.F.; Rose, C.; Layton, D.M.; Galactéros, F.; Barcellini, W.; Morton, D.H.; van Beers, E.J.; Yaish, H.; Ravindranath, Y.; Kuo, K.H.M.; et al. Safety and Efficacy of Mitapivat in Pyruvate Kinase Deficiency. N. Engl. J. Med. 2019, 381, 933–944. [Google Scholar] [CrossRef]
- Kuo, K.H.M.; Layton, D.M.; Lal, A.; AL-Samkari, H.; Tai, F.; Lynch, M.; Uhlig, K.; Vichinsky, E.P. Proof of concept for the oral pyruvate kinase activator mitapivat in adults with non–transfusion-dependent thalassemia: Interim results from an ongoing, phase 2, open-label, multicenter study. Blood 2020, 136, abst-2600. [Google Scholar]
- Casu, C.; Oikonomidou, P.R.; Chen, H.; Nandi, V.; Ginzburg, Y.; Prasad, P.; Fleming, R.E.; Shah, Y.M.; Valore, E.V.; Nemeth, E.; et al. Minihepcidin peptides as disease modifiers in mice affected by β-thalassemia and polycythemia vera. Blood 2016, 128, 265–276. [Google Scholar] [CrossRef] [Green Version]
- Casu, C.; Chessa, R.; Liu, A.; Gupta, R.; Drakesmith, H.; Fleming, R.; Ginzburg, Y.Z.; MacDonald, B.; Rivella, S. Minihepcidins improve ineffective erythropoiesis and splenomegaly in a new mouse model of adult β-thalassemia major. Haematologica 2020, 105, 1835–1844. [Google Scholar] [CrossRef] [Green Version]
- Nicholls, A.; Likliter, J.; Tozzi, L.; Liu, D.; Shames, R. Hepcidin mimetic PTG-300 induces dose-related and sustained reductions in serum iron and transferrin saturation in healthy. In Proceedings of the 23rd Congress of the European Hematology Association, Stockholm, Sweden, 14–17 June 2018; p. S895. [Google Scholar]
- Lal, A.; Piga, A.; Viprakasit, V.; Maynard, J.; Kattamis, A.; Yaeger, D.; Byrnes, B.; Chawla, L.; Tidmarsh, G. A Phase 1, Open-Label Study to Determine the Safety, Tolerability, and Pharmacokinetics of Escalating Doses of LJPC-401 (Synthetic Human Hepcidin) in Patients with Iron Overload. In Proceedings of the 23rd Congress of the European Hematology Association, Stockholm, Sweden, 14–17 June 2018; p. S894. [Google Scholar]
- Bourne, G.; Li, Z.; Brandari, A.; Frederick, B.; McMahon, J.; Tran, V. Hepcidin mimetic PTG-300 for treatment of ineffective erythropoiesis and chronic anemia in hemoglobinopathy diseases. In Proceedings of the 23rd Congress of the European Hematology Association, Stockholm, Sweden, 14–17 June 2018; p. S843. [Google Scholar]
- Lal, A.; Voskaridou, E.; Flevari, P.; Taher, A.; Chew, L.-P.; Valone, F.; Gupta, S.; Viprakasit, V. A hepcidin mimetic, PTG-300, demonstrates pharmacodynamic effects indicating reduced iron availability in transfusion-dependent beta-thalassemia subjects. In Proceedings of the 25th Congress of the European Hematology Association, Virtual Congress, Stockholm, Sweden, 11–21 June 2020; p. S298. [Google Scholar]
- Li, H.; Rybicki, A.C.; Suzuka, S.M.; von Bonsdorff, L.; Breuer, W.; Hall, C.B.; Cabantchik, Z.I.; Bouhassira, E.E.; Fabry, M.E.; Ginzburg, Y.Z. Transferrin therapy ameliorates disease in β-thalassemic mice. Nat. Med. 2010, 16, 177–182. [Google Scholar] [CrossRef]
- Goya, N.; Miyazaki, S.; Kodate, S.; Ushio, B. A family of congenital atransferrinemia. Blood 1972, 40, 239–245. [Google Scholar] [CrossRef] [Green Version]
- Gelderman, M.P.; Baek, J.H.; Yalamanoglu, A.; Puglia, M.; Vallelian, F.; Burla, B.; Vostal, J.; Schaer, D.J.; Buehler, P.W. Reversal of hemochromatosis by apotransferrin in non-transfused and transfused Hbbth3/+ (Heterozygous b1/b2 globin gene deletion) mice. Haematologica 2015, 100, 611–622. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, P.J.; Toudjarska, I.; Sendamarai, A.K.; Racie, T.; Milstein, S.; Bettencourt, B.R.; Hettinger, J.; Bumcrot, D.; Fleming, M.D. An RNAi therapeutic targeting Tmprss6 decreases iron overload in Hfe−/− mice and ameliorates anemia and iron overload in murine β-thalassemia intermedia. Blood 2013, 121, 1200–1208. [Google Scholar] [CrossRef]
- Guo, S.; Casu, C.; Gardenghi, S.; Booten, S.; Aghajan, M.; Peralta, R.; Watt, A.; Freier, S.; Monia, B.P.; Rivella, S. Reducing TMPRSS6 ameliorates hemochromatosis and β-thalassemia in mice. J. Clin. Investig. 2013, 123, 1531–1541. [Google Scholar] [CrossRef] [Green Version]
- McCaleb, M.; Lickliter, J.; Dibble, A.; Schneider, E.; Aghajan, M.; Guo, S.; Hughes, S.; Geary, R.S.; Monia, B.P. Transmembrane Protease, Serine 6 (TMPRSS6) Antisense Oligonucleotide (IONIS-TMPRSS6-LRX) Reduces Plasma Iron Levels of Healthy Volunteers in a Phase 1 Clinical Study. Blood 2018, 132, 3634. [Google Scholar] [CrossRef]
- Manolova, V.; Nyffenegger, N.; Flace, A.; Altermatt, P.; Varol, A.; Doucerain, C.; Sundstrom, H.; Dürrenberger, F. Oral ferroportin inhibitor ameliorates ineffective erythropoiesis in a model of β-thalassemia. J. Clin. Investig. 2019, 130, 491–506. [Google Scholar] [CrossRef] [PubMed]
- Richard, F.; van Lier, J.J.; Roubert, B.; Haboubi, T.; Göhring, U.-M.; Dürrenberger, F. Oral ferroportin inhibitor VIT-2763: First-in-human, phase 1 study in healthy volunteers. Am. J. Hematol. 2020, 95, 68–77. [Google Scholar] [CrossRef]
- Dover, G.J.; Boyer, S.H. Fetal hemoglobin-containing cells have the same mean corpuscular hemoglobin as cells without fetal hemoglobin: A reciprocal relationship between gamma- and beta-globin gene expression in normal subjects and in those with high fetal hemoglobin production. Blood 1987, 69, 1109–1113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Babbs, C.; Higgs, D.R. Variable cells with identical genetic codes. Blood 2020, 135, 1921–1922. [Google Scholar] [CrossRef]
- Crona, M.; Codó, P.; Jonna, V.R.; Hofer, A.; Fernandes, A.P.; Tholander, F. A ribonucleotide reductase inhibitor with deoxyribonucleoside-reversible cytotoxicity. Mol. Oncol. 2016, 10, 1375–1386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pule, G.D.; Mowla, S.; Novitzky, N.; Wiysonge, C.S.; Wonkam, A. A systematic review of known mechanisms of hydroxyurea-induced fetal hemoglobin for treatment of sickle cell disease. Expert Rev. Hematol. 2015, 8, 669–679. [Google Scholar] [CrossRef] [Green Version]
- Wang, M.; Tang, D.C.; Liu, W.; Chin, K.; Zhu, J.G.; Fibach, E.; Rodgers, G.P. Hydroxyurea exerts bi-modal dose-dependent effects on erythropoiesis in human cultured erythroid cells via distinct pathways. Br. J. Haematol. 2002, 119, 1098–1105. [Google Scholar] [CrossRef]
- Flanagan, J.M.; Steward, S.; Howard, T.A.; Mortier, N.A.; Kimble, A.C.; Aygun, B.; Hankins, J.S.; Neale, G.A.; Ware, R.E. Hydroxycarbamide alters erythroid gene expression in children with sickle cell anaemia. Br. J. Haematol. 2012, 157, 240–248. [Google Scholar] [CrossRef]
- Zohaib, M.; Ansari, S.H.; Shamsi, T.S.; Zubarev, R.A.; Zarina, S. Pharmacoproteomics Profiling of Plasma From β-Thalassemia Patients in Response to Hydroxyurea Treatment. J. Clin. Pharmacol. 2019, 59, 98–106. [Google Scholar] [CrossRef] [Green Version]
- Yasara, N.; Premawardhena, A.; Mettananda, S. A comprehensive review of hydroxyurea for β-haemoglobinopathies : The role revisited during COVID-19 pandemic. Orphanet J. Rare Dis. 2021, 16, 1–12. [Google Scholar] [CrossRef]
- Meo, A.; Cassinerio, E.; Castelli, R.; Bignamini, D.; Perego, L.; Cappellini, M.D. Effect of hydroxyurea on extramedullary haematopoiesis in thalassaemia intermedia: Case reports and literature review. Int. J. Lab. Hematol. 2008, 30, 425–431. [Google Scholar] [CrossRef]
- Yasara, N.; Wickramarathne, N.; Mettananda, C.; Manamperi, A.; Premawardhena, A.; Mettananda, S. Efficacy and safety of oral hydroxyurea in transfusion-dependent β-thalassaemia: A protocol for randomised double-blind controlled clinical trial. BMJ Open 2020, 10, 1–6. [Google Scholar] [CrossRef]
- Amare, G.G.; Meharie, B.G.; Belayneh, Y.M. A drug repositioning success: The repositioned therapeutic applications and mechanisms of action of thalidomide. J. Oncol. Pharm. Pract. 2021, 27, 673–678. [Google Scholar] [CrossRef]
- Dulmovits, B.M.; Appiah-Kubi, A.O.; Papoin, J.; Hale, J.; He, M.; Al-Abed, Y.; Didier, S.; Gould, M.; Husain-Krautter, S.; Singh, S.A.; et al. Pomalidomide reverses γ-globin silencing through the transcriptional reprogramming of adult hematopoietic progenitors. Blood 2016, 127, 1481–1492. [Google Scholar] [CrossRef]
- Masera, N.; Tavecchia, L.; Capra, M.; Cazzaniga, G.; Vimercati, C.; Pozzi, L.; Biondi, A.; Masera, G. Optimal response to thalidomide in a patient with thalassaemia major resistant to conventional therapy. Blood Transfus. 2010, 8, 63–65. [Google Scholar] [CrossRef]
- Aguilar-Lopez, L.B.; Delgado-Lamas, J.L.; Rubio-Jurado, B.; Perea, F.J.; Ibarra, B. Thalidomide therapy in a patient with thalassemia major. Blood Cells. Mol. Dis. 2008, 41, 136–137. [Google Scholar] [CrossRef]
- Chen, J.; Zhu, W.; Cai, N.; Bu, S.; Li, J.; Huang, L. Thalidomide induces haematologic responses in patients with β-thalassaemia. Eur. J. Haematol. 2017, 99, 437–441. [Google Scholar] [CrossRef]
- Ren, Q.; Zhou, Y.-L.; Wang, L.; Chen, Y.-S.; Ma, Y.-N.; Li, P.-P.; Yin, X.-L. Clinical trial on the effects of thalidomide on hemoglobin synthesis in patients with moderate thalassemia intermedia. Ann. Hematol. 2018, 97, 1933–1939. [Google Scholar] [CrossRef] [PubMed]
- Bhurani, D.; Kapoor, J.; Yadav, N.; Khushoo, V.; Agrawal, N.; Ahmed, R.; Arora, J.S.; Mehta, P. Experience with combination of hydroxyurea and low-dose thalidomide in transfusion-dependent beta thalassemia patients. Ann. Hematol. 2021, 100, 1417–1427. [Google Scholar] [CrossRef]
- Palumbo, A.; Palladino, C. Venous and arterial thrombotic risks with thalidomide: Evidence and practical guidance. Ther. Adv. Drug Saf. 2012, 3, 255–266. [Google Scholar] [CrossRef] [Green Version]
- Andemariam, B.; Scheele, W.; Gordeuk, V.; Howard, J.; Kanter, J.; Eleftheriou, P.; Pancham, S.; Hagar, R.; Clarke, L.; Gershwin, B.; et al. IMR-687, a highly selective phosphodiesterase 9 inhibitor (PDE9I), increases F-cells and fetal hemoglobin in a PH-2A interim analysis. In Proceedings of the 25th Congress of the European Hematology Association, Hague, The Netherlands, 11–21 June 2020; p. S290. Available online: https://library.ehaweb.org/eha/2020/eha25th/295109/ (accessed on 26 April 2021).
- Fibach, E.; Bianchi, N.; Borgatti, M.; Zuccato, C.; Finotti, A.; Lampronti, I.; Prus, E.; Mischiati, C.; Gambari, R. Effects of rapamycin on accumulation of α-, β- and γ-globin mRNAs in erythroid precursor cells from β-thalassaemia patients. Eur. J. Haematol. 2006, 77, 437–441. [Google Scholar] [CrossRef]
- Santos, M.E.H.P.; Olops, L.; Vendrame, F.; Tavares, A.H.J.; Leonardo, D.P.; de Azevedo, P.C.; Piovesana, L.G.; Costa, F.F.; Fertrin, K.Y. Benserazide as a potential novel fetal hemoglobin inducer: An observational study in non-carriers of hemoglobin disorders. Blood Cells Mol. Dis. 2021, 87, 102511. [Google Scholar] [CrossRef]
- Pace, B.S.; Perrine, S.; Li, B.; Makala, L.; Xu, H.; Takezaki, M.; Wolf, R.F.; Wang, A.; Xu, X.; Huang, J.; et al. Benserazide racemate and enantiomers induce fetal globin gene expression in vivo: Studies to guide clinical development for beta thalassemia and sickle cell disease. Blood Cells Mol. Dis. 2021, 89, 102561. [Google Scholar] [CrossRef] [PubMed]
- Singer, S.T.; Kuypers, F.A.; Olivieri, N.F.; Weatherall, D.J.; Mignacca, R.; Coates, T.D.; Davies, S.; Sweeters, N.; Vichinsky, E.P. Single and combination drug therapy for fetal hemoglobin augmentation in hemoglobin E-β0-thalassemia: Considerations for treatment. Ann. N. Y. Acad. Sci. 2005, 1054, 250–256. [Google Scholar] [CrossRef]
- Nişli, G.; Kavakli, K.; Aydinok, Y.; Oztop, S.; Cetingül, N. Termination of transfusion dependence in beta-thalassemia: Two-year experience with recombinant human erythropoietin. Pediatr. Hematol. Oncol. 1997, 14, 285–287. [Google Scholar]
- Casu, C.; Pettinato, M.; Liu, A.; Aghajan, M.; Lo Presti, V.; Lidonnici, M.R.; Munoz, K.A.; O’Hara, E.; Olivari, V.; Di Modica, S.M.; et al. Correcting β-thalassemia by combined therapies that restrict iron and modulate erythropoietin activity. Blood 2020, 136, 1968–1979. [Google Scholar] [CrossRef]
- Schmidt, P.J.; Fitzgerald, K.; Butler, J.S.; Fleming, M.D. Global loss of Tfr2 with concomitant induced iron deficiency greatly ameliorates the phenotype of a murine thalassemia intermedia model. Am. J. Hematol. 2021, 96, 251–257. [Google Scholar] [CrossRef]
- Crippa, S.; Rossella, V.; Aprile, A.; Silvestri, L.; Rivis, S.; Scaramuzza, S.; Pirroni, S.; Avanzini, M.A.; Basso-Ricci, L.; Hernandez, R.J.; et al. Bone marrow stromal cells from β-thalassemia patients have impaired hematopoietic supportive capacity. J. Clin. Investig. 2019, 129, 1566–1580. [Google Scholar] [CrossRef] [Green Version]
- Aprile, A.; Gulino, A.; Storto, M.; Villa, I.; Beretta, S.; Merelli, I.; Rubinacci, A.; Ponzoni, M.; Marktel, S.; Tripodo, C.; et al. Hematopoietic stem cell function in β-thalassemia is impaired and is rescued by targeting the bone marrow niche. Blood 2020, 136, 610–622. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Longo, F.; Piolatto, A.; Ferrero, G.B.; Piga, A. Ineffective Erythropoiesis in ?-Thalassaemia: Key Steps and Therapeutic Options by Drugs. Int. J. Mol. Sci. 2021, 22, 7229. https://doi.org/10.3390/ijms22137229
Longo F, Piolatto A, Ferrero GB, Piga A. Ineffective Erythropoiesis in ?-Thalassaemia: Key Steps and Therapeutic Options by Drugs. International Journal of Molecular Sciences. 2021; 22(13):7229. https://doi.org/10.3390/ijms22137229
Chicago/Turabian StyleLongo, Filomena, Andrea Piolatto, Giovanni Battista Ferrero, and Antonio Piga. 2021. "Ineffective Erythropoiesis in ?-Thalassaemia: Key Steps and Therapeutic Options by Drugs" International Journal of Molecular Sciences 22, no. 13: 7229. https://doi.org/10.3390/ijms22137229
APA StyleLongo, F., Piolatto, A., Ferrero, G. B., & Piga, A. (2021). Ineffective Erythropoiesis in ?-Thalassaemia: Key Steps and Therapeutic Options by Drugs. International Journal of Molecular Sciences, 22(13), 7229. https://doi.org/10.3390/ijms22137229