
Molecular Basis of Late-Life Depression


{kind=link}
Abstract
:1. Introduction
2. Depression and Late-Life Depression
3. Late-Life Depression and Suicide
4. Depression and Dementia
5. Mechanisms Underlying Depression/Late-Life Depression
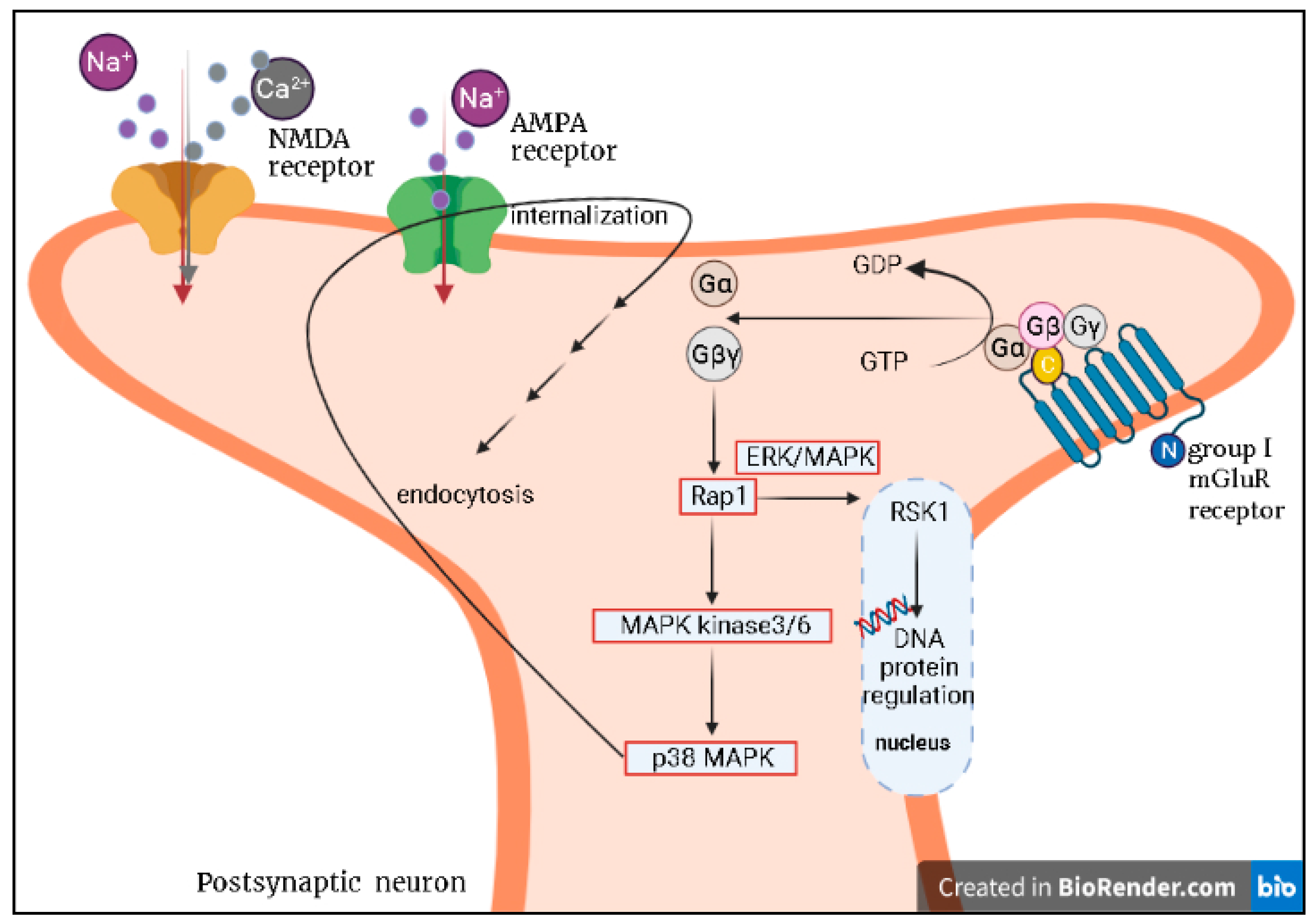
5.1. Glutamatergic System
5.2. Hypothalamic–Pituitary–Adrenal (HPA) Axis and Immunological Biomarkers
5.3. Brain-Derived Neurotrophic Factor
6. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Parmelee, P.A.; Katz, I.R.; Lawton, M.P. Depression Among Institutionalized Aged: Assessment and Prevalence Estimation. J. Gerontol. 1989, 44, M22–M29. [Google Scholar] [CrossRef] [PubMed]
- Kok, R.M.; Nolen, W.A.; Heeren, T.J. Efficacy of treatment in older depressed patients: A systematic review and meta-analysis of double-blind randomized controlled trials with antidepressants. J. Affect. Disord. 2012, 141, 103–115. [Google Scholar] [CrossRef] [PubMed]
- Alexopoulos, G.S. Depression in the elderly. Lancet 2005, 365, 1961–1970. [Google Scholar] [CrossRef]
- Rot, M.A.H.; Mathew, S.J.; Charney, D.S. Neurobiological mechanisms in major depressive disorder. Can. Med. Assoc. J. 2009, 180, 305–313. [Google Scholar] [CrossRef] [Green Version]
- Maletic, V. Neurobiology of depression, fibromyalgia and neuropathic pain. Front. Biosci. 2009, 14, 5291–5338. [Google Scholar] [CrossRef]
- Kennedy, S.H.; Lam, R.W.; McIntyre, R.S.; Tourjman, S.V.; Bhat, V.; Blier, P.; Hasnain, M.; Jollant, F.; Levitt, A.J.; MacQueen, G.M.; et al. Canadian Network for Mood and Anxiety Treatments (CANMAT) 2016 Clinical Guidelines for the Management of Adults with Major Depressive Disorder: Section 3. Pharmacological Treatments. Can. J. Psychiatry 2016, 61, 540–560. [Google Scholar] [CrossRef]
- MacQueen, G.M.; Frey, B.N.; Ismail, Z.; Jaworska, N.; Steiner, M.; Van Lieshout, R.J.; Kennedy, S.; Lam, R.W.; Milev, R.V.; Parikh, S.V.; et al. Canadian Network for Mood and Anxiety Treatments (CANMAT) 2016 Clinical Guidelines for the Management of Adults with Major Depressive Disorder: Section 6. Special Populations: Youth, Women, and the Elderly. Can. J. Psychiatry 2016, 61, 588–603. [Google Scholar] [CrossRef] [Green Version]
- Unützer, J.; Katon, W.; Callahan, C.M.; Williams, J.J.W.; Hunkeler, E.; Harpole, L.; Hoffing, M.; Della Penna, R.D.; Noel, P.; Lin, E.H.B.; et al. Collaborative Care Management of Late-Life Depression in the Primary Care Setting. JAMA 2002, 288, 2836–2845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Unützer, J.; Katon, W.; Williams, J.W.; Callahan, C.M.; Harpole, L.; Hunkeler, E.M.; Hoffing, M.; Arean, P.; Hegel, M.T.; Schoenbaum, M.; et al. Improving Primary Care for Depression in Late Life: The Design of a Multicenter Randomized Trial. Med. Care 2001, 39, 785–799. [Google Scholar] [CrossRef]
- Alexopoulos, G.S.; Katz, I.R.; Bruce, M.L.; Heo, M.; Have, T.T.; Raue, P.; Bogner, H.R.; Schulberg, H.C.; Mulsant, B.H.; Reynolds, C.F.; et al. Remission in Depressed Geriatric Primary Care Patients: A Report from the PROSPECT Study. Am. J. Psychiatry 2005, 162, 718–724. [Google Scholar] [CrossRef] [Green Version]
- Mulsant, B.H.; Alexopoulos, G.S.; Reynolds, C.F.; Katz, I.R.; Abrams, R.; Oslin, D.; Schulberg, H.C.; the PROSPECT Study Group. Pharmacological treatment of depression in older primary care patients: The PROSPECT algorithm. Int. J. Geriatr. Psychiatry 2001, 16, 585–592. [Google Scholar] [CrossRef]
- Blazer, D.G. Depression in the elderly: Myths and Misconceptions. Psychiatr. Clin. N. Am. 1997, 20, 111–119. [Google Scholar] [CrossRef]
- Hegeman, J.M.; Kok, R.M.; Van Der Mast, R.C.; Giltay, E. Phenomenology of depression in older compared with younger adults: Meta-analysis. Br. J. Psychiatry 2012, 200, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Wilkowska-Chmielewska, J.; Szelenberger, W.; Wojnar, M. Age-dependent symptomatology of depression in hospitalized patients and its implications for DSM-5. J. Affect. Disord. 2013, 150, 142–145. [Google Scholar] [CrossRef]
- Schaakxs, R.; Comijs, H.C.; Lamers, F.; Beekman, A.T.F.; Penninx, B.W.J.H. Age-related variability in the presentation of symptoms of major depressive disorder. Psychol. Med. 2016, 47, 543–552. [Google Scholar] [CrossRef]
- Nelson, J.C.; Delucchi, K.; Schneider, L.S. Efficacy of Second Generation Antidepressants in Late-Life Depression: A Meta-Analysis of the Evidence. Am. J. Geriatr. Psychiatry 2008, 16, 558–567. [Google Scholar] [CrossRef]
- Tedeschini, E.; Levkovitz, Y.; Iovieno, N.; Ameral, V.E.; Nelson, J.C.; Papakostas, G.I. Efficacy of Antidepressants for Late-Life Depression: A meta-analysis and meta-regression of placebo-controlled randomized trials. J. Clin. Psychiatry 2011, 72, 1660–1668. [Google Scholar] [CrossRef] [PubMed]
- Rush, A.J.; Trivedi, M.H.; Wisniewski, S.R.; Nierenberg, A.A.; Stewart, J.W.; Warden, D.; Niederehe, G.; Thase, M.E.; Lavori, P.W.; Lebowitz, B.; et al. Acute and Longer-Term Outcomes in Depressed Outpatients Requiring One or Several Treatment Steps: A STAR*D Report. Am. J. Psychiatry 2006, 163, 1905–1917. [Google Scholar] [CrossRef] [PubMed]
- Lenze, E.J.; Mulsant, B.H.; Blumberger, D.M.; Karp, J.F.; Newcomer, J.W.; Anderson, S.; Dew, M.A.; Butters, M.A.; Stack, J.A.; Begley, A.E.; et al. Efficacy, safety, and tolerability of augmentation pharmacotherapy with aripiprazole for treatment-resistant depression in late life: A randomised, double-blind, placebo-controlled trial. Lancet 2015, 386, 2404–2412. [Google Scholar] [CrossRef] [Green Version]
- Cooper, C.; Katona, C.; Lyketsos, K.; Blazer, D.; Brodaty, H.; Rabins, P.; Lima, C.A.D.M.; Livingston, G. A Systematic Review of Treatments for Refractory Depression in Older People. Am. J. Psychiatry 2011, 168, 681–688. [Google Scholar] [CrossRef]
- Maust, D.T.; Oslin, D.W.; Thase, M.E. Going Beyond Antidepressant Monotherapy for Incomplete Response in Nonpsychotic Late-Life Depression: A Critical Review. Am. J. Geriatr. Psychiatry 2013, 21, 973–986. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ochs-Ross, R.; Daly, E.J.; Zhang, Y.; Lane, R.; Lim, P.; Morrison, R.L.; Hough, D.; Manji, H.; Drevets, W.C.; Sanacora, G.; et al. Efficacy and Safety of Esketamine Nasal Spray Plus an Oral Antidepressant in Elderly Patients With Treatment-Resistant Depression—TRANSFORM-3. Am. J. Geriatr. Psychiatry 2020, 28, 121–141. [Google Scholar] [CrossRef] [PubMed]
- Copeland, J.R.M.; Dewey, M.E.; Wood, N.; Searle, R.; Davidson, I.A.; McWilliam, C. Range of Mental Illness Among the Elderly in the Community. Br. J. Psychiatry 1987, 150, 815–823. [Google Scholar] [CrossRef] [PubMed]
- Steffens, D.C.; Skoog, I.; Norton, M.C.; Hart, A.D.; Tschanz, J.T.; Plassman, B.L.; Wyse, B.W.; Welsh-Bohmer, K.A.; Breitner, J.C. Prevalence of Depression and Its Treatment in an Elderly Population: The Cache County Study. Arch. Gen. Psychiatry 2000, 57, 601–607. [Google Scholar] [CrossRef]
- Spicer, R.S.; Miller, T. Suicide acts in 8 states: Incidence and case fatality rates by demographics and method. Am. J. Public Health 2000, 90, 1885–1891. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hedna, K.; Hensing, G.; Skoog, I.; Fastbom, J.; Waern, M. Sociodemographic and gender determinants of late-life suicide in users and non-users of antidepressants. Eur. J. Public Health 2020, 30, 958–964. [Google Scholar] [CrossRef]
- Bickford, D.; Morin, R.T.; Nelson, J.C.; Mackin, R.S. Determinants of Suicide-related Ideation in Late Life Depression: Associations with Perceived Stress. Clin. Gerontol. 2020, 43, 37–45. [Google Scholar] [CrossRef]
- Voshaar, R.C.O.; Kapur, N.; Bickley, H.; Williams, A.; Purandare, N. Suicide in later life: A comparison between cases with early-onset and late-onset depression. J. Affect. Disord. 2011, 132, 185–191. [Google Scholar] [CrossRef]
- Saczynski, J.S.; Beiser, A.; Seshadri, S.; Auerbach, S.; Wolf, P.A.; Au, R. Depressive symptoms and risk of dementia: The Framingham Heart Study. Neurology 2010, 75, 35–41. [Google Scholar] [CrossRef] [Green Version]
- Deuschle, M.; Gilles, M.; Scharnholz, B.; Lederbogen, F.; Lang, U.E.; Hellweg, R. Changes of Serum Concentrations of Brain-Derived Neurotrophic Factor (BDNF) during Treatment with Venlafaxine and Mirtazapine: Role of Medication and Response to Treatment. Pharmacopsychiatry 2012, 46, 54–58. [Google Scholar] [CrossRef] [Green Version]
- Cereseto, M.; Reinés, A.; Ferrero, A.; Sifonios, L.; Rubio, M.; Wikinski, S. Chronic treatment with high doses of corticosterone decreases cytoskeletal proteins in the rat hippocampus. Eur. J. Neurosci. 2006, 24, 3354–3364. [Google Scholar] [CrossRef]
- Gould, E.; Tanapat, P.; Rydel, T.; Hastings, N. Regulation of hippocampal neurogenesis in adulthood. Biol. Psychiatry 2000, 48, 715–720. [Google Scholar] [CrossRef]
- Laske, C.; Stransky, E.; Fritsche, A.; Eschweiler, G.W.; Leyhe, T. Inverse association of cortisol serum levels with T-tau, P-tau 181 and P-tau 231 peptide levels and T-tau/Aβ 1–42 ratios in CSF in patients with mild Alzheimer’s disease dementia. Eur. Arch. Psychiatry Clin. Neurosci. 2008, 259, 80–85. [Google Scholar] [CrossRef]
- Alexopoulos, G.S. Mechanisms and treatment of latelife depression. Transl. Psychiatry 2019, 9, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kita, T.; Morrison, P.F.; Heyes, M.P.; Markey, S.P. Effects of systemic and central nervous system localized inflammation on the contributions of metabolic precursors to the l-kynurenine and quinolinic acid pools in brain. J. Neurochem. 2002, 82, 258–268. [Google Scholar] [CrossRef] [PubMed]
- Setiawan, E.; Wilson, A.A.; Mizrahi, R.; Rusjan, P.M.; Miler, L.; Rajkowska, G.; Suridjan, I.; Kennedy, J.L.; Rekkas, P.V.; Houle, S.; et al. Role of Translocator Protein Density, a Marker of Neuroinflammation, in the Brain During Major Depressive Episodes. JAMA Psychiatry 2015, 72, 268–275. [Google Scholar] [CrossRef] [PubMed]
- Gimeno, D.; Kivimaki, M.; Brunner, E.; Elovainio, M.; De Vogli, R.; Steptoe, A.; Kumari, M.; Lowe, G.D.; Rumley, A.; Marmot, M.; et al. Associations of C-reactive protein and interleukin-6 with cognitive symptoms of depression: 12-year follow-up of the Whitehall II study. Psychol. Med. 2008, 39, 413–423. [Google Scholar] [CrossRef]
- Brunoni, A.R.; Lopes, M.; Fregni, F. A systematic review and meta-analysis of clinical studies on major depression and BDNF levels: Implications for the role of neuroplasticity in depression. Int. J. Neuropsychopharmacol. 2008, 11, 1169–1180. [Google Scholar] [CrossRef]
- Laske, C.; Sopova, K.; Gkotsis, C.; Eschweiler, G.W.; Straten, G.; Gawaz, M.; Leyhe, T.; Stellos, K. Amyloid-β Peptides in Plasma and Cognitive Decline After 1 Year Follow-Up in Alzheimer’s Disease Patients. J. Alzheimer’s Dis. 2010, 21, 1263–1269. [Google Scholar] [CrossRef]
- Laske, C.; Stellos, K.; Hoffmann, N.; Stransky, E.; Straten, G.; Eschweiler, G.W.; Leyhe, T. Higher BDNF serum levels predict slower cognitive decline in Alzheimer’s disease patients. Int. J. Neuropsychopharmacol. 2010, 14, 399–404. [Google Scholar] [CrossRef]
- Banerjee, S.; Hellier, J.; Romeo, R.; Dewey, M.; Knapp, M.; Ballard, C.; Baldwin, R.; Bentham, P.; Fox, C.; Holmes, C.; et al. Study of the use of antidepressants for depression in dementia: The HTA-SADD trial—A multicentre, randomised, double-blind, placebo-controlled trial of the clinical effectiveness and cost-effectiveness of sertraline and mirtazapine. Health Technol. Assess. 2013, 17, 1–166. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Culang, M.E.; Sneed, J.R.; Keilp, J.G.; Rutherford, B.R.; Pelton, G.H.; Devanand, D.P.; Roose, S.P. Change in Cognitive Functioning Following Acute Antidepressant Treatment in Late-Life Depression. Am. J. Geriatr. Psychiatry 2009, 17, 881–888. [Google Scholar] [CrossRef] [Green Version]
- Ford, A.H.; Almeida, O.P. Management of Depression in Patients with Dementia: Is Pharmacological Treatment Justified? Drugs Aging 2017, 34, 89–95. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, P.B.; Martin, B.K.; Frangakis, C.; Mintzer, J.E.; Weintraub, D.; Porsteinsson, A.; Schneider, L.S.; Rabins, P.V.; Munro, C.A.; Meinert, C.L.; et al. Sertraline for the Treatment of Depression in Alzheimer Disease. Am. J. Geriatr. Psychiatry 2010, 18, 136–145. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Newport, D.J.; Carpenter, L.L.; McDonald, W.M.; Potash, J.B.; Tohen, M.; Nemeroff, C.B. The APA Council of Research Task Force on Novel Biomarkers and Treatments Ketamine and Other NMDA Antagonists: Early Clinical Trials and Possible Mechanisms in Depression. Am. J. Psychiatry 2015, 172, 950–966. [Google Scholar] [CrossRef] [Green Version]
- Khachaturian, Z.S. Introduction and Overview. Ann. N. Y. Acad. Sci. 1989, 568, 1–4. [Google Scholar] [CrossRef] [PubMed]
- Coultrap, S.J.; Bickford, P.C.; Browning, M.D. Blueberry-enriched diet ameliorates age-related declines in NMDA receptor-dependent LTP. AGE 2008, 30, 263–272. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magnusson, K.R.; Nelson, S.E.; Young, A.B. Age-related changes in the protein expression of subunits of the NMDA receptor. Mol. Brain Res. 2002, 99, 40–45. [Google Scholar] [CrossRef]
- Cao, X.; Cui, Z.; Feng, R.; Tang, Y.-P.; Qin, Z.; Mei, B.; Tsien, J.Z. Maintenance of superior learning and memory function in NR2B transgenic mice during ageing. Eur. J. Neurosci. 2007, 25, 1815–1822. [Google Scholar] [CrossRef]
- Duman, R.S.; Li, N.; Liu, R.-J.; Duric, V.; Aghajanian, G. Signaling pathways underlying the rapid antidepressant actions of ketamine. Neuropharmacology 2012, 62, 35–41. [Google Scholar] [CrossRef] [Green Version]
- Dwyer, J.M.; Lepack, A.E.; Duman, R.S. mTOR activation is required for the antidepressant effects of mGluR2/3 blockade. Int. J. Neuropsychopharmacol. 2011, 15, 429–434. [Google Scholar] [CrossRef] [Green Version]
- Wei, Y.; Chang, L.; Hashimoto, K. Molecular mechanisms underlying the antidepressant actions of arketamine: Beyond the NMDA receptor. Mol. Psychiatry 2021, 1–15. [Google Scholar] [CrossRef]
- Lin, C.-H.; Huang, M.-W.; Lin, C.-H.; Huang, C.-H.; Lane, H.-Y. Altered mRNA expressions for N-methyl-D-aspartate receptor-related genes in WBC of patients with major depressive disorder. J. Affect. Disord. 2019, 245, 1119–1125. [Google Scholar] [CrossRef]
- Levin, R.; Dor-Abarbanel, A.E.; Edelman, S.; Durrant, A.R.; Hashimoto, K.; Javitt, D.C.; Heresco-Levy, U. Behavioral and cognitive effects of the N-methyl-D-aspartate receptor co-agonist D-serine in healthy humans: Initial findings. J. Psychiatr. Res. 2015, 61, 188–195. [Google Scholar] [CrossRef]
- Wei, I.-H.; Chen, K.-T.; Tsai, M.-H.; Wu, C.-H.; Lane, H.-Y.; Huang, C.-C. Acute Amino Acidd-Serine Administration, Similar to Ketamine, Produces Antidepressant-like Effects through Identical Mechanisms. J. Agric. Food Chem. 2017, 65, 10792–10803. [Google Scholar] [CrossRef]
- Scoriels, L.; Avellar, M. The Co-agonist Site of NMDA-glutamate Receptors: A Novel Therapeutic Target for Age-related Cognitive Decline. Curr. Pharm. Des. 2014, 20, 5160–5168. [Google Scholar] [CrossRef]
- Lane, H.Y.; Lin, C.H.; Green, M.F.; Hellemann, G.; Huang, C.C.; Chen, P.W.; Tun, R.; Chang, Y.C.; Tsai, G.E. Add-on treatment of benzoate for schizophrenia: A randomized, double-blind, placebo-controlled trial of D-amino acid oxidase inhibitor. JAMA Psychiatry 2013, 70, 1267–1275. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-H.; Chen, P.-K.; Chang, Y.-C.; Chuo, L.-J.; Chen, Y.-S.; Tsai, G.E.; Lane, H.-Y. Benzoate, a D-Amino Acid Oxidase Inhibitor, for the Treatment of Early-Phase Alzheimer Disease: A Randomized, Double-Blind, Placebo-Controlled Trial. Biol. Psychiatry 2014, 75, 678–685. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-H.; Chen, P.-K.; Wang, S.-H.; Lane, H.-Y. Effect of Sodium Benzoate on Cognitive Function Among Patients With Behavioral and Psychological Symptoms of Dementia. JAMA Netw. Open 2021, 4, e216156. [Google Scholar] [CrossRef] [PubMed]
- Lane, H.-Y.; Tu, C.-H.; Lin, W.-C.; Lin, C.-H. Brain Activity of Benzoate, a D-Amino Acid Oxidase Inhibitor, in Patients With Mild Cognitive Impairment in a Randomized, Double-Blind, Placebo Controlled Clinical Trial. Int. J. Neuropsychopharmacol. 2021, 24, 392–399. [Google Scholar] [CrossRef]
- McQuail, J.; Frazier, C.J.; Bizon, J.L. Molecular aspects of age-related cognitive decline: The role of GABA signaling. Trends Mol. Med. 2015, 21, 450–460. [Google Scholar] [CrossRef] [Green Version]
- Różycka, A.; Liguz-Lecznar, M. The space where aging acts: Focus on the GABAergic synapse. Aging Cell 2017, 16, 634–643. [Google Scholar] [CrossRef]
- Sanacora, G.; Gueorguieva, R.; Epperson, C.N.; Wu, Y.-T.; Appel, M.; Rothman, D.L.; Krystal, J.H.; Mason, G.F. Subtype-Specific Alterations of γ-Aminobutyric Acid and Glutamatein Patients with Major Depression. Arch. Gen. Psychiatry 2004, 61, 705–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, K.; Nabeshima, T. Changes in NMDA receptor/nitric oxide signaling pathway in the brain with aging. Microsc. Res. Tech. 1998, 43, 68–74. [Google Scholar] [CrossRef]
- Ghosh, A.; Carnahan, J.; Greenberg, M.; Bodmer, H.; Viville, S.; Benoist, C.; Mathis, D. Requirement for BDNF in activity-dependent survival of cortical neurons. Science 1994, 263, 1618–1623. [Google Scholar] [CrossRef]
- Réus, G.Z.; Stringari, R.B.; Kirsch, T.R.; Fries, G.R.; Kapczinski, F.; Roesler, R.; de Quevedo, J.L. Neurochemical and behavioural effects of acute and chronic memantine administration in rats: Further support for NMDA as a new pharmacological target for the treatment of depression? Brain Res. Bull. 2010, 81, 585–589. [Google Scholar] [CrossRef] [PubMed]
- Pelton, G.H.; Harper, O.L.; Roose, S.P.; Marder, K.; D’Antonio, K.; Devanand, D.P. Combined treatment with memantine/es-citalopram for older depressed patients with cognitive impairment: A pilot study. Int. J. Geriatr. Psychiatry 2016, 31, 648–655. [Google Scholar] [CrossRef] [PubMed]
- Lüscher, C.; Huber, K.M. Group 1 mGluR-Dependent Synaptic Long-Term Depression: Mechanisms and Implications for Circuitry and Disease. Neuron 2010, 65, 445–459. [Google Scholar] [CrossRef] [Green Version]
- Conn, P.J.; Pin, J.-P. Pharmacology and functions of metabotropic glutamate receptors. Annu. Rev. Pharmacol. Toxicol. 1997, 37, 205–237. [Google Scholar] [CrossRef] [Green Version]
- Nakanishi, S. Molecular diversity of glutamate receptors and implications for brain function. Science 1992, 258, 597–603. [Google Scholar] [CrossRef]
- Gladding, C.M.; Fitzjohn, S.; Molnár, E. Metabotropic Glutamate Receptor-Mediated Long-Term Depression: Molecular Mechanisms. Pharmacol. Rev. 2009, 61, 395–412. [Google Scholar] [CrossRef] [Green Version]
- Lorrain, D.; Baccei, C.; Bristow, L.; Anderson, J.; Varney, M. Effects of ketamine and n-methyl-d-aspartate on glutamate and dopamine release in the rat prefrontal cortex: Modulation by a group II selective metabotropic glutamate receptor agonist LY379268. Neuroscience 2003, 117, 697–706. [Google Scholar] [CrossRef]
- Wang, H.; Ardiles, A.O.; Yang, S.; Tran, T.; Posada-Duque, R.; Valdivia, G.; Baek, M.; Chuang, Y.-A.; Palacios, A.G.; Gallagher, M.; et al. Metabotropic Glutamate Receptors Induce a Form of LTP Controlled by Translation and Arc Signaling in the Hippocampus. J. Neurosci. 2016, 36, 1723–1729. [Google Scholar] [CrossRef] [Green Version]
- Spooren, W.; Lesage, A.; Lavreysen, H.; Gasparini, F.; Steckler, T. Metabotropic Glutamate Receptors: Their Therapeutic Potential in Anxiety. Curr. Top. Behav. Neurosci. 2010, 2, 391–413. [Google Scholar] [CrossRef] [PubMed]
- Bocchio, M.; Lukacs, I.P.; Stacey, R.; Plaha, P.; Apostolopoulos, V.; Livermore, L.; Sen, A.; Ansorge, O.; Gillies, M.J.; Somogyi, P.; et al. Group II Metabotropic Glutamate Receptors Mediate Presynaptic Inhibition of Excitatory Transmission in Pyramidal Neurons of the Human Cerebral Cortex. Front. Cell. Neurosci. 2019, 12, 508. [Google Scholar] [CrossRef]
- Kiritoshi, T.; Neugebauer, V. Group II mGluRs modulate baseline and arthritis pain-related synaptic transmission in the rat medial prefrontal cortex. Neuropharmacology 2015, 95, 388–394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marek, G.; Wright, R.; Gewirtz, J.; Schoepp, D. A major role for thalamocortical afferents in serotonergic hallucinogen receptor function in the rat neocortex. Neuroscience 2001, 105, 379–392. [Google Scholar] [CrossRef]
- Joffe, M.E.; Santiago, C.; Engers, J.L.; Lindsley, C.W.; Conn, P.J. Metabotropic glutamate receptor subtype 3 gates acute stress-induced dysregulation of amygdalo-cortical function. Mol. Psychiatry 2019, 24, 916–927. [Google Scholar] [CrossRef] [PubMed]
- Joffe, M.E.; Santiago, C.; Stansley, B.J.; Maksymetz, J.; Gogliotti, R.G.; Engers, J.L.; Nicoletti, F.; Lindsley, C.W.; Conn, P.J. Mechanisms underlying prelimbic prefrontal cortex mGlu3/mGlu5-dependent plasticity and reversal learning deficits following acute stress. Neuropharmacology 2019, 144, 19–28. [Google Scholar] [CrossRef]
- Witkin, J.; Mitchell, S.; Wafford, K.; Carter, G.; Gilmour, G.; Li, J.; Eastwood, B.; Overshiner, C.; Li, X.; Rorick-Kehn, L.; et al. Comparative Effects of LY3020371, a Potent and Selective Metabotropic Glutamate (mGlu) 2/3 Receptor Antagonist, and Ketamine, a Noncompetitive N-Methyl-d-Aspartate Receptor Antagonist in Rodents: Evidence Supporting the Use of mGlu2/3 Antagonists, for the Treatment of Depression. J. Pharmacol. Exp. Ther. 2017, 361, 68–86. [Google Scholar] [CrossRef] [Green Version]
- Lowy, M.T.; Wittenberg, L.; Yamamoto, B.K. Effect of Acute Stress on Hippocampal Glutamate Levels and Spectrin Proteolysis in Young and Aged Rats. J. Neurochem. 2002, 65, 268–274. [Google Scholar] [CrossRef]
- Magarinos, A.; McEwen, B. Stress-induced atrophy of apical dendrites of hippocampal CA3c neurons: Involvement of glucocorticoid secretion and excitatory amino acid receptors. Neuroscience 1995, 69, 89–98. [Google Scholar] [CrossRef]
- Bocchio-Chiavetto, L.; Bagnardi, V.; Zanardini, R.; Molteni, R.; Nielsen, M.G.; Placentino, A.; Giovannini, C.; Rillosi, L.; Ventriglia, M.; Riva, M.A.; et al. Serum and plasma BDNF levels in major depression: A replication study and meta-analyses. World J. Biol. Psychiatry 2010, 11, 763–773. [Google Scholar] [CrossRef]
- Duman, R.S.; Monteggia, L.M. A Neurotrophic Model for Stress-Related Mood Disorders. Biol. Psychiatry 2006, 59, 1116–1127. [Google Scholar] [CrossRef]
- Krishnan, V.; Nestler, E.J. The molecular neurobiology of depression. Nat. Cell Biol. 2008, 455, 894–902. [Google Scholar] [CrossRef] [PubMed]
- Adzic, M.; Brkic, Z.; Mitic, M.; Francija, E.; Jovicic, M.J.; Radulovic, J.; Maric, N. Therapeutic Strategies for Treatment of Inflammation-related Depression. Curr. Neuropharmacol. 2018, 16, 176–209. [Google Scholar] [CrossRef] [PubMed]
- Pace, T.W.; Hu, F.; Miller, A.H. Cytokine-effects on glucocorticoid receptor function: Relevance to glucocorticoid resistance and the pathophysiology and treatment of major depression. Brain Behav. Immun. 2007, 21, 9–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, H.-J.; Bae, K.-Y.; Kim, S.-W.; Shin, I.-S.; Kim, H.-R.; Shin, M.-G.; Yoon, J.-S.; Kim, J.-M. Longitudinal associations between glucocorticoid receptor methylation and late-life depression. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2018, 84, 56–62. [Google Scholar] [CrossRef]
- Coull, J.A.M.; Beggs, S.; Boudreau, D.; Boivin, D.; Tsuda, M.; Inoue, K.; Gravel, C.; Salter, M.W.; De Koninck, Y. BDNF from microglia causes the shift in neuronal anion gradient underlying neuropathic pain. Nature 2005, 438, 1017–1021. [Google Scholar] [CrossRef] [PubMed]
- Parkhurst, C.N.; Yang, G.; Ninan, I.; Savas, J.N.; Yates, J.R., III; Lafaille, J.J.; Hempstead, B.L.; Littman, D.R.; Gan, W.-B. Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 2013, 155, 1596–1609. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trang, T.; Beggs, S.; Wan, X.; Salter, M.W. P2X4-Receptor-Mediated Synthesis and Release of Brain-Derived Neurotrophic Factor in Microglia Is Dependent on Calcium and p38-Mitogen-Activated Protein Kinase Activation. J. Neurosci. 2009, 29, 3518–3528. [Google Scholar] [CrossRef] [PubMed]
- Coyle, J.T.; Puttfarcken, P. Oxidative stress, glutamate, and neurodegenerative disorders. Science 1993, 262, 689–695. [Google Scholar] [CrossRef]
- Sherman, A.D. l-Kynurenine Its synthesis and possible regulatory function in brain. Neurochem. Res. 1980, 5, 223–239. [Google Scholar] [CrossRef]
- Hayaishi, O. Properties and Function of Indoleamine 2, 3-Dioxygenase1. J. Biochem. 1976, 79, 13–21p. [Google Scholar] [CrossRef] [PubMed]
- Stone, T.; Perkins, M. Quinolinic acid: A potent endogenous excitant at amino acid receptors in CNS. Eur. J. Pharmacol. 1981, 72, 411–412. [Google Scholar] [CrossRef]
- Erhardt, S.; Lim, E.; Linderholm, K.R.; Janelidze, S.; Lindqvist, D.; Samuelsson, M.; Lundberg, K.; Postolache, T.T.; Träskman-Bendz, L.; Guillemin, G.; et al. Connecting inflammation with glutamate agonism in suicidality. Neuropsychopharmacology 2012, 38, 743–752. [Google Scholar] [CrossRef] [PubMed]
- Dowlati, Y.; Herrmann, N.; Swardfager, W.; Liu, H.; Sham, L.; Reim, E.K.; Lanctôt, K.L. A Meta-Analysis of Cytokines in Major Depression. Biol. Psychiatry 2010, 67, 446–457. [Google Scholar] [CrossRef]
- Maes, M.; Bosmans, E.; De Jongh, R.; Kenisbc, G.; Vandoolaeghea, E.; Neels, H. Increased serum Il-6 and Il-1 receptor antagonist concentrations in major depression and treatment resistant depression. Cytokine 1997, 9, 853–858. [Google Scholar] [CrossRef]
- Sutcigil, L.; Oktenli, C.; Musabak, U.H.; Bozkurt, A.; Cansever, A.; Uzun, O.; Sanisoglu, S.Y.; Yesilova, Z.; Ozmenler, N.; Ozsahin, A.; et al. Pro- and Anti-Inflammatory Cytokine Balance in Major Depression: Effect of Sertraline Therapy. Clin. Dev. Immunol. 2007, 2007, 1–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosenblat, J.; Cha, D.S.; Mansur, R.B.; McIntyre, R.S. Inflamed moods: A review of the interactions between inflammation and mood disorders. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2014, 53, 23–34. [Google Scholar] [CrossRef]
- Bremmer, M.; Beekman, A.; Deeg, D.; Penninx, B.; Dik, M.; Hack, C.; Hoogendijk, W. Inflammatory markers in late-life depression: Results from a population-based study. J. Affect. Disord. 2008, 106, 249–255. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-M.; Stewart, R.; Kim, J.-W.; Kang, H.-J.; Bae, K.-Y.; Kim, S.-W.; Shin, I.-S.; Yoon, J.-S. Changes in pro-inflammatory cytokine levels and late-life depression: A two year population based longitudinal study. Psychoneuroendocrinology 2018, 90, 85–91. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Charlton, R.A.; Lamar, M.; Zhang, A.; Ren, X.; Ajilore, O.; Pandey, G.N.; Kumar, A. Associations between pro-inflammatory cytokines, learning, and memory in late-life depression and healthy aging. Int. J. Geriatr. Psychiatry 2017, 33, 104–112. [Google Scholar] [CrossRef] [Green Version]
- Gorba, T.; Wahle, P. Expression of TrkB and TrkC but not BDNF mRNA in neurochemically identified interneurons in rat visual cortexin vivoand in organotypic cultures. Eur. J. Neurosci. 1999, 11, 1179–1190. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Kastner, R.; Wetmore, C.; Olson, L. Comparative study of brain-derived neurotrophic factor messenger rna and protein at the cellular level suggests multiple roles in hippocampus, striatum and cortex. Neuroscience 1996, 74, 161–183. [Google Scholar] [CrossRef]
- Barde, Y.A. Trophic factors and neuronal survival. Neuron 1989, 2, 1525–1534. [Google Scholar] [CrossRef]
- Parpura, V.; Zorec, R. Gliotransmission: Exocytotic release from astrocytes. Brain Res. Rev. 2010, 63, 83–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harward, S.C.; Hedrick, N.G.; Hall, C.E.; Parra-Bueno, P.; Milner, T.A.; Pan, E.; Laviv, T.; Hempstead, B.L.; Yasuda, R.; McNamara, J.O. Autocrine BDNF-TrkB signalling within a single dendritic spine. Nature 2016, 538, 99–103. [Google Scholar] [CrossRef] [Green Version]
- Wong, Y.-H.; Lee, C.-M.; Xie, W.; Cui, B.; Poo, M.-M. Activity-dependent BDNF release via endocytic pathways is regulated by synaptotagmin-6 and complexin. Proc. Natl. Acad. Sci. USA 2015, 112, E4475–E4484. [Google Scholar] [CrossRef] [Green Version]
- Fernandez, P.J.C.; Säuberli, K.; Colzani, M.; Moreau, T.; Ghevaert, C.; Barde, Y.-A. Brain-derived Neurotrophic Factor in Megakaryocytes. J. Biol. Chem. 2016, 291, 9872–9881. [Google Scholar] [CrossRef] [Green Version]
- Autry, A.E.; Monteggia, L.M. Brain-Derived Neurotrophic Factor and Neuropsychiatric Disorders. Pharmacol. Rev. 2012, 64, 238–258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zafra, F.; Castrén, E.; Thoenen, H.; Lindholm, D. Interplay between glutamate and gamma-aminobutyric acid transmitter systems in the physiological regulation of brain-derived neurotrophic factor and nerve growth factor synthesis in hippocampal neurons. Proc. Natl. Acad. Sci. USA 1991, 88, 10037–10041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- George, D.; Gálvez, V.; Martin, D.; Kumar, D.; Leyden, J.; Hadzi-Pavlovic, D.; Harper, S.; Brodaty, H.; Glue, P.; Taylor, R.; et al. Pilot Randomized Controlled Trial of Titrated Subcutaneous Ketamine in Older Patients with Treatment-Resistant Depression. Am. J. Geriatr. Psychiatry 2017, 25, 1199–1209. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Martinowich, K.; Lee, F.S. BDNF at the synapse: Why location matters. Mol. Psychiatry 2017, 22, 1370–1375. [Google Scholar] [CrossRef] [Green Version]
- Carlberg, L.; Scheibelreiter, J.; Hassler, M.R.; Schloegelhofer, M.; Schmoeger, M.; Ludwig, B.; Kasper, S.; Aschauer, H.; Egger, G.; Schosser, A. Brain-derived neurotrophic factor (BDNF)—Epigenetic regulation in unipolar and bipolar affective disorder. J. Affect. Disord. 2014, 168, 399–406. [Google Scholar] [CrossRef] [PubMed]
- Dell’osso, B.; D’addario, C.; Palazzo, M.; Benatti, B.; Camuri, G.; Galimberti, D.; Fenoglio, C.; Scarpini, E.; Di Francesco, A.; Maccarrone, M.; et al. Epigenetic modulation of BDNF gene: Differences in DNA methylation between unipolar and bipolar patients. J. Affect. Disord. 2014, 166, 330–333. [Google Scholar] [CrossRef]
- Alfonso, J.; Frick, L.R.; Silberman, D.M.; Palumbo, M.L.; Genaro, A.M.; Frasch, A.C. Regulation of Hippocampal Gene Expression Is Conserved in Two Species Subjected to Different Stressors and Antidepressant Treatments. Biol. Psychiatry 2006, 59, 244–251. [Google Scholar] [CrossRef] [PubMed]
- Niswender, C.M.; Conn, P.J. Metabotropic Glutamate Receptors: Physiology, Pharmacology, and Disease. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 295–322. [Google Scholar] [CrossRef] [Green Version]
- Aziz, R.; Steffens, D.C. What Are the Causes of Late-Life Depression? Psychiatr. Clin. N. Am. 2013, 36, 497–516. [Google Scholar] [CrossRef] [Green Version]
- Garcia, L.S.; Comim, C.M.; Valvassori, S.S.; Réus, G.Z.; Barbosa, L.M.; Andreazza, A.C.; Stertz, L.; Fries, G.R.; Gavioli, E.; Kapczinski, F.; et al. Acute administration of ketamine induces antidepressant-like effects in the forced swimming test and increases BDNF levels in the rat hippocampus. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2008, 32, 140–144. [Google Scholar] [CrossRef]
- Diniz, B.S.; Reynolds, C.F.; Begley, A.; Dew, M.A.; Anderson, S.; Lotrich, F.; Erickson, K.I.; Lopez, O.; Aizenstein, H.; Sibille, E.L.; et al. Brain-derived neurotrophic factor levels in late-life depression and comorbid mild cognitive impairment: A longitudinal study. J. Psychiatr. Res. 2014, 49, 96–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kuo, C.-Y.; Lin, C.-H.; Lane, H.-Y. Molecular Basis of Late-Life Depression. Int. J. Mol. Sci. 2021, 22, 7421. https://doi.org/10.3390/ijms22147421
Kuo C-Y, Lin C-H, Lane H-Y. Molecular Basis of Late-Life Depression. International Journal of Molecular Sciences. 2021; 22(14):7421. https://doi.org/10.3390/ijms22147421
Chicago/Turabian StyleKuo, Chien-Yi, Chieh-Hsin Lin, and Hsien-Yuan Lane. 2021. "Molecular Basis of Late-Life Depression" International Journal of Molecular Sciences 22, no. 14: 7421. https://doi.org/10.3390/ijms22147421