
The Structural Effect of FLT3 Mutations at 835th Position and Their Interaction with Acute Myeloid Leukemia Inhibitors: In Silico Approach


Abstract
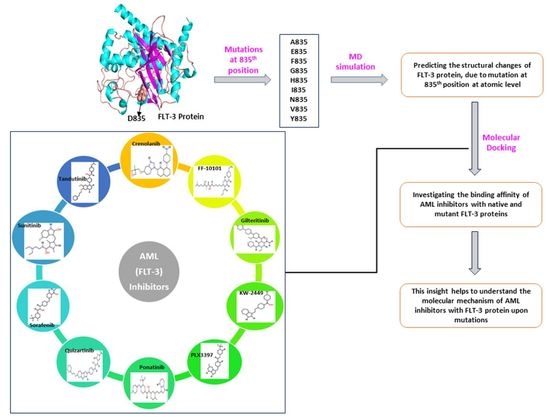
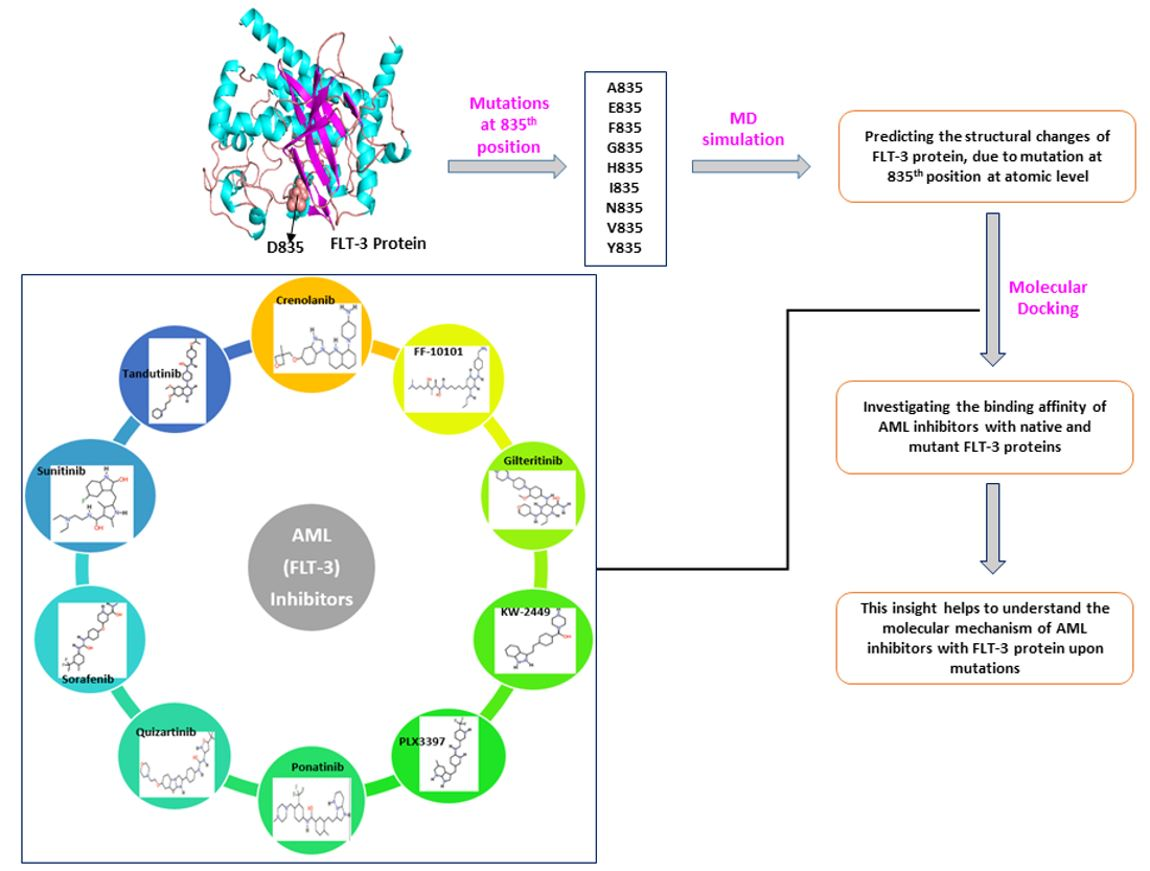
1. Introduction
2. Results
2.1. Re-Modeling of Native FLT3 Protein and Build the Mutant Structures
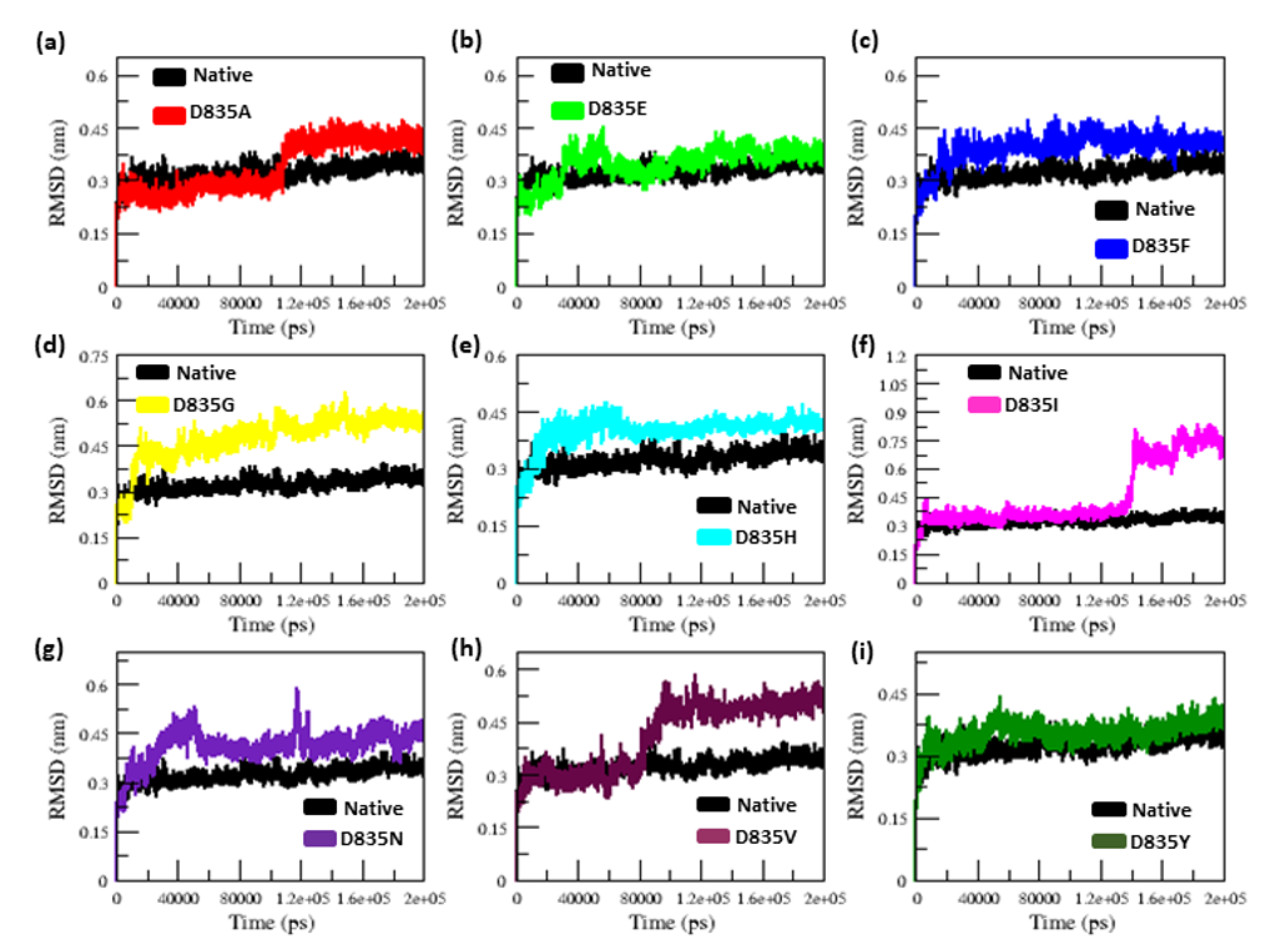
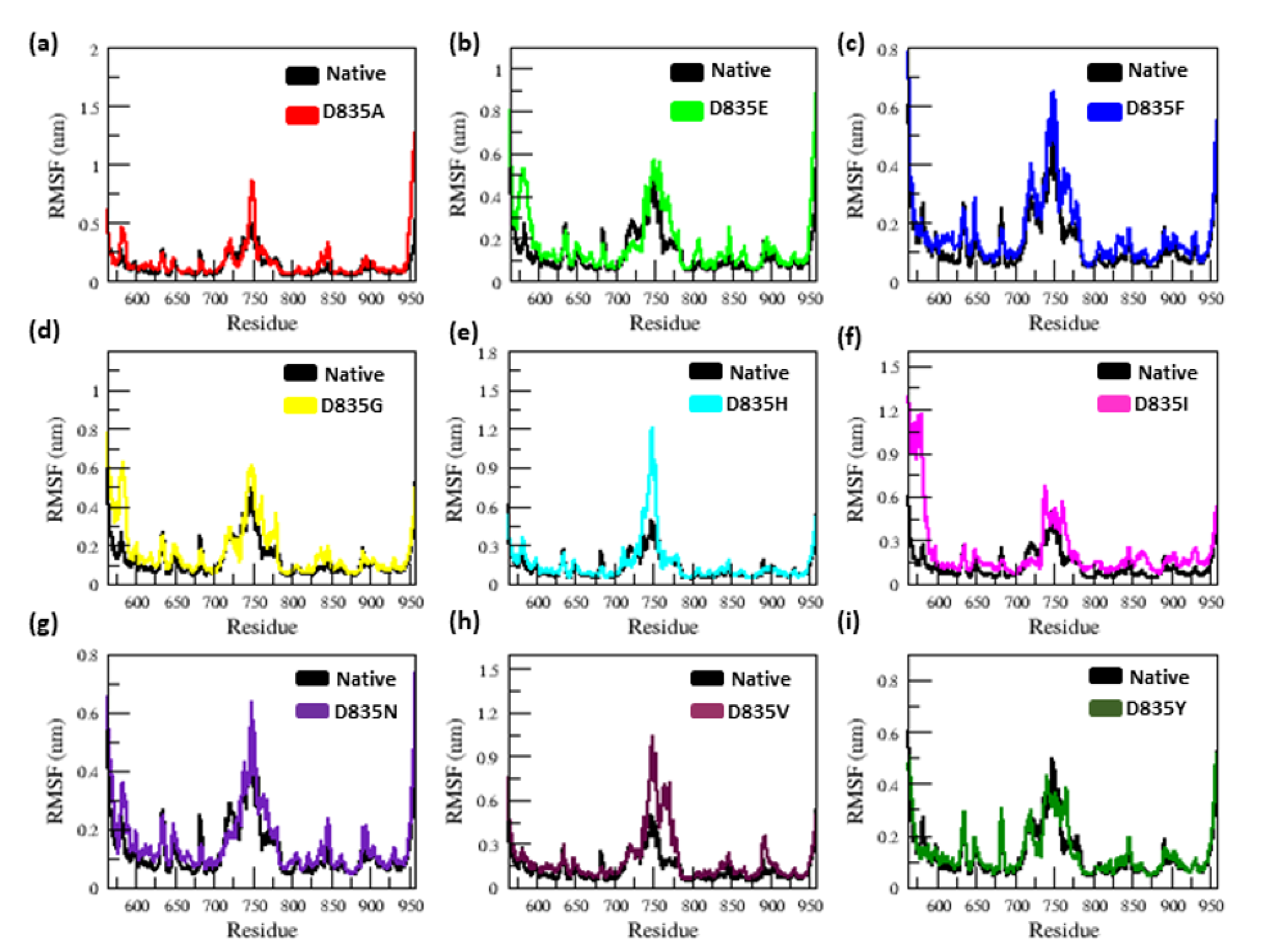
2.2. MD Simulations
2.2.1. RMSD and RMSF Analysis
2.2.2. Secondary Structure Analysis of Native and Mutant FLT3 Proteins
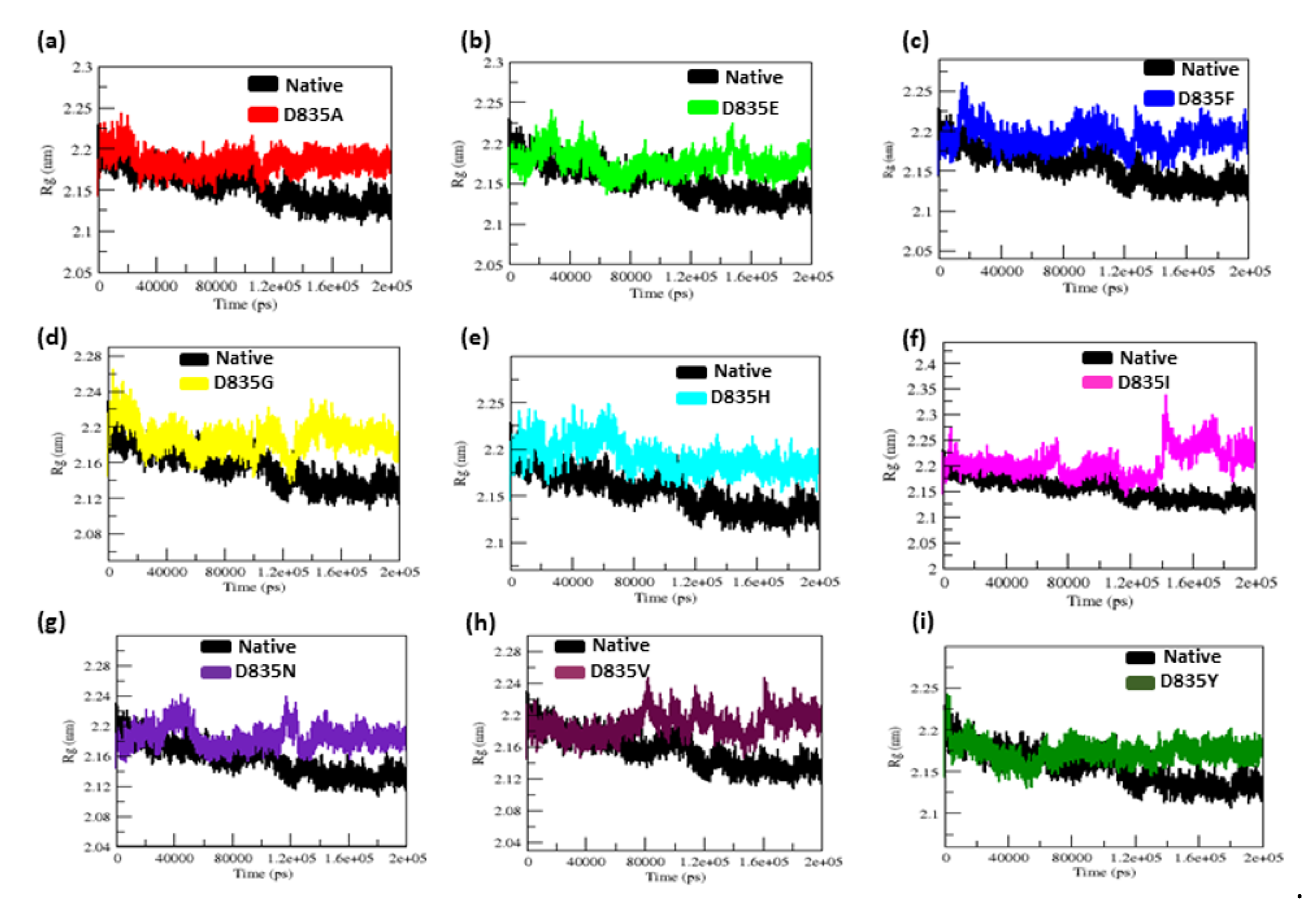
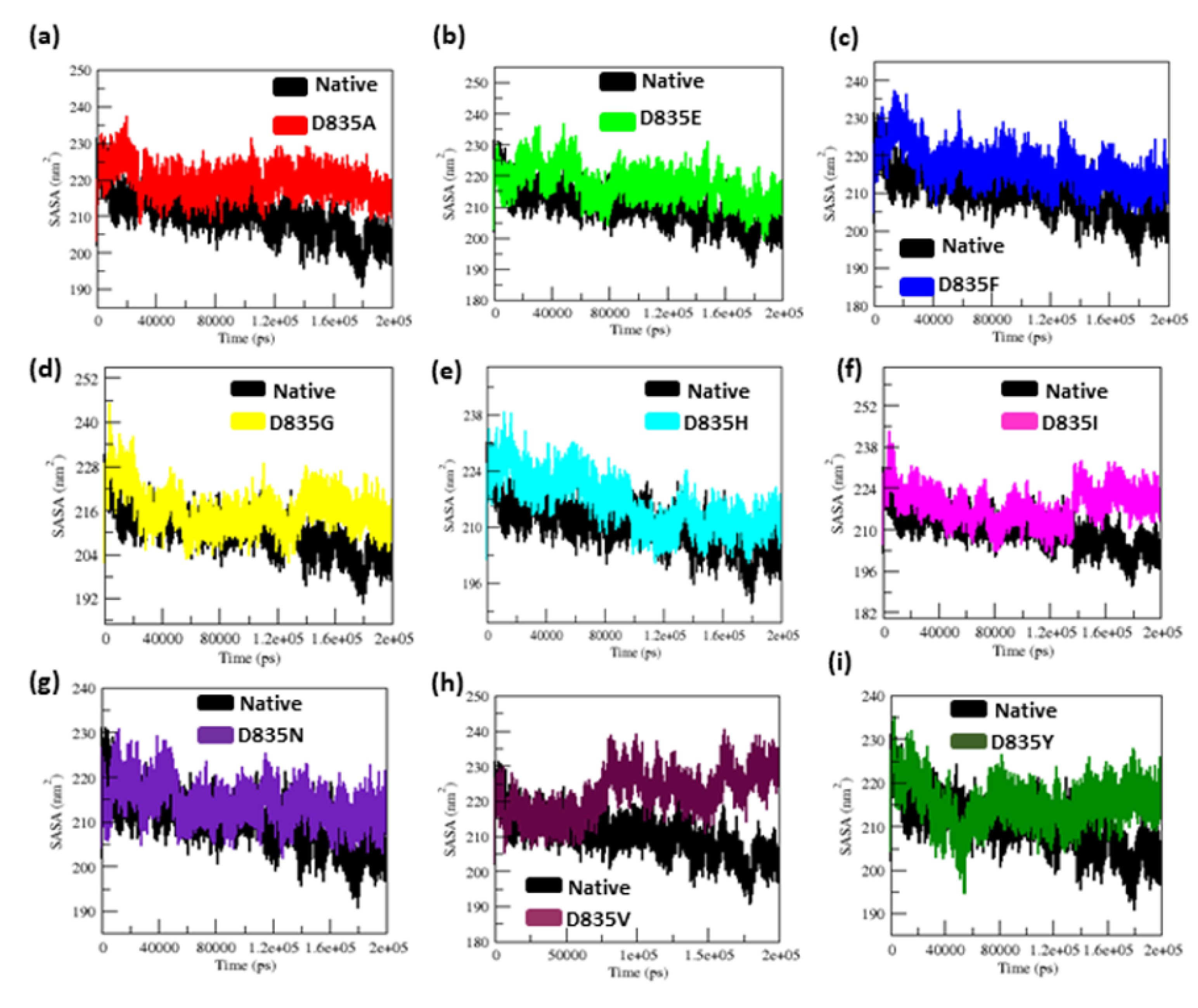
2.2.3. Geometry and Surface Analysis of Native and Mutants FLT3 Protein
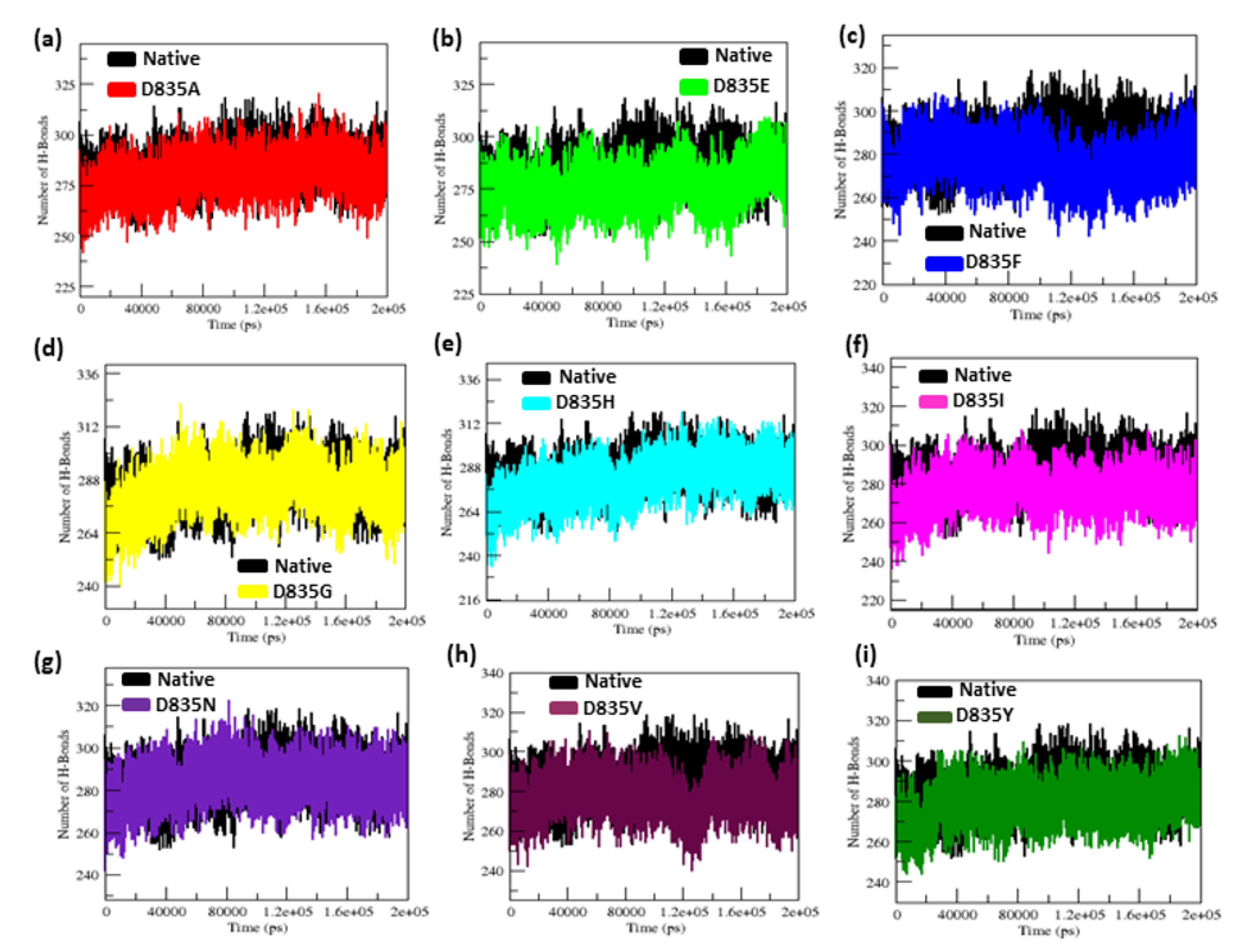
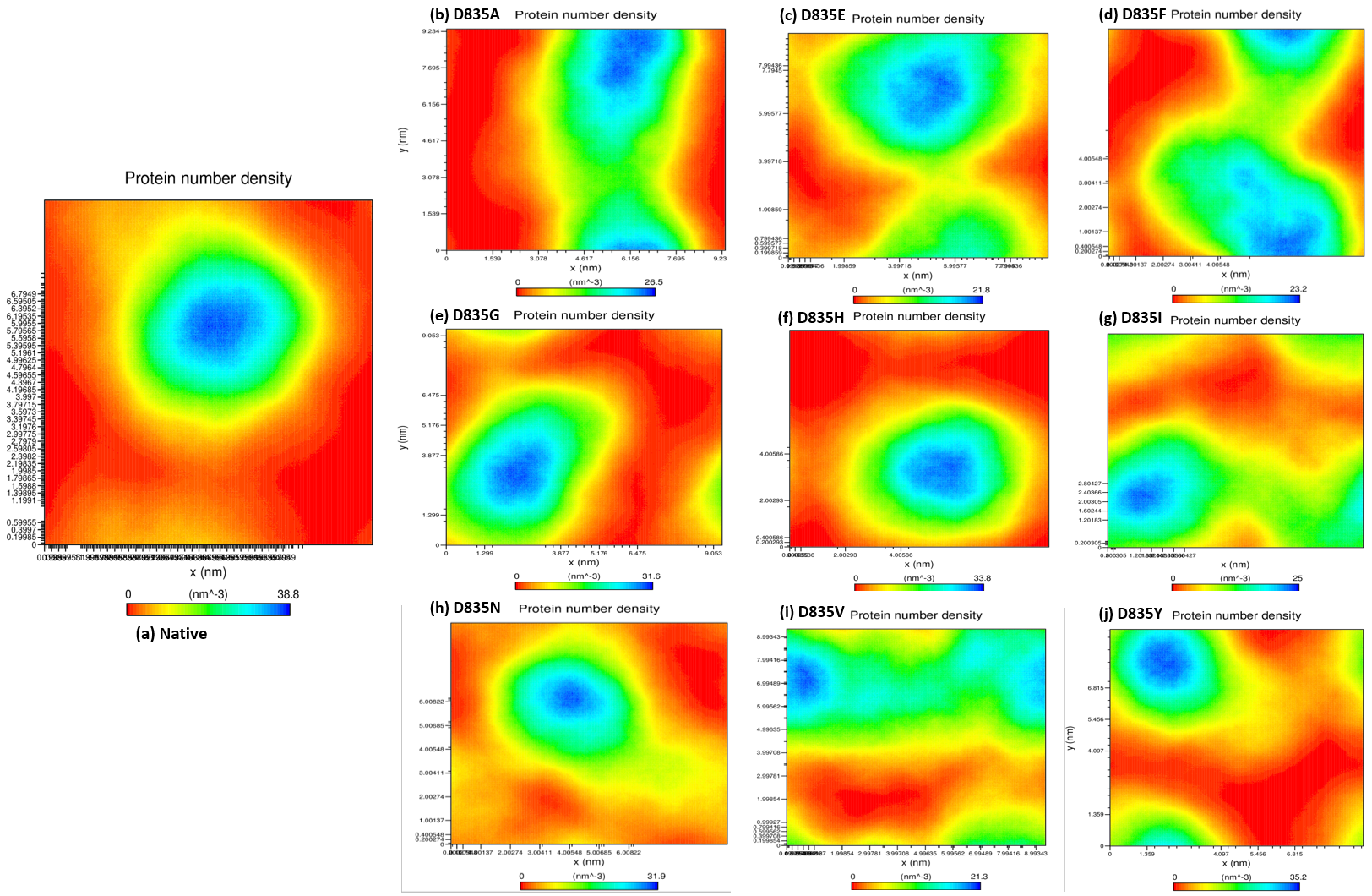
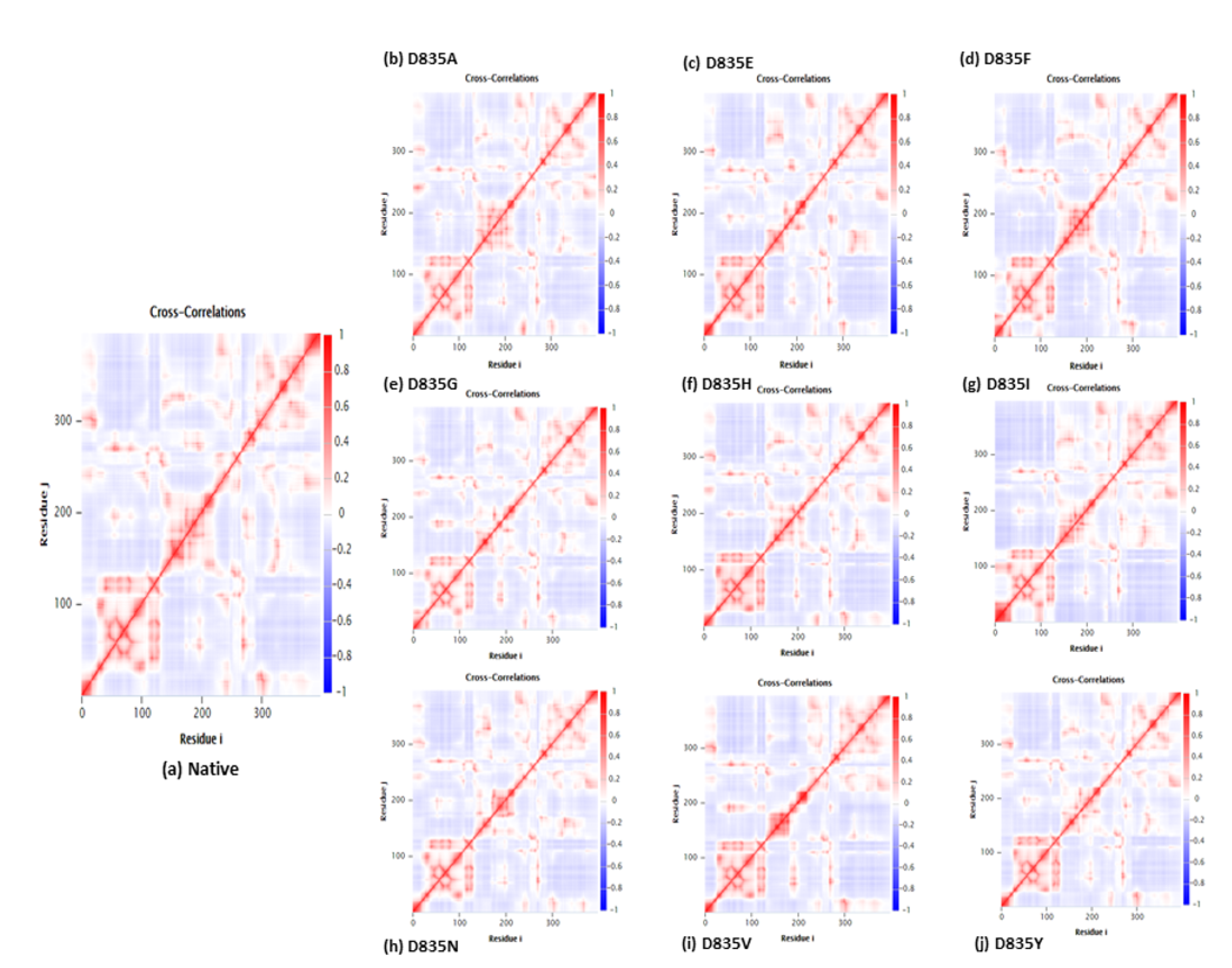
2.2.4. NH-Bonds, Density and DCCM Plot Analysis
2.3. Docking Analysis of AML Inhibitors with Native and Mutants FLT3 Proteins
3. Discussions
4. Methods
4.1. Datasets
4.2. Re-Modeling of FLT3 Protein
4.3. MD Simulation
4.4. Molecular Docking Using AutoDock Vina 4.2
4.4.1. Preparation of Native and Mutant FLT3 Proteins and Inhibitors for Docking
4.4.2. Docking of Native and Mutant FLT3 Proteins with AML Inhibitors
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Van Galen, P.; Hovestadt, V.; Wadsworth, M.H., II; Hughes, T.K.; Griffin, G.K.; Battaglia, S.; Verga, J.A.; Stephansky, J.; Pastika, T.J.; Story, J.L.; et al. Single-Cell RNA-Seq reveals AML hierarchies relevant to disease progression and immunity. Cell 2019, 176, 1265–1281. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.; Schlenk, R.F.; Russell, N.H.; Levis, M.J. Targeting FLT3 mutations in AML: Review of current knowledge and evidence. Leukemia 2019, 33, 299–312. [Google Scholar] [CrossRef] [PubMed]
- Carow, C.E.; Levenstein, M.; Kaufmann, S.H.; Chen, J.; Amin, S.; Rockwell, P.; Witte, L.; Borowitz, M.J.; Civin, C.I.; Small, D. Expression of the hematopoietic growth factor receptor FLT3 (STK-1/Flk2) in human leukemias. Blood 1996, 87, 1089–1096. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Zhang, Y.; Wang, F.; Wang, M.; Liu, H.; Chen, X.; Cao, P.; Ma, X.; Teng, W.; Zhang, X.; et al. The mutational spectrum of FLT3 gene in acute lymphoblastic leukemia is different from acute myeloid leukemia. Cancer Gene Ther. 2020, 27, 81–88. [Google Scholar] [CrossRef] [PubMed]
- Kayser, S.; Levis, M.J. FLT3 tyrosine kinase inhibitors in acute myeloid leukemia: Clinical implications and limitations. Leuk. Lymphoma 2014, 55, 243–255. [Google Scholar] [CrossRef] [PubMed]
- Gilliland, D.G.; Griffin, J.D. The roles of FLT3 in hematopoiesis and leukemia. Blood 2002, 100, 1532–1542. [Google Scholar] [CrossRef]
- Levis, M.; Small, D. FLT3: ITDoes matter in leukemia. Leukemia 2003, 17, 1738–1752. [Google Scholar] [CrossRef]
- Pratz, K.W.; Levis, M. How I treat FLT3-mutated AML. Blood 2017, 129, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Yamamoto, Y.; Kiyoi, H.; Nakano, Y.; Suzuki, R.; Kodera, Y.; Miyawaki, S.; Asou, N.; Kuriyama, K.; Yagasaki, F.; Shimazaki, C.; et al. Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood 2001, 97, 2434–2439. [Google Scholar] [CrossRef]
- Grafone, T.; Palmisano, M.; Nicci, C.; Storti, S. An overview on the role of FLT3-tyrosine kinase receptor in acute myeloid leukemia: Biology and treatment. Oncol. Rev. 2012, 6, e8. [Google Scholar] [CrossRef]
- Man, C.H.; Fung, T.K.; Ho, C.; Han, H.H.; Chow, H.C.; Ma, A.C.; Choi, W.W.; Lok, S.; Cheung, A.M.; Eaves, C.; et al. Sorafenib treatment of FLT3-ITD (+) acute myeloid leukemia: Favorable initial outcome and mechanisms of subsequent non-responsiveness associated with the emergence of a D835 mutation. Blood 2012, 119, 5133–5143. [Google Scholar] [CrossRef]
- Nakao, M.; Yokota, S.; Iwai, T.; Kaneko, H.; Horiike, S.; Kashima, K.; Sonoda, Y.; Fujimoto, T.; Misawa, S. Internal tandem duplication of the flt3 gene found in acute myeloid leukemia. Leukemia 1996, 10, 1911–1918. [Google Scholar] [PubMed]
- Smith, C.C.; Lin, K.; Stecula, A.; Sali, A.; Shah, N.P. FLT3 D835 mutations confer differential resistance to type II FLT3 inhibitors. Leukemia 2015, 29, 2390–2392. [Google Scholar] [CrossRef]
- Morley, G.M.; Uden, M.; Gullick, W.J.; Dibb, N.J. Cell specific transformation by c-fms activating loop mutations is attributable to constitutive receptor degradation. Oncogene 1999, 18, 3076–3084. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fenski, R.; Flesch, K.; Serve, S.; Mizuki, M.; Oelmann, E.; Kratz-Albers, K.; Kienast, J.; Leo, R.; Schwartz, S.; Berdel, W.E.; et al. Constitutive activation of FLT3 in acute myeloid leukaemia and its consequences for growth of 32D cells. Br. J. Haematol. 2000, 108, 322–330. [Google Scholar] [CrossRef]
- Abu-Duhier, F.M.; Goodeve, A.C.; Wilson, G.A.; Gari, M.A.; Peake, I.R.; Rees, D.C.; Vandenberghe, E.A.; Winship, P.R.; Reilly, J.T. Flt3 internal tandem duplication mutations in adult acute myeloid leukaemia define a high-risk group. Br. J. Haematol. 2000, 111, 190–195. [Google Scholar] [PubMed]
- Kottaridis, P.D.; Gale, R.E.; Frew, M.E.; Harrison, G.; Langabeer, S.E.; Belton, A.A.; Walker, H.; Wheatley, K.; Bowen, D.T.; Burnett, A.K.; et al. The presence of flt3 internal tandem duplication in patients with acute myeloid leukemia (AML) adds important prognostic information to cytogenetic risk group and response to the RST cycle of chemotherapy: Analysis of 854 patients from the United Kingdom medical research council AML 10 and 12 trials. Blood 2001, 98, 1752–1759. [Google Scholar]
- Fröhling, S.; Schlenk, R.F.; Breitruck, J.; Benner, A.; Kreitmeier, S.; Tobis, K.; Döhner, H.; Döhner, K.; AML Study Group Ulm. Acute Myeloid Leukemia. Prognostic significance of activating t3 mutations in younger adults (16 to 60 years) with acute myeloid leukemia and normal cytogenetics: A study of the AML study group ULM. Blood 2001, 100, 4372–4380. [Google Scholar] [CrossRef]
- Thiede, C.; Steudel, C.; Mohr, B.; Schaich, M.; Schäkel, U.; Platzbecker, U.; Wermke, M.; Bornhäuser, M.; Ritter, M.; Neubauer, A.; et al. Analysis of t3-activating mutations in 979 patients with acute myelogenous leukemia: Association with fab subtypes and identification of subgroups with poor prognosis: Presented in part at the 42nd annual meeting of the American Society of Hematology, December 1–5, 2000, San Francisco, CA (abstract 2334). Blood 2002, 99, 4326–4335. [Google Scholar]
- Yanada, M.; Matsuo, K.; Suzuki, T.; Kiyoi, H.; Naoe, T. Prognostic significance of t3 internal tandem duplication and tyrosine kinase domain mutations for acute myeloid leukemia: A meta-analysis. Leukemia 2005, 19, 1345. [Google Scholar] [CrossRef]
- Naoe, T.; Kiyoi, H.; Yamamoto, Y.; Minami, Y.; Yamamoto, K.; Ueda, R.; Saito, H. Flt3 tyrosine kinase as a target molecule for selective antileukemia therapy. Cancer Chemother. Pharmacol. 2001, 48, S27–S30. [Google Scholar] [CrossRef]
- Sawyers, C.L. Finding the next gleevec: Flt3 targeted kinase inhibitor therapy for acute myeloid leukemia. Cancer Cell 2002, 1, 413–415. [Google Scholar] [CrossRef]
- Levis, M.; Small, D. Novel t3 tyrosine kinase inhibitors. Expert Opin. Investig. Drugs 2003, 12, 1951–1962. [Google Scholar] [CrossRef] [PubMed]
- Stone, R.M.; DeAngelo, D.J.; Klimek, V.; Galinsky, I.; Estey, E.; Nimer, S.D.; Grandin, W.; Lebwohl, D.; Wang, Y.; Cohen, P.; et al. Patients with acute myeloid leukemia and an activating mutation in t3 respond to a small-molecule t3 tyrosine kinase inhibitor, pkc412. Blood 2005, 105, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Daver, N.; Cortes, J.; Ravandi, F.; Patel, K.P.; Burger, J.A.; Konopleva, M.; Kantarjian, H. Secondary mutations as mediators of resistance to targeted therapy in leukemia. Blood 2015, 125, 3236–3245. [Google Scholar] [CrossRef] [PubMed]
- Smith, C.C.; Paguirigan, A.; Jeschke, G.R.; Lin, K.C.; Massi, E.; Tarver, T.; Chin, C.S.; Asthana, S.; Olshen, A.; Travers, K.J.; et al. Heterogeneous resistance to quizartinib in acute myeloid leukemia revealed by single-cell analysis. Blood 2017, 130, 48–58. [Google Scholar] [CrossRef]
- Smith, C.C.; Wang, Q.; Chin, C.S.; Salerno, S.; Damon, L.E.; Levis, M.J.; Perl, A.E.; Travers, K.J.; Wang, S.; Hunt, J.P.; et al. Validation of ITD mutations in FLT3 as a therapeutic target in human acute myeloid leukaemia. Nature 2012, 485, 260–263. [Google Scholar] [CrossRef]
- Kiyoi, H.; Kawashima, N.; Ishikawa, Y. FLT3 mutations in acute myeloid leukemia: Therapeutic paradigm beyond inhibitor development. Cancer Sci. 2020, 111, 312–322. [Google Scholar] [CrossRef]
- Grunwald, M.R.; Levis, M.J. Flt3 inhibitors for acute myeloid leukemia: A review of their efficacy and mechanisms of resistance. Int. J. Hematol. 2013, 97, 683–694. [Google Scholar] [CrossRef]
- Gill, H.; Leung, A.Y.H.; Kwong, Y.L. Molecularly targeted therapy in acute myeloid leukemia. Future Oncol. 2016, 12, 827–838. [Google Scholar] [CrossRef] [PubMed]
- Hassanein, M.; Almahayni, M.H.; Ahmed, S.O.; Gaballa, S.; El Fakih, R. Flt3 inhibitors for treating acute myeloid leukemia. Clin. Lymphoma Myeloma Leuk. 2016, 16, 543–549. [Google Scholar] [CrossRef]
- Fathi, A.T.; Chen, Y.B. The role of t3 inhibitors in the treatment of t 3-mutated acute myeloid leukemia. Eur. J. Haematol. 2017, 98, 330–336. [Google Scholar] [CrossRef] [PubMed]
- Leick, M.B.; Levis, M.J. The future of targeting t3 activation in AML. Curr. Hematol. Malig. Rep. 2017, 12, 153–167. [Google Scholar] [CrossRef] [PubMed]
- Larrosa-Garcia, M.; Baer, M.R. Flt3 inhibitors in acute myeloid leukemia: Current status and future directions. Mol. Cancer Ther. 2017, 16, 991–1001. [Google Scholar] [CrossRef] [PubMed]
- Kiyoi, H.; Naoe, T. Biology, clinical relevance, and molecularly targeted therapy in acute leukemia with FLT3 mutation. Int. J. Hematol. 2006, 83, 301–308. [Google Scholar] [CrossRef]
- Weisberg, E.; Sattler, M.; Ray, A.; Griffin, J.D. Drug resistance in mutant FLT3-positive AML. Oncogene 2010, 29, 5120–5134. [Google Scholar] [CrossRef] [PubMed]
- Kamaraj, B.; Purohit, R. Mutational Analysis on Membrane Associated Transporter Protein (MATP) and their structural consequences in oculocutaeous albinism Type 4 (OCA4)-a molecular dynamics approach. J. Cell. Biochem. 2016, 117, 2608–2619. [Google Scholar] [CrossRef]
- Zaki, O.K.; Krishnamoorthy, N.; El Abd, H.S.; Harche, S.A.; Mattar, R.A.; Nofal, M.Y.; El Bekay, R.; Ahmed, K.A.; Doss, C.G.; Zayed, H. Two patients with Canavan disease and structural modeling of a novel mutation. Metab. Brain Dis. 2017, 32, 171–177. [Google Scholar] [CrossRef]
- Zaki, O.K.; Priya Doss, C.G.; Ali, S.A.; Murad, G.G.; Elashi, S.A.; Ebnou, M.S.; Kumar, D.T.; Khalifa, O.; Gamal, R.; El Abd, H.S.; et al. Genotype-phenotype correlation in patients with isovaleric acidaemia: Comparative structural modelling and computational analysis of novel variants. Hum. Mol. Genet. 2017, 26, 3105–3115. [Google Scholar] [CrossRef]
- Smith, C.C.; Zhang, C.; Lin, K.C.; Lasater, E.A.; Zhang, Y.; Massi, E.; Damon, L.E.; Pendleton, M.; Bashir, A.; Sebra, R.; et al. Characterizing and overriding the structural mechanism of the quizartinib-resistant FLT3 “gatekeeper” F691L mutation with PLX3397. Cancer Discov. 2015, 5, 668–679. [Google Scholar] [CrossRef]
- Zorn, J.A.; Wang, Q.; Fujimura, E.; Barros, T.; Kuriyan, J. Crystal structure of the FLT3 kinase domain bound to the inhibitor quizartinib (AC220). PLoS ONE 2015, 10, e0121177. [Google Scholar] [CrossRef]
- Yamaura, T.; Nakatani, T.; Uda, K.; Ogura, H.; Shin, W.; Kurokawa, N.; Saito, K.; Fujikawa, N.; Date, T.; Takasaki, M.; et al. A novel irreversible FLT3 inhibitor, FF-10101, shows excellent efficacy against AML cells with FLT3 mutations. Blood 2018, 131, 426–438. [Google Scholar] [CrossRef] [PubMed]
- Kawase, T.; Nakazawa, T.; Eguchi, T.; Tsuzuki, H.; Ueno, Y.; Amano, Y.; Suzuki, T.; Mori, M.; Yoshida, T. Effect of Fms-like tyrosine kinase 3 (FLT3) ligand (FL) on antitumor activity of gilteritinib, a FLT3 inhibitor, in mice xenografted with FL-overexpressing cells. Oncotarget 2019, 10, 6111–6123. [Google Scholar] [CrossRef] [PubMed]
- Gaffney, B.J. Lipoxygenases: Structural principles and spectroscopy. Annu. Rev. Biophys. Biomol. Struct. 1996, 25, 431–459. [Google Scholar] [CrossRef] [PubMed]
- Grantcharova, V.P.; Riddle, D.S.; Santiago, J.V.; Baker, D. Important role of hydrogen bonds in the structurally polarized transition state for folding of the src SH3 domain. Nat. Struct. Biol. 1998, 5, 714–720. [Google Scholar] [CrossRef] [PubMed]
- Kamaraj, B.; Purohit, R. Computational screening of disease-associated mutations in OCA2 gene. Cell Biochem. Biophys. 2014, 68, 97–109. [Google Scholar] [CrossRef]
- Lobanov, M.Y.; Bogatyreva, N.S.; Galzitskaya, O.V. Radius of gyration as an indicator of protein structure compactness. Mol. Biol. 2008, 42, 623–628. [Google Scholar] [CrossRef]
- Pace, C.N.; Fu, H.; Lee Fryar, K.; Landua, J.; Trevino, S.R.; Schell, D.; Thurlkill, R.L.; Imura, S.; Scholtz, J.M.; Gajiwala, K.; et al. Contribution of hydrogen bonds to protein stability. Protein Sci. 2014, 23, 652–661. [Google Scholar] [CrossRef]
- Gopalakrishnan, C.; Al-Subaie, A.M.; Yeh, H.Y.; Tayubi, I.A.; Kamaraj, B. Prioritization of SNPs in y+ LAT-1 culpable of Lysinuric protein intolerance and their mutational impacts using protein-protein docking and molecular dynamics simulation studies. J. Cell. Biochem. 2019, 120, 18496–18508. [Google Scholar] [CrossRef]
- Kamaraj, B.; Bogaerts, A. Structure and function of p53-DNA complexes with inactivation and rescue mutations: A molecular dynamics simulation study. PLoS ONE 2015, 10, e0134638. [Google Scholar] [CrossRef]
- Balu, K.; Rajendran, V.; Sethumadhavan, R.; Purohit, R. Investigation of binding phenomenon of NSP3 and p130Cas mutants and their effect on cell signalling. Cell Biochem. Biophys. 2013, 67, 623–633. [Google Scholar] [CrossRef] [PubMed]
- Kamaraj, B.; Purohit, R. In silico screening and molecular dynamics simulation of disease-associated nsSNP in TYRP1 gene and its structural consequences in OCA3. BioMed Res. Int. 2013, 2013. [Google Scholar] [CrossRef] [PubMed]
- Kamaraj, B.; Al-Subaie, A.M.; Ahmad, F.; Surapaneni, K.M.; Alsamman, K. Effect of novel leukemia mutations (K75E & E222K) on interferon regulatory factor 1 and its interaction with DNA: Insights from molecular dynamics simulations and docking studies. J. Biomol. Struct. Dyn. 2020, 1–13. [Google Scholar] [CrossRef]
- Chasman, D.; Adams, R.M. Predicting the functional consequences of non-synonymous single nucleotide polymorphisms: Structure-based assessment of amino acid variation. J. Mol. Biol. 2001, 307, 683–706. [Google Scholar] [CrossRef] [PubMed]
- Sunyaev, S.; Ramensky, V.; Bork, P. Towards a structural basis of human non-synonymous single nucleotide polymorphisms. Trends Genet. 2000, 16, 198–200. [Google Scholar] [CrossRef]
- Nagarajan, N.; Chellam, J.; Kannan, R.R. Exploring the functional impact of mutational drift in LRRK2 gene and identification of specific inhibitors for the treatment of Parkinson disease. J. Cell. Biochem. 2018, 119, 4878–4889. [Google Scholar] [CrossRef]
- Nagasundaram, N.; Wilson Alphonse, C.R.; Samuel Gnana, P.V.; Rajaretinam, R.K. Molecular dynamics validation of crizotinib resistance to ALK mutations (L1196M and G1269A) and identification of specific inhibitors. J. Cell. Biochem. 2017, 118, 3462–3471. [Google Scholar] [CrossRef] [PubMed]
- Nagasundaram, N.; Chakraborty, C.; Karthick, V.; Balaji, V.; Siva, R.; Lu, A.; Ge, Z.; Zhu, H. Mechanism of artemisinin resistance for malaria PfATP6 L263 mutations and discovering potential antimalarials: An integrated computational approach. Sci. Rep. 2016, 6, 1–12. [Google Scholar]
- UniProt Consortium. UniProt: A hub for protein information. Nucleic Acids Res. 2015, 43, D204–D212. [Google Scholar] [CrossRef]
- Tate, J.G.; Bamford, S.; Jubb, H.C.; Sondka, Z.; Beare, D.M.; Bindal, N.; Boutselakis, H.; Cole, C.G.; Creatore, C.; Dawson, E.; et al. COSMIC: The catalogue of somatic mutations in cancer. Nucleic Acids Res. 2019, 47, D941–D947. [Google Scholar] [CrossRef]
- Griffith, J.; Black, J.; Faerman, C.; Swenson, L.; Wynn, M.; Lu, F.; Lippke, J.; Saxena, K. The structural basis for autoinhibition of FLT3 by the juxtamembrane domain. Mol. Cell 2004, 13, 169–178. [Google Scholar] [CrossRef]
- Berman, H.M.; Battistuz, T.; Bhat, T.N.; Bluhm, W.F.; Bourne, P.E.; Burkhardt, K.; Feng, Z.; Gilliland, G.L.; Iype, L.; Jain, S.; et al. The protein data bank. Acta Crystallogr. D Biol. Crystallogr. 2002, 58, 899–907. [Google Scholar] [CrossRef]
- Kim, S.; Thiessen, P.A.; Bolton, E.E.; Chen, J.; Fu, G.; Gindulyte, A.; Han, L.; He, J.; He, S.; Shoemaker, B.A.; et al. PubChem substance and compound databases. Nucleic Acids Res. 2016, 44, D1202–D1213. [Google Scholar] [CrossRef]
- Zhang, Y. I-TASSER server for protein 3D structure prediction. BMC Bioinform. 2008, 9, 40. [Google Scholar] [CrossRef]
- Laskowski, R.A.; Rullmannn, J.A.; MacArthur, M.W.; Kaptein, R.; Thornton, J.M. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J. Biomol. NMR 1996, 8, 477–486. [Google Scholar] [CrossRef]
- Wiederstein, M.; Sippl, M.J. ProSA-web: Interactive web service for the recognition of errors in three-dimensional structures of proteins. Nucleic Acids Res. 2007, 35, W407–W410. [Google Scholar] [CrossRef]
- Abraham, M.; Van Der Spoel, D.; Lindahl, E.; Hess, B.; Spoel, D.; van der Lindahl, E. GROMACS User Manual Version 5.0.4. 2014. Available online: www.gromacs.org (accessed on 21 December 2020).
- Fliege, J.; Svaiter, B.F. Steepest descent methods for multicriteria optimization. Math. Methods Oper. Res. 2000, 51, 479–494. [Google Scholar] [CrossRef]
- Darden, T.; York, D.; Pedersen, L. Particle mesh Ewald: An N log (N) method for Ewald sums in large systems. J. Chem. Phys. 1993, 98, 10089–10092. [Google Scholar] [CrossRef]
- Berendsen, H.J.; Postma, J.V.; van Gunsteren, W.F.; DiNola, A.R.H.J.; Haak, J.R. Molecular dynamics with coupling to an external bath. J. Chem. Phys. 1984, 81, 3684–3690. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Polymorphic transitions in single crystals: A new molecular dynamics method. J. App. Phys. 1981, 52, 7182–7190. [Google Scholar] [CrossRef]
- Hess, B.; Bekker, H.; Berendsen, H.J.; Fraaije, J.G. LINCS: A linear constraint solver for molecular simulations. J. Comput. Chem. 1997, 18, 1463–1472. [Google Scholar] [CrossRef]
- Turner, P.J. XMGRACE, Version 5.1. 19; Center for Coastal and Land-Margin Research, Oregon Graduate Institute of Science and Technology: Beaverton, OR, USA, 2005. [Google Scholar]
- DeLano, W.L. The PyMOL molecular graphics system 2002. Available online: http://www.pymol.org (accessed on 21 December 2020).
- Morris, G.M.; Huey, R.; Lindstrom, W.; Sanner, M.F.; Belew, R.K.; Goodsell, D.S.; Olson, A.J. AutoDock4 and AutoDockTools4: Automated docking with selective receptor flexibility. J. Comput. Chem. 2009, 30, 2785–2791. [Google Scholar] [CrossRef] [PubMed]
- Trott, O.; Olson, A.J. AutoDock Vina: Improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput. Chem. 2010, 31, 455–461. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Type of Protein | Parameters | |||||
|---|---|---|---|---|---|---|
| RMSD (nm) | RMSF | RG (nm) | SASA | Density | NH-Bond | |
| Native | 0.32 ± 0.02 | 0.12 ± 0.09 | 2.15 ± 0.02 | 210.25 ± 5.50 | 38.8 | 284.85 ± 9.20 |
| D835A | 0.34 ± 0.07 | 0.16 ± 0.15 | 2.18 ± 0.01 | 219.51 ± 3.77 | 26.5 | 279.91 ± 9.17 |
| D835E | 0.35 ± 0.04 | 0.17 ± 0.14 | 2.18 ± 0.02 | 217.02 ± 5.13 | 21.8 | 276.11 ± 9.09 |
| D835F | 0.39 ± 0.04 | 0.16 ± 0.12 | 2.19 ± 0.01 | 217.02 ± 4.94 | 23.2 | 276.58 ± 8.99 |
| D835G | 0.47 ± 0.07 | 0.17 ± 0.13 | 2.19 ± 0.02 | 216.16 ± 5.28 | 31.6 | 283.47 ± 9.95 |
| D835H | 0.40 ± 0.04 | 0.16 ± 0.16 | 2.20 ± 0.02 | 215.94 ± 6.51 | 33.8 | 281.06 ± 11.23 |
| D835I | 0.45 ± 0.17 | 0.22 ± 0.22 | 2.21 ± 0.03 | 218.12 ± 5.48 | 25 | 275.78 ± 9.15 |
| D835N | 0.41 ± 0.05 | 0.15 ± 0.11 | 2.18 ± 0.04 | 214.24 ± 3.98 | 31.9 | 283.65 ± 8.96 |
| D835V | 0.41 ± 0.09 | 0.19 ±0.17 | 2.19 ± 0.01 | 222.08 ± 5.96 | 21.3 | 277.54 ± 9.04 |
| D835Y | 0.35 ± 0.03 | 0.13 ± 0.09 | 2.17 ± 0.01 | 215.21 ± 4.66 | 35.2 | 279.04 ± 8.85 |
| Protein Type | Secondary Structures of Protein | |||
|---|---|---|---|---|
| Helix | Beta-Sheets | Coil | Turn | |
| Native | 155 (39%) | 74 (18%) | 168 (42%) | 80 (2%) |
| D835A | 139 (35%) | 60 (15%) | 196 (49%) | 104 (26%) |
| D835E | 145 (36%) | 68 (17%) | 182 (46%) | 124 (31%) |
| D835F | 152 (38%) | 74 (18%) | 169 (46%) | 88 (22%) |
| D835G | 147 (37%) | 66 (16%) | 182 (46%) | 120 (30%) |
| D835H | 153 (38%) | 69 (17%) | 173 (44%) | 96 (24%) |
| D835I | 141 (35%) | 67 (16%) | 187 (47%) | 88 (22%) |
| D835N | 152 (38%) | 63 (15%) | 176 (44%) | 96 (24%) |
| D835V | 150 (37%) | 68 (17%) | 177 (44%) | 88 (22%) |
| D835Y | 150 (37%) | 74 (18%) | 175 (44%) | 100 (25%) |
| Protein Type (0 ns) | Inhibitor’s Name | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Crenolanib | FF-10101 | Gilteritinib | KW-2449 | PLX3397 | Ponatinib | Quizartinib | Sorafenib | Sunitinib | Tandutinib | |
| Native | −8.9 | −8.3 | −8.5 | −9.9 | −9.7 | −10.3 | −9.5 | −10.4 | −8.3 | −9.6 |
| D835A | −8.5 | −5.8 | −6.7 | −8.8 | −9.6 | −7.5 | −7.3 | −8.3 | −6.4 | −7.1 |
| D835E | −10.4 | −7.7 | −7.3 | −10.7 | −10.1 | −7.9 | −6.4 | −8.3 | −9.3 | −7.5 |
| D835F | −9.7 | −7.8 | −8.1 | −9.2 | −9.2 | −8.4 | −8.4 | −9.2 | −8.1 | −7.4 |
| D835G | −10.2 | −5.9 | −7.6 | −9.9 | −9.9 | −8.4 | −3.8 | −10.5 | −9.4 | −7.2 |
| D835H | −9.2 | −7.6 | −9.3 | −8.3 | −9.0 | −10.1 | −9.5 | −9.5 | −8.0 | −8.7 |
| D835I | −9.0 | −8.6 | −8.0 | −8.7 | −9.9 | −10.0 | −8.2 | −10.8 | −8.4 | −9.8 |
| D835N | −9.6 | −8.2 | −4.8 | −9.5 | −9.8 | −7.7 | −5.0 | −9.7 | −8.3 | −8.6 |
| D835V | −10.4 | −8.8 | −9.6 | −10.6 | −10.1 | −12.2 | −10.5 | −10.8 | −9.0 | −9.9 |
| D835Y | −8.9 | −8.3 | −8.9 | −9.5 | −10.1 | −9.9 | −11.1 | −10.4 | −8.7 | −9.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Al-Subaie, A.M.; Kamaraj, B. The Structural Effect of FLT3 Mutations at 835th Position and Their Interaction with Acute Myeloid Leukemia Inhibitors: In Silico Approach. Int. J. Mol. Sci. 2021, 22, 7602. https://doi.org/10.3390/ijms22147602
Al-Subaie AM, Kamaraj B. The Structural Effect of FLT3 Mutations at 835th Position and Their Interaction with Acute Myeloid Leukemia Inhibitors: In Silico Approach. International Journal of Molecular Sciences. 2021; 22(14):7602. https://doi.org/10.3390/ijms22147602
Chicago/Turabian StyleAl-Subaie, Abeer M., and Balu Kamaraj. 2021. "The Structural Effect of FLT3 Mutations at 835th Position and Their Interaction with Acute Myeloid Leukemia Inhibitors: In Silico Approach" International Journal of Molecular Sciences 22, no. 14: 7602. https://doi.org/10.3390/ijms22147602
APA StyleAl-Subaie, A. M., & Kamaraj, B. (2021). The Structural Effect of FLT3 Mutations at 835th Position and Their Interaction with Acute Myeloid Leukemia Inhibitors: In Silico Approach. International Journal of Molecular Sciences, 22(14), 7602. https://doi.org/10.3390/ijms22147602