
Preconditioning-Activated AKT Controls Neuronal Tolerance to Ischemia through the MDM2–p53 Pathway

 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
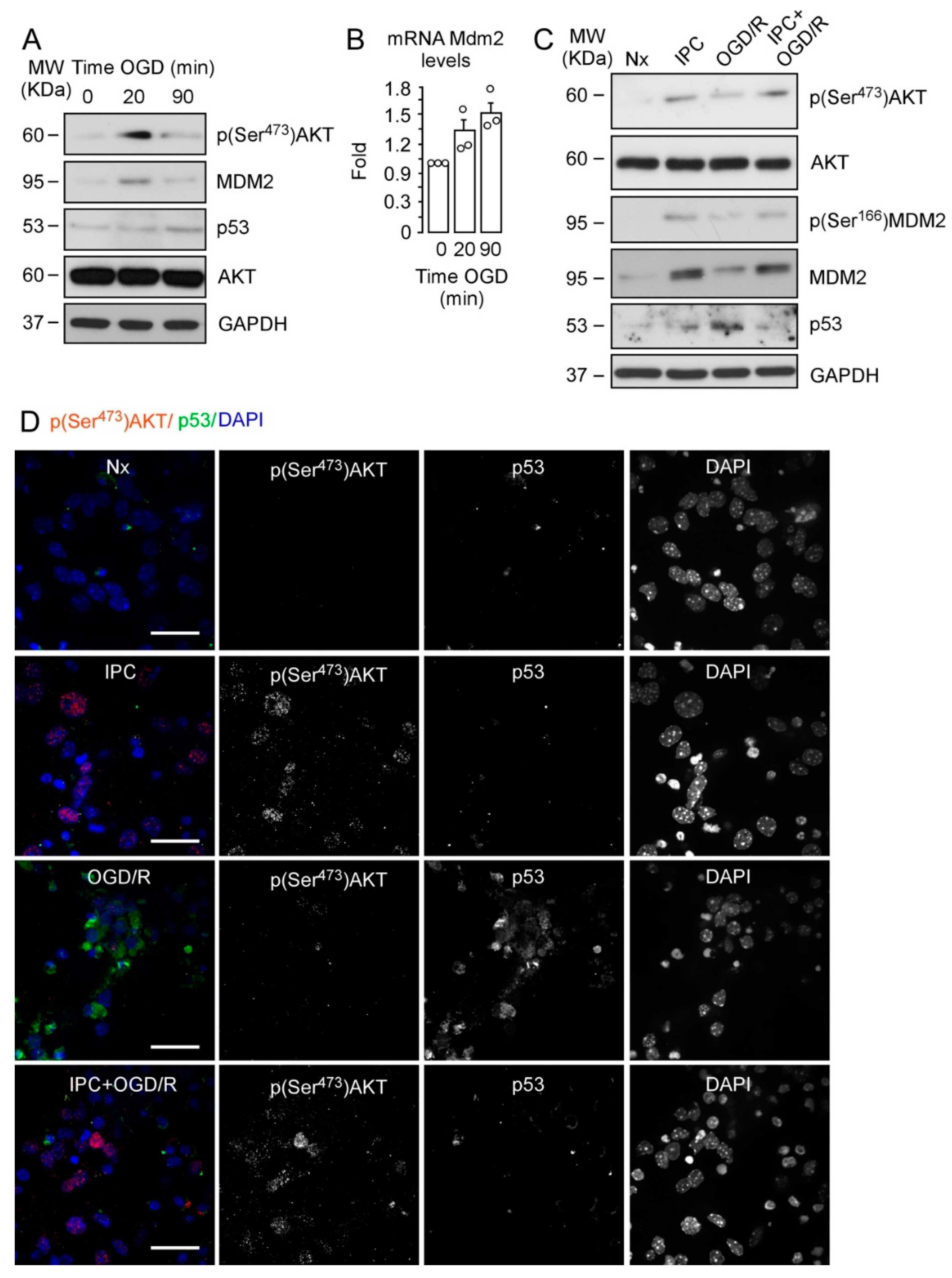
2.1. IPC-Promoted Neuroprotection Is Mediated by Phosphorylation of AKT at Ser473, Phosphorylation of MDM2 at Ser166, and p53 Destabilization
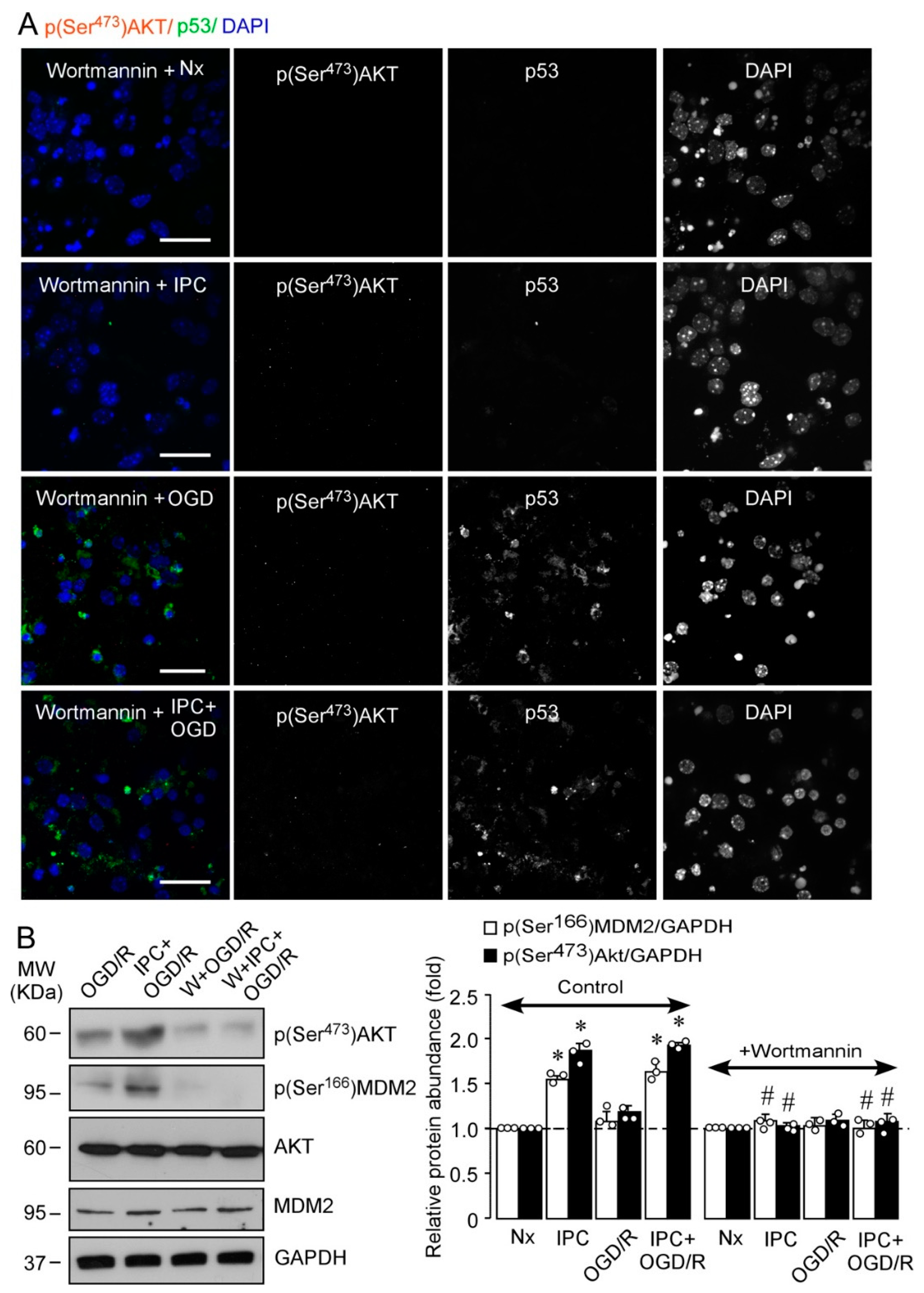
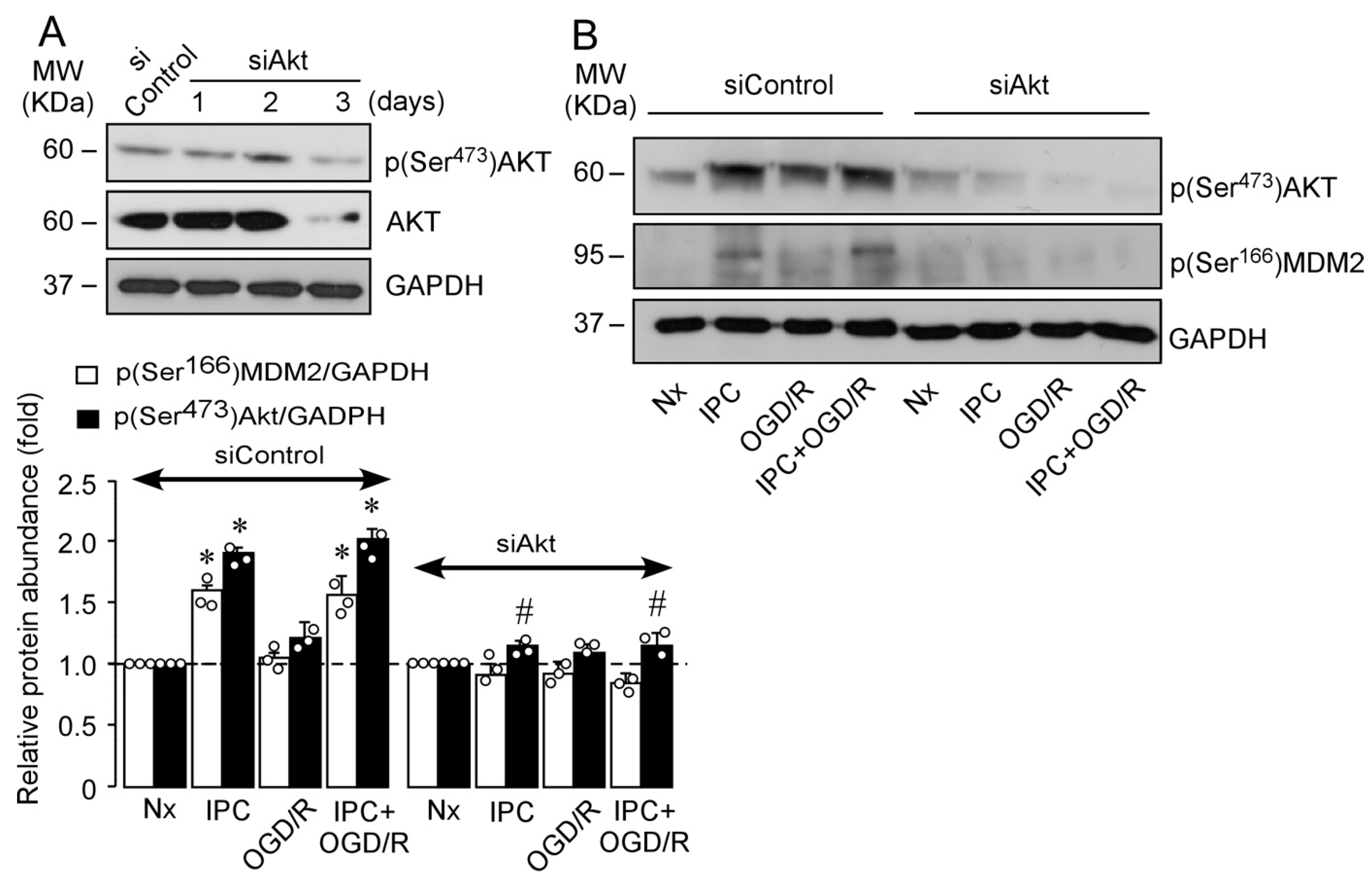
2.2. IPC Triggers MDM2 Phosphorylation at Ser166 via the PI3K/AKT Pathway
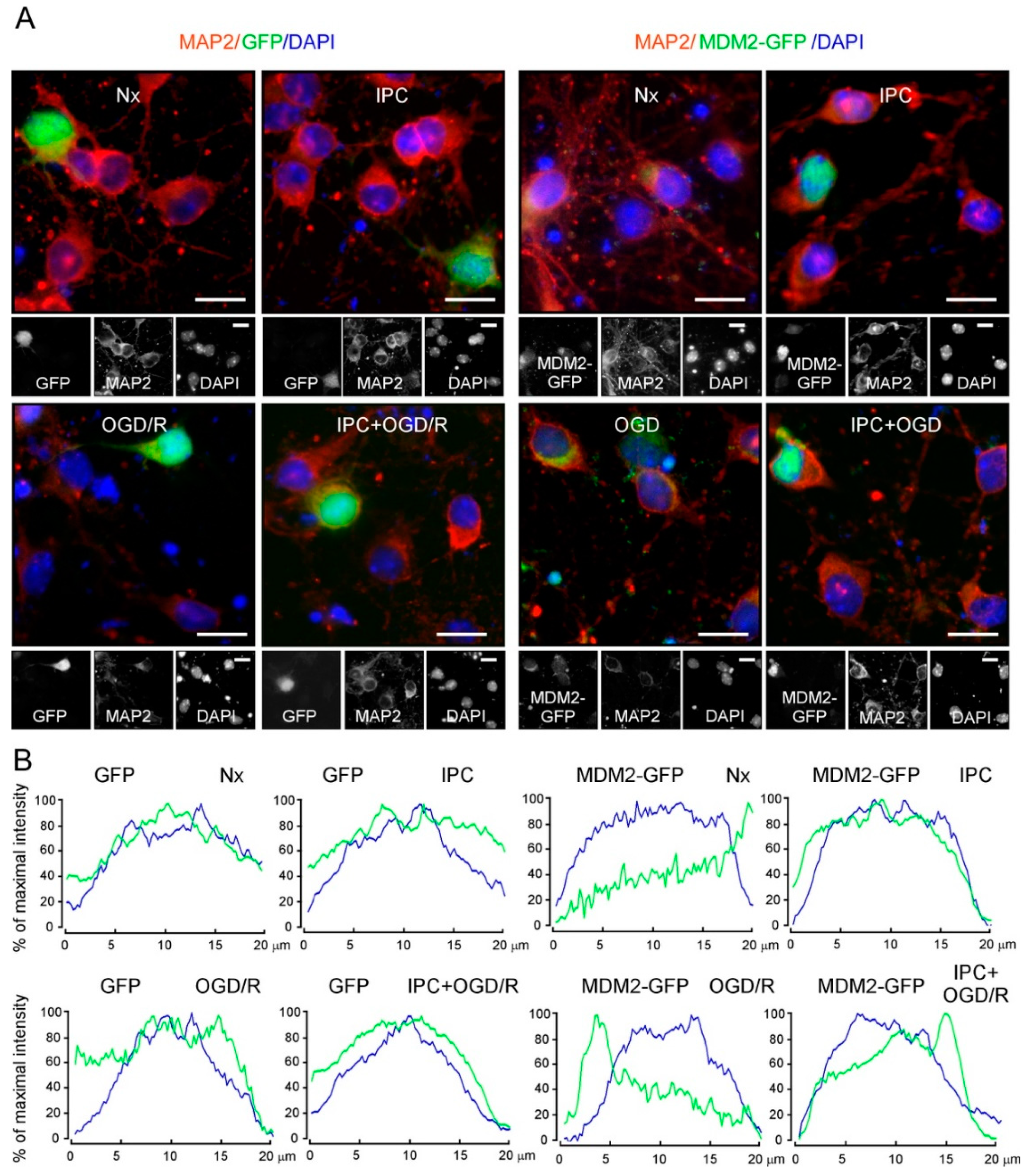
2.3. IPC-Activated AKT Triggers Nuclear MDM2 Protein Stabilization after Ischemia
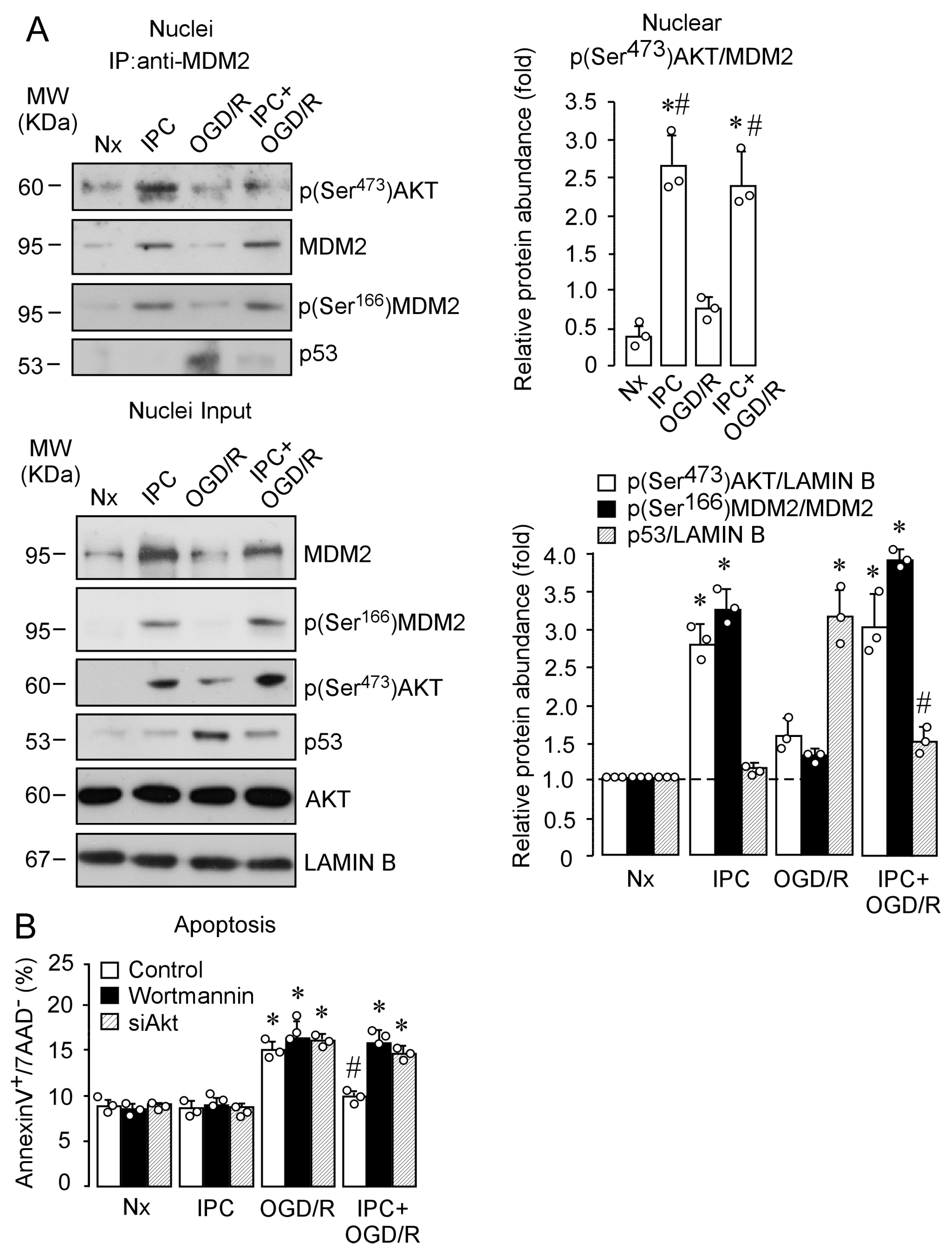
2.4. IPC Promotes p(Ser473)AKT and MDM2 Interaction, Which Enhances MDM2 Stabilization in the Nucleus and Reduces Induced Neuronal Apoptosis upon Ischemia
3. Discussion
4. Materials and Methods
4.1. Primary Cultures of Cortical Neurons
4.2. Oxygen Glucose Deprivation and Preconditioning Models
4.3. Cell Transfections
4.4. Flow Cytometric Detection of Apoptotic Cell Death
4.5. Caspase-3 Activity Assay
4.6. Immunoblots and Co-Immunoprecipitation Assay
4.7. Immunocytochemistry and Image Analysis
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Acknowledgments
Conflicts of Interest
References
- Emberson, J.; Lees, K.R.; Lyden, P.; Blackwell, L.; Albers, G.; Bluhmki, E.; Brott, T.; Cohen, G.; Davis, S.; Donnan, G.; et al. Effect of treatment delay, age, and stroke severity on the effects of intravenous thrombolysis with alteplase for acute ischaemic stroke: A meta-analysis of individual patient data from randomised trials. Lancet 2014, 384, 1929–1935. [Google Scholar] [CrossRef] [Green Version]
- Wang, W.-W.; Chen, D.-Z.; Zhao, M.; Yang, X.-F.; Gong, D.-R. Prior transient ischemic attacks may have a neuroprotective effect in patients with ischemic stroke. Arch. Med. Sci. 2017, 5, 1057–1061. [Google Scholar] [CrossRef] [PubMed]
- Ramos-Araque, M.E.; Rodriguez, C.; Vecino, R.; Garcia, E.C.; Alfonso, M.D.L.; Barba, M.S.; Colàs-Campàs, L.; Purroy, F.; Arenillas, J.F.; Almeida, A.; et al. The Neuronal Ischemic Tolerance Is Conditioned by the Tp53 Arg72Pro Polymorphism. Transl. Stroke Res. 2019, 10, 204–215. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Iadecola, C.; Anrather, J. Stroke research at a crossroad: Asking the brain for directions. Nat. Neurosci. 2011, 14, 1363–1368. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, C.; Jiang, M.; Zhang, L.; Hu, Y.; Hu, Z.; Zhang, M.; Qi, J.; Su, A.; Lou, N.; Xian, X.; et al. Peroxisome proliferator-activated receptor gamma participates in the acquisition of brain ischemic tolerance induced by ischemic preconditioning via glial glutamate transporter 1 in vivo and in vitro. J. Neurochem. 2019, 151, 608–625. [Google Scholar] [CrossRef]
- Rodriguez, C.; Agulla, J.; Delgado-Esteban, M. Refocusing the Brain: New Approaches in Neuroprotection against Ischemic Injury. Neurochem. Res. 2021, 46, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Stenzel-Poore, M.P.; Stevens, S.L.; Xiong, Z.; Lessov, N.S.; Harrington, A.C.; Mori, M.; Meller, R.; Rosenzweig, H.L.; Tobar, E.; Shaw, E.T.; et al. Effect of ischaemic preconditioning on genomic response to cerebral ischaemia: Similarity to neuroprotective strategies in hibernation and hypoxia-tolerant states. Lancet 2003, 362, 1028–1037. [Google Scholar] [CrossRef]
- Gidday, J.M. Cerebral preconditioning and ischaemic tolerance. Nat. Rev. Neurosci. 2006, 7, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Stetler, R.A.; Leak, R.; Gan, Y.; Li, P.; Zhang, F.; Hu, X.; Jing, Z.; Chen, J.; Zigmond, M.J.; Gao, Y. Preconditioning provides neuroprotection in models of CNS disease: Paradigms and clinical significance. Prog. Neurobiol. 2014, 114, 58–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koch, S.; Della Morte, D.; Dave, K.R.; Sacco, R.L.; Perez-Pinzon, A.M. Biomarkers for Ischemic Preconditioning: Finding the Responders. Br. J. Pharmacol. 2014, 34, 933–941. [Google Scholar] [CrossRef] [Green Version]
- La Russa, D.; Frisina, M.; Secondo, A.; Bagetta, G.; Amantea, D. Modulation of Cerebral Store-operated Calcium Entry-regulatory Factor (SARAF) and Peripheral Orai1 Following Focal Cerebral Ischemia and Preconditioning in Mice. Neuroscience 2020, 441, 8–21. [Google Scholar] [CrossRef]
- Sisalli, M.J.; Annunziato, L.; Scorziello, A. Novel Cellular Mechanisms for Neuroprotection in Ischemic Preconditioning: A View from Inside Organelles. Front. Neurol. 2015, 6, 115. [Google Scholar] [CrossRef] [Green Version]
- Durukan, A.; Tatlisumak, T. Preconditioning-induced ischemic tolerance: A window into endogenous gearing for cerebroprotection. Exp. Transl. Stroke Med. 2010, 2, 2. [Google Scholar] [CrossRef] [Green Version]
- Broughton, B.R.; Reutens, D.; Sobey, C.G.; Sims, K.; Politei, J.; Banikazemi, M.; Lee, P. Apoptotic Mechanisms after Cerebral Ischemia. Stroke 2009, 40, 788–794. [Google Scholar] [CrossRef] [Green Version]
- Zhao, H.; Sapolsky, R.M.; Steinberg, G.K. Phosphoinositide-3-Kinase/Akt Survival Signal Pathways Are Implicated in Neuronal Survival After Stroke. Mol. Neurobiol. 2006, 34, 249–270. [Google Scholar] [CrossRef]
- Uzdensky, A.B. Apoptosis regulation in the penumbra after ischemic stroke: Expression of pro- and antiapoptotic proteins. Apoptosis 2019, 24, 687–702. [Google Scholar] [CrossRef] [PubMed]
- Fukunaga, K.; Kawano, T. Akt is a molecular target for signal transduction therapy in brain ischemic insult. J. Pharmacol. Sci. 2003, 92, 317–327. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, E.Y.; Efendizade, A.; Cai, L.; Ding, Y. The role of Akt (protein kinase B) and protein kinase C in ischemia–reperfusion injury. Neurol. Res. 2016, 38, 301–308. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Esteban, M.; Martín-Zanca, D.; Andres-Martin, L.; Almeida, A.; Bolanos, J.P. Inhibition of PTEN by peroxynitrite activates the phosphoinositide-3-kinase/Akt neuroprotective signaling pathway. J. Neurochem. 2007, 102, 194–205. [Google Scholar] [CrossRef]
- Manning, B.D.; Toker, A. AKT/PKB Signaling: Navigating the Network. Cell 2017, 169, 381–405. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diez, H.; Garrido, J.J.; Wandosell, F. Specific Roles of Akt iso Forms in Apoptosis and Axon Growth Regulation in Neurons. PLoS ONE 2012, 7, e32715. [Google Scholar] [CrossRef] [Green Version]
- Santi, S.A.; Lee, H. The Akt isoforms are present at distinct subcellular locations. Am. J. Physiol. Physiol. 2010, 298, C580–C591. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Talukder, M.H.; Varadharaj, S.; Velayutham, M.; Zweier, J.L. Early ischaemic preconditioning requires Akt- and PKA-mediated activation of eNOS via serine1176 phosphorylation. Cardiovasc. Res. 2012, 97, 33–43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ouyang, Y.-B.; Tan, Y.; Comb, M.; Liu, C.-L.; Martone, M.E.; Siesjö, B.K.; Hu, B.-R. Survival- and Death-Promoting Events after Transient Cerebral Ischemia: Phosphorylation of Akt, Release of Cytochrome C, and Activation of Caspase-Like Proteases. Br. J. Pharmacol. 1999, 19, 1126–1135. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, S.; Hafeez, A.; Noorulla, F.; Geng, X.; Shao, G.; Ren, C.; Lu, G.; Zhao, H.; Ding, Y.; Ji, X. Preconditioning in neuroprotection: From hypoxia to ischemia. Prog. Neurobiol. 2017, 157, 79–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mayo, L.D.; Donner, D.B. A phosphatidylinositol 3-kinase/Akt pathway promotes translocation of Mdm2 from the cytoplasm to the nucleus. Proc. Natl. Acad. Sci. USA 2001, 98, 11598–11603. [Google Scholar] [CrossRef] [Green Version]
- Ashcroft, M.; Ludwig, R.L.; Woods, D.B.; Copeland, T.D.; Weber, H.O.; Macrae, E.J.; Vousden, K.H. Phosphorylation of HDM2 by Akt. Oncogene 2002, 21, 1955–1962. [Google Scholar] [CrossRef] [Green Version]
- Xhou, B.P.; Liao, J.; Xia, W. HER-2/neu induces p53 ubiquitination via Akt-mediated MDM2 phosphorylation. Nat. Cell Biol. 2001, 3, 973–982. [Google Scholar] [CrossRef]
- Grossman, S.R.; Perez, M.; Kung, A.L.; Joseph, M.; Mansur, C.; Xiao, Z.-X.; Kumar, S.; Howley, P.; Livingston, D.M. p300/MDM2 Complexes Participate in MDM2-Mediated p53 Degradation. Mol. Cell 1998, 2, 405–415. [Google Scholar] [CrossRef]
- Toth, A.; Nickson, P.; Qin, L.L.; Erhardt, P. Differential Regulation of Cardiomyocyte Survival and Hypertrophy by MDM2, an E3 Ubiquitin Ligase. J. Biol. Chem. 2006, 281, 3679–3689. [Google Scholar] [CrossRef] [Green Version]
- Hausenloy, D.J.; Tsang, A.; Mocanu, M.M.; Yellon, D.M. Ischemic preconditioning protects by activating prosurvival kinases at reperfusion. Am. J. Physiol. Circ. Physiol. 2005, 288, H971–H976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mocanu, M.M.; Yellon, D.M. p53 down-regulation: A new molecular mechanism involved in ischaemic preconditioning. FEBS Lett. 2003, 555, 302–306. [Google Scholar] [CrossRef] [Green Version]
- Vecino, R.; Burguete, M.C.; Jover-Mengual, T.; Agulla, J.; Bobo-Jiménez, V.; Salom, J.B.; Almeida, A.; Delgado-Esteban, M. The MDM2-p53 pathway is involved in preconditioning-induced neuronal tolerance to ischemia. Sci. Rep. 2018, 8, 1610. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodriguez, C.; Ramos-Araque, M.E.; Domínguez-Martínez, M.; Sobrino, T.; Sánchez-Morán, I.; Agulla, J.; Delgado-Esteban, M.; Gómez-Sánchez, J.C.; Bolaños, J.P.; Castillo, J.; et al. Single-Nucleotide Polymorphism 309T>G in the MDM2 Promoter Determines Functional Outcome After Stroke. Stroke 2018, 49, 2437–2444. [Google Scholar] [CrossRef]
- Feng, J.; Park, J.; Cron, P.; Hess, D.; Hemmings, B.A. Identification of a PKB/Akt Hydrophobic Motif Ser-473 Kinase as DNA-dependent Protein Kinase. J. Biol. Chem. 2004, 279, 41189–41196. [Google Scholar] [CrossRef] [Green Version]
- Lai, T.W.; Zhang, S.; Wang, Y.T. Excitotoxicity and stroke: Identifying novel targets for neuroprotection. Prog. Neurobiol. 2014, 115, 157–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Constantino, L.C.; Binder, L.B.; Vandresen-Filho, S.; Viola, G.G.; Ludka, F.K.; Lopes, M.W.; Leal, R.B.; Tasca, C.I. Role of Phosphatidylinositol-3 Kinase Pathway in NMDA Preconditioning: Different Mechanisms for Seizures and Hippocampal Neuronal Degeneration Induced by Quinolinic Acid. Neurotox. Res. 2018, 34, 452–462. [Google Scholar] [CrossRef]
- Xie, R.; Cheng, M.; Li, M.; Xiong, X.; Daadi, M.; Sapolsky, R.M.; Zhao, H. Akt Isoforms Differentially Protect against Stroke-Induced Neuronal Injury by Regulating mTOR Activities. Br. J. Pharmacol. 2013, 33, 1875–1885. [Google Scholar] [CrossRef] [Green Version]
- Soriano, F.X.; Papadia, S.; Hofmann, F.; Hardingham, N.R.; Bading, H.; Hardingham, G.E. Preconditioning doses of NMDA promote neuroprotection by enhancing neuronal excitability. J. Neurosci. 2006, 26, 4509–4518. [Google Scholar] [CrossRef] [Green Version]
- Grabb, M.C.; Choi, D.W. Ischemic Tolerance in Murine Cortical Cell Culture: Critical Role for NMDA Receptors. J. Neurosci. 1999, 19, 1657–1662. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.; Lu, T.-J.; Chen, X.-J.; Zhou, Y.; Chen, Q.; Feng, X.-Y.; Xu, L.; Duan, W.-H.; Xiong, Z.-Q. Differential Roles of NMDA Receptor Subtypes in Ischemic Neuronal Cell Death and Ischemic Tolerance. Stroke 2008, 39, 3042–3048. [Google Scholar] [CrossRef]
- Gomez-Sanchez, J.C.; Esteban, M.D.; Rodriguez-Hernandez, I.; Sobrino, T.; De La Ossa, N.P.; Reverte, S.; Bolaños, J.P.; Gonzalez-Sarmiento, R.; Castillo, J.; Almeida, A. The human Tp53 Arg72Pro polymorphism explains different functional prognosis in stroke. J. Exp. Med. 2011, 208, 429–437. [Google Scholar] [CrossRef] [Green Version]
- Xu, W.; Gao, L.; Li, T.; Zheng, J.; Shao, A.; Zhang, J. Mesencephalic Astrocyte-Derived Neurotrophic Factor (MANF) Protects Against Neuronal Apoptosis via Activation of Akt/MDM2/p53 Signaling Pathway in a Rat Model of Intracerebral Hemorrhage. Front. Mol. Neurosci. 2018, 11, 176. [Google Scholar] [CrossRef] [PubMed]
- Sánchez-Morán, I.; Rodríguez, C.; Lapresa, R.; Agulla, J.; Sobrino, T.; Castillo, J.; Bolaños, J.P.; Almeida, A. Nuclear WRAP53 promotes neuronal survival and functional recovery after stroke. Sci. Adv. 2020, 6, eabc5702. [Google Scholar] [CrossRef]
- Burmistrova, O.; Olias-Arjona, A.; Lapresa, R.; Jimenez-Blasco, D.; Eremeeva, T.; Shishov, D.; Romanov, S.; Zakurdaeva, K.; Almeida, A.; Fedichev, P.O.; et al. Targeting PFKFB3 alleviates cerebral ischemia-reperfusion injury in mice. Sci. Rep. 2019, 9, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Tu, Y.; Kim, E.; Gao, Y.; Rankin, G.O.; Li, B.; Chen, Y.C. Theaflavin-3, 3′-digallate induces apoptosis and G2 cell cycle arrest through the Akt/MDM2/p53 pathway in cisplatin-resistant ovarian cancer A2780/CP70 cells. Int. J. Oncol. 2016, 48, 2657–2665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wan, W.; Hou, Y.; Wang, K.; Cheng, Y.; Pu, X.; Ye, X. The LXR-623-induced long non-coding RNA LINC01125 suppresses the proliferation of breast cancer cells via PTEN/AKT/p53 signaling pathway. Cell Death Dis. 2019, 10, 248. [Google Scholar] [CrossRef] [PubMed]
- Tao, J.; Cui, Y.; Duan, Y.; Zhang, N.; Wang, C.; Zhang, F. Puerarin attenuates locomotor and cognitive deficits as well as hippocampal neuronal injury through the PI3K/Akt1/GSK-3β signaling pathway in an in vivo model of cerebral ischemia. Oncotarget 2017, 8, 106283–106295. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Janelidze, S.; Hu, B.-R.; Siesjö, P.; Siesjö, B.K. Alterations of Akt1 (PKBα) and p70S6K in Transient Focal Ischemia. Neurobiol. Dis. 2001, 8, 147–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pignataro, G.; Boscia, F.; Esposito, E.; Sirabella, R.; Cuomo, O.; Vinciguerra, A.; Di Renzo, G.; Annunziato, L. NCX1 and NCX3: Two new effectors of delayed preconditioning in brain ischemia. Neurobiol. Dis. 2012, 45, 616–623. [Google Scholar] [CrossRef]
- Li, J.; Kurokawa, M. Regulation of MDM2 Stability after DNA Damage. J. Cell. Physiol. 2015, 230, 2318–2327. [Google Scholar] [CrossRef] [Green Version]
- Olson, D.C.; Marechal, V.; Momand, J.; Chen, J.; Romocki, C.; Levine, A.J. Identification and characterization of multiple mdm-2 proteins and mdm-2-p53 protein complexes. Oncogene 1993, 8, 2353–2360. [Google Scholar] [PubMed]
- Uranga, R.M.; Katz, S.; Salvador, G.A. Enhanced Phosphatidylinositol 3-kinase (PI3K)/Akt Signaling Has Pleiotropic Targets in Hippocampal Neurons Exposed to Iron-induced Oxidative Stress. J. Biol. Chem. 2013, 288, 19773–19784. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Almeida, A.; Sánchez-Morán, I.; Rodríguez, C. Mitochondrial–nuclear p53 trafficking controls neuronal susceptibility in stroke. IUBMB Life 2021, 73, 582–591. [Google Scholar] [CrossRef] [PubMed]
- Delgado-Esteban, M.; Garcia-Higuera, I.; Maestre, C.; Moreno, S.; Almeida, A. APC/C-Cdh1 coordinates neurogenesis and cortical size during development. Nat. Commun. 2013, 4, 2879. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Constantino, L.C. The Role of NMDA Receptors in the Development of Brain Resistance through Pre- and Postconditioning. Aging Dis. 2014, 5, 430–441. [Google Scholar] [CrossRef]
- Almeida, A.; Esteban, M.D.; Bolaños, J.P.; Medina, J.M. Oxygen and glucose deprivation induces mitochondrial dysfunction and oxidative stress in neurones but not in astrocytes in primary culture. J. Neurochem. 2002, 81, 207–217. [Google Scholar] [CrossRef]
- Lahav, G.; Rosenfeld, N.; Sigal, A.; Geva-Zatorsky, N.; Levine, A.J.; Elowitz, M.B.; Alon, U. Dynamics of the p53-Mdm2 feedback loop in individual cells. Nat. Genet. 2004, 36, 147–150. [Google Scholar] [CrossRef]
- Li, J.; Karaplis, A.C.; Huang, D.C.; Siegel, P.M.; Camirand, A.; Yang, X.F.; Muller, W.J.; Kremer, R. PTHrP drives breast tumor initiation, progression, and metastasis in mice and is a potential therapy target. J. Clin. Investig. 2011, 121, 4655–4669. [Google Scholar] [CrossRef] [Green Version]
- Maestre, C.; Esteban, M.D.; Gomez-Sanchez, J.C.; Bolaños, J.P.; Almeida, A. Cdk5 phosphorylates Cdh1 and modulates cyclin B1 stability in excitotoxicity. EMBO J. 2008, 27, 2736–2745. [Google Scholar] [CrossRef] [Green Version]
- De Tudela, M.V.-P.; Esteban, M.D.; Maestre, C.; Bobo-Jiménez, V.; Jiménez-Blasco, D.; Vecino, R.; Bolaños, J.P.; Almeida, A. Regulation of Bcl-xL-ATP Synthase Interaction by Mitochondrial Cyclin B1-Cyclin-Dependent Kinase-1 Determines Neuronal Survival. J. Neurosci. 2015, 35, 9287–9301. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bobo-Jiménez, V.; Esteban, M.D.; Angibaud, J.; Sánchez-Morán, I.; de la Fuente, A.; Yajeya, J.; Nägerl, U.V.; Castillo, J.; Bolaños, J.P.; Almeida, A. APC/CCdh1-Rock2 pathway controls dendritic integrity and memory. Proc. Natl. Acad. Sci. USA 2017, 114, 4513–4518. [Google Scholar] [CrossRef] [PubMed] [Green Version]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Barrio, E.; Vecino, R.; Sánchez-Morán, I.; Rodríguez, C.; Suárez-Pindado, A.; Bolaños, J.P.; Almeida, A.; Delgado-Esteban, M. Preconditioning-Activated AKT Controls Neuronal Tolerance to Ischemia through the MDM2–p53 Pathway. Int. J. Mol. Sci. 2021, 22, 7275. https://doi.org/10.3390/ijms22147275
Barrio E, Vecino R, Sánchez-Morán I, Rodríguez C, Suárez-Pindado A, Bolaños JP, Almeida A, Delgado-Esteban M. Preconditioning-Activated AKT Controls Neuronal Tolerance to Ischemia through the MDM2–p53 Pathway. International Journal of Molecular Sciences. 2021; 22(14):7275. https://doi.org/10.3390/ijms22147275
Chicago/Turabian StyleBarrio, Emilia, Rebeca Vecino, Irene Sánchez-Morán, Cristina Rodríguez, Alberto Suárez-Pindado, Juan P. Bolaños, Angeles Almeida, and Maria Delgado-Esteban. 2021. "Preconditioning-Activated AKT Controls Neuronal Tolerance to Ischemia through the MDM2–p53 Pathway" International Journal of Molecular Sciences 22, no. 14: 7275. https://doi.org/10.3390/ijms22147275
APA StyleBarrio, E., Vecino, R., Sánchez-Morán, I., Rodríguez, C., Suárez-Pindado, A., Bolaños, J. P., Almeida, A., & Delgado-Esteban, M. (2021). Preconditioning-Activated AKT Controls Neuronal Tolerance to Ischemia through the MDM2–p53 Pathway. International Journal of Molecular Sciences, 22(14), 7275. https://doi.org/10.3390/ijms22147275