
Innate Lymphoid Cells in Intestinal Homeostasis and Inflammatory Bowel Disease

 ,
, 
Abstract
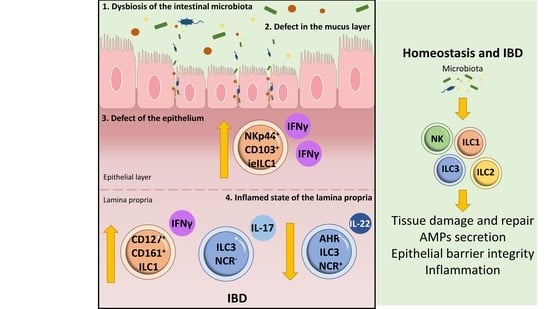
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
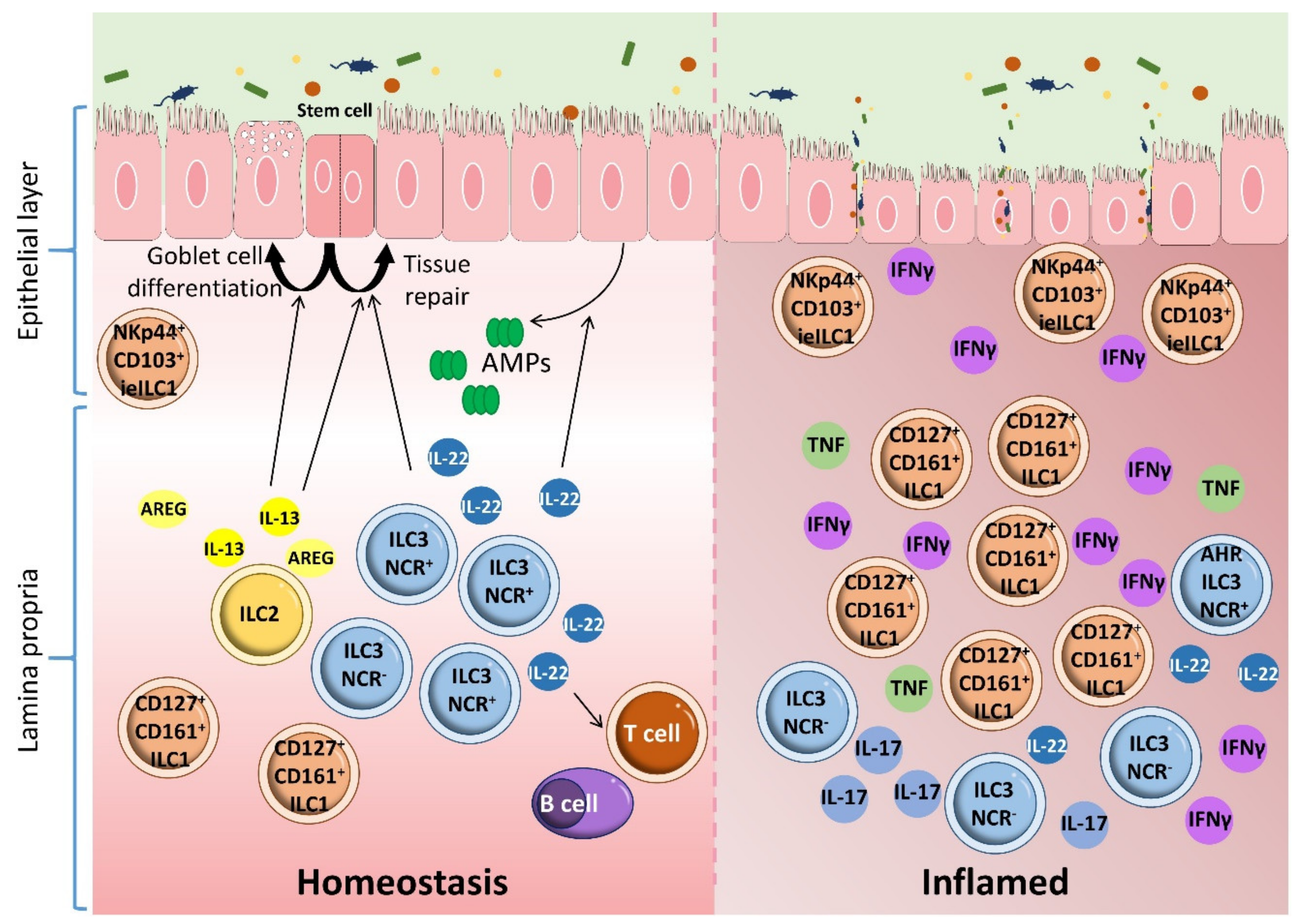{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Inflammatory Bowel Disease (IBD)
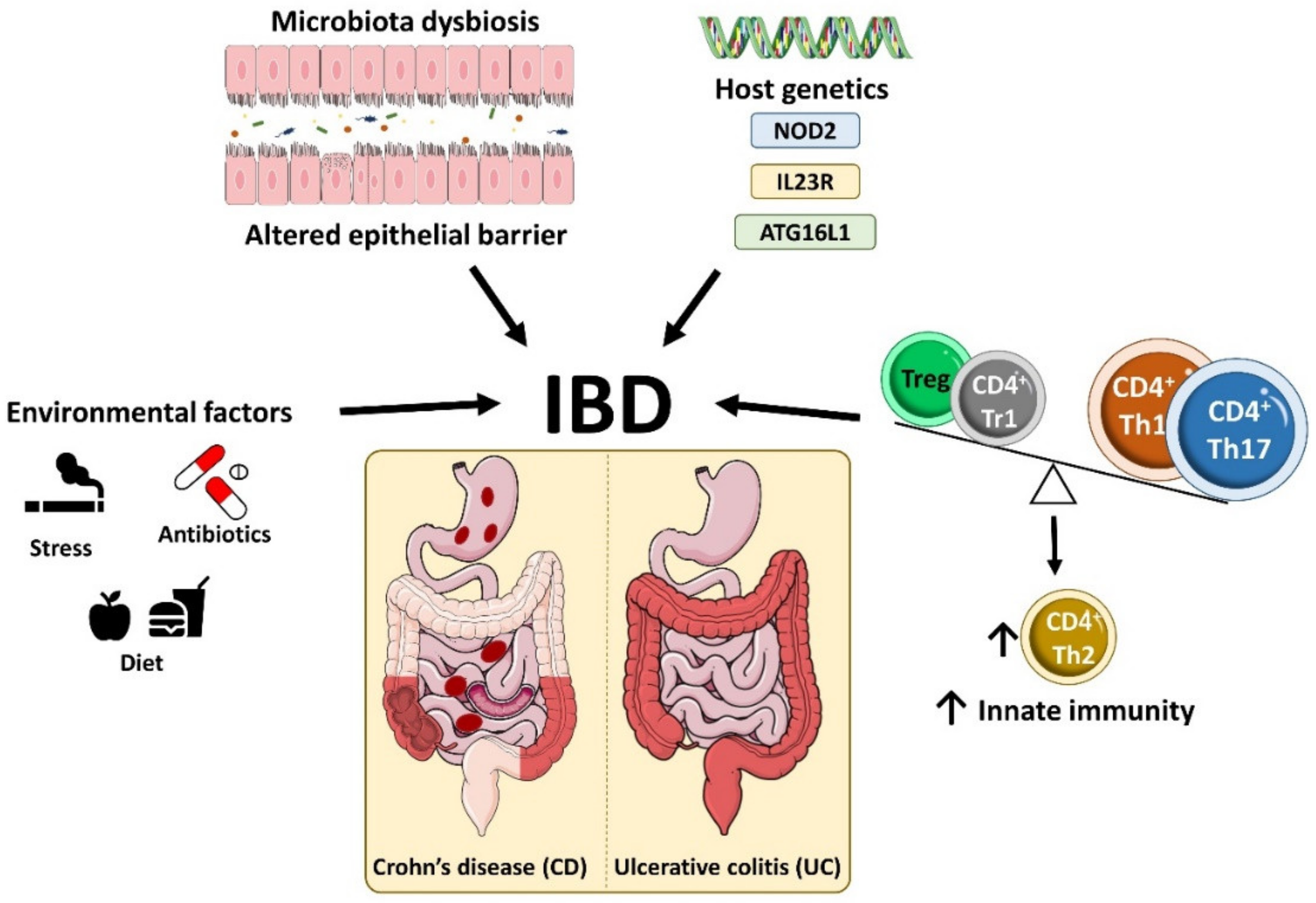
2. Inflammatory Bowel Disease Etiology and Pathophysiology
3. Innate Lymphoid Cells (ILCs)
4. Type 1 ILCs
5. Type 2 ILCs
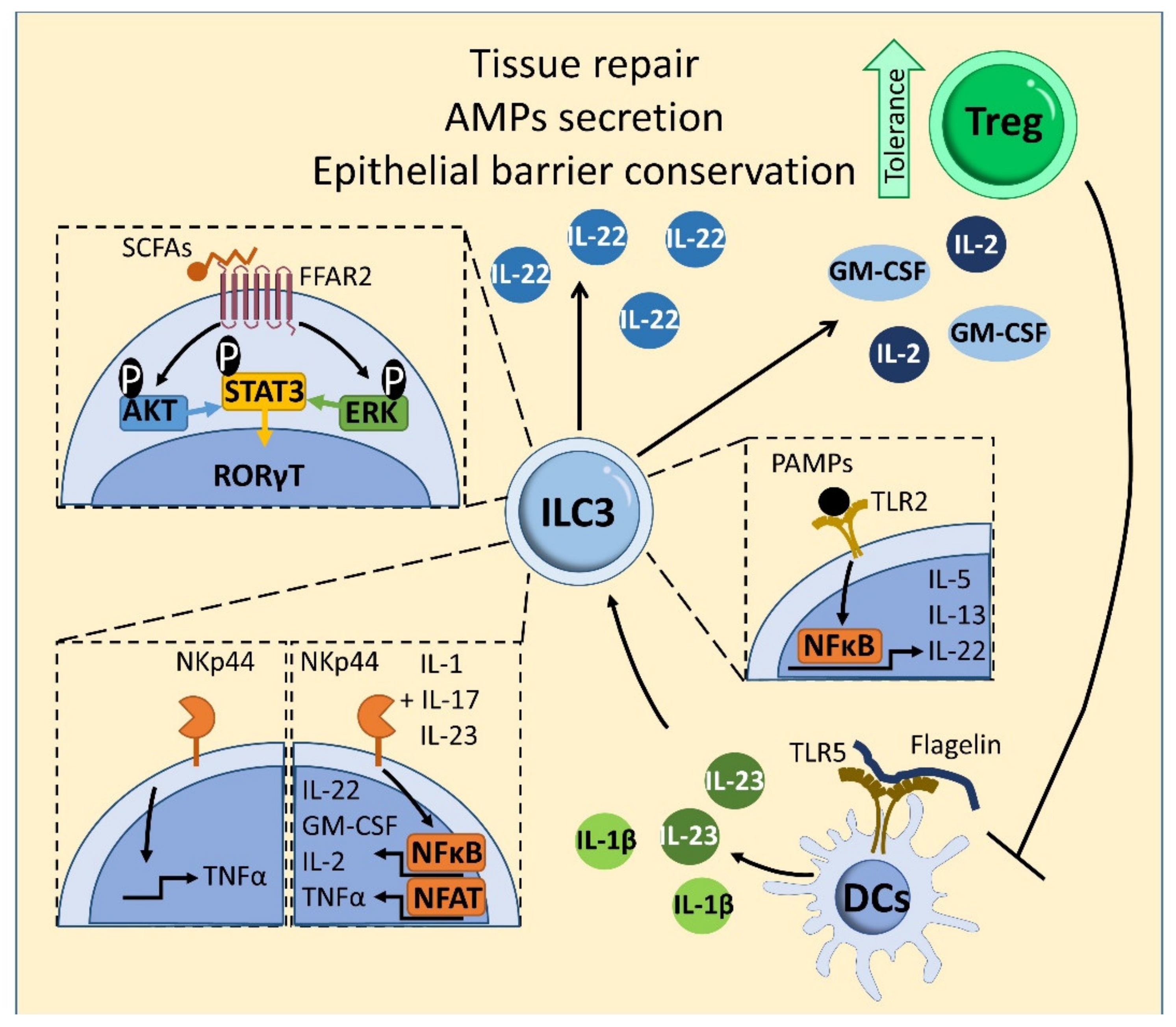
6. Type 3 ILCs
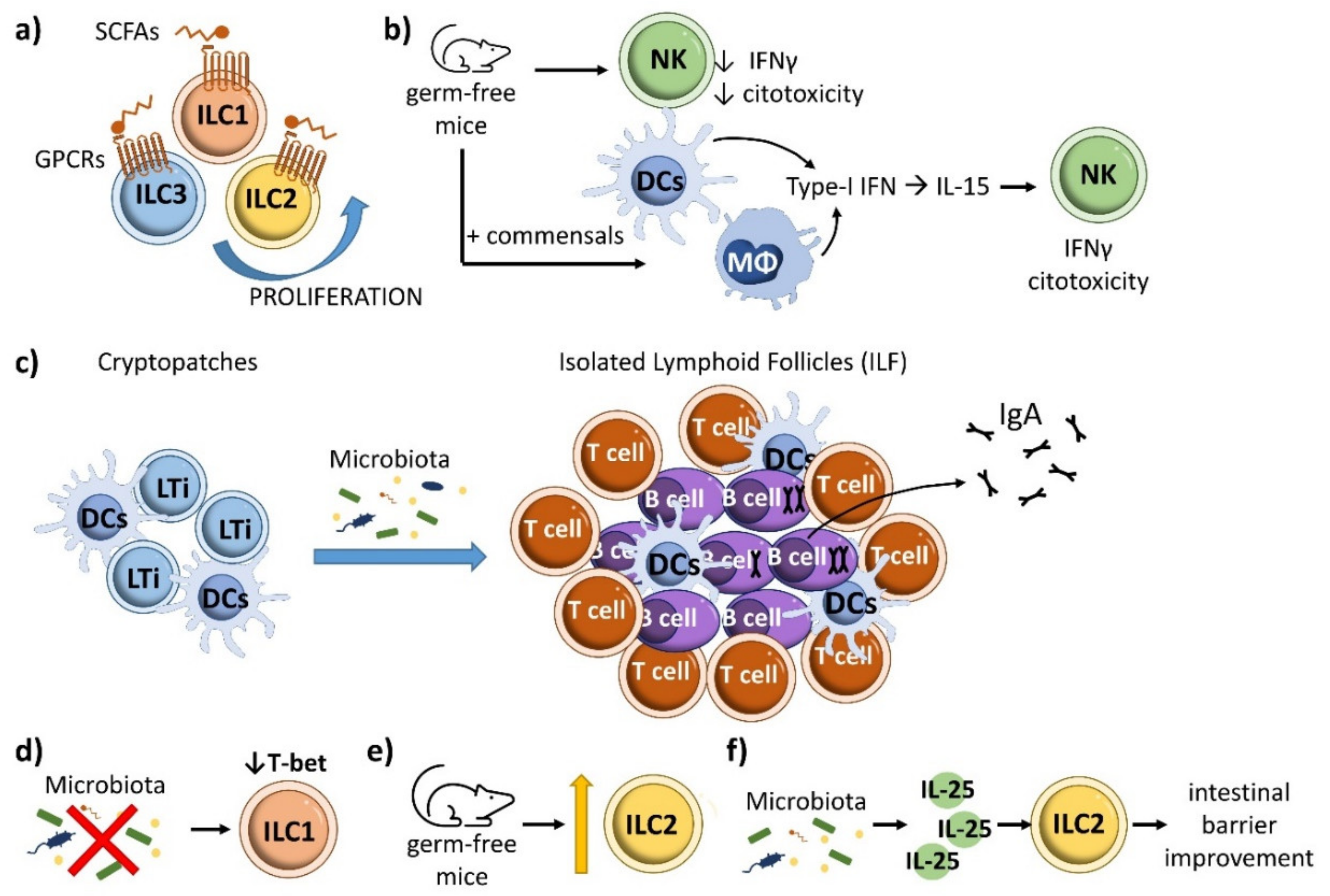
7. ILCs and Intestinal Homeostasis
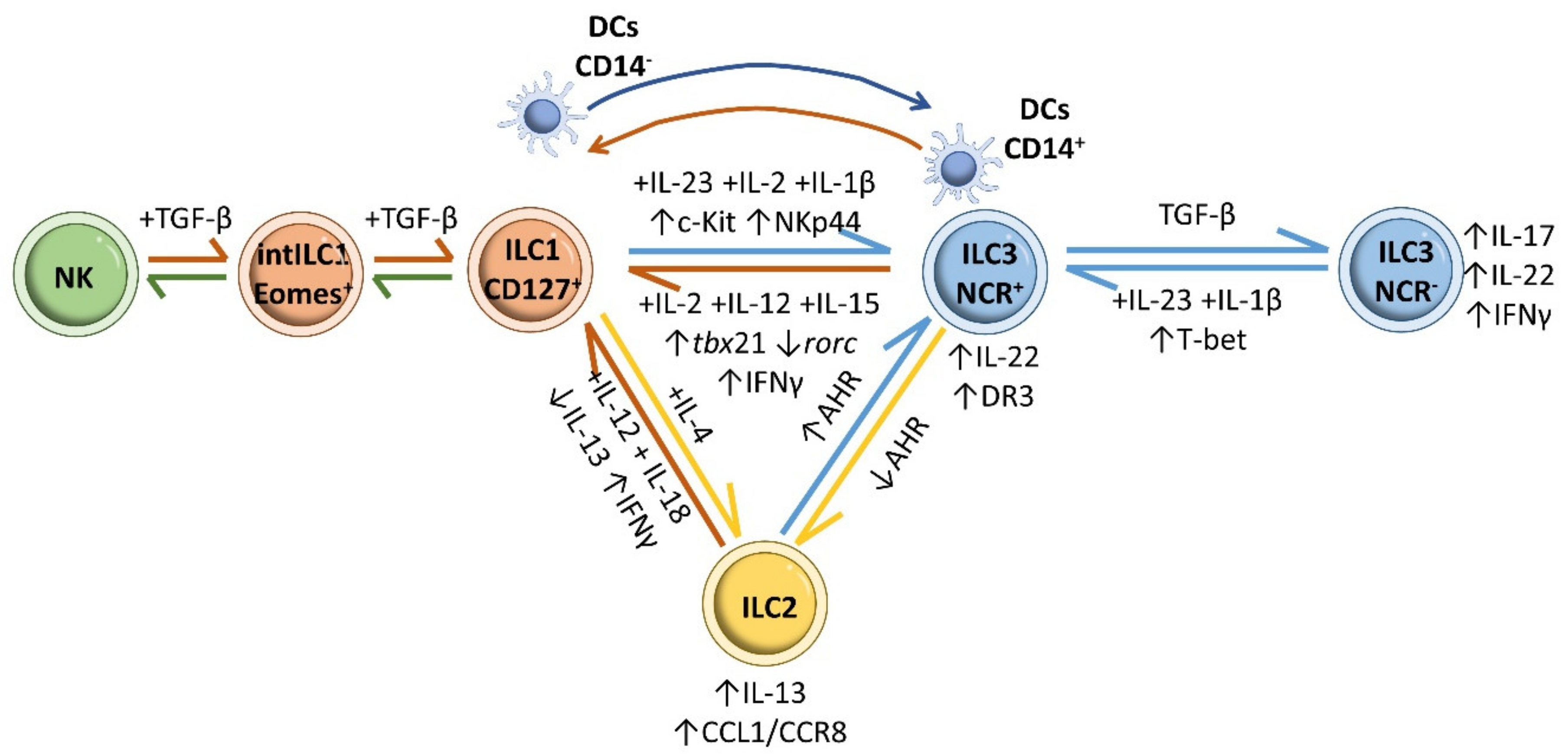
8. ILCs and Inflammatory Bowel Disease
9. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| AHR | Aryl hydrocarbon receptor |
| CCL1 | C-C chemokine ligand 1 |
| CCR8 | C-C motif chemokine receptor 8 |
| CD | Crohn’s disease |
| IBD | Inflammatory bowel disease |
| ILCs | Innate lymphoid cells |
| EGFR | Epidermal growth factor receptor |
| Fut2 | Fucosyl transferase 2 |
| GPCRs | G protein-coupled receptors |
| NCR | Natural cytotoxicity receptor |
| NK | Natural killer |
| NOD2 | Nucleotide binding oligomerization domain containing 2 |
| LTis | Lymphoid tissue-inducers |
| PAMPs | Pathogen-associated molecular patterns |
| PGD2 | Prostaglandin D2 |
| PTGDR2 | Prostaglandin D2 receptor 2 |
| RORγt | RAR-related orphan receptor gamma t |
| SCFAs | Short-chain fatty acids |
| SLAMF1 | Signaling lymphocytic activation molecule family member 1 |
| SPF | Specific-pathogen-free |
| Th | T-helper |
| TL1A | TNF-like ligand 1 A |
| TLRs | Toll-like receptors |
| Treg | T regulatory |
| Tr1 | T regulatory type 1 |
| UC | Ulcerative colitis |
References
- Satsangi, J.; Silverberg, M.S.; Vermeire, S.; Colombel, J.F. The Montreal classification of inflammatory bowel disease: Controversies, consensus, and implications. Gut 2006, 55, 749–753. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Global burden of inflammatory bowel disease 2017 Collaborators. The global, regional, and national burden of inflammatory bowel disease in 195 countries and territories, 1990–2017: A systematic analysis for the Global Burden of Disease Study 2017. Lancet Gastroenterol. Hepatol. 2020, 5, 17–30. [Google Scholar] [CrossRef] [Green Version]
- Peloquin, J.M.; Goel, G.; Villablanca, E.J.; Xavier, R.J. Mechanisms of pediatric inflammatory bowel disease. Annu. Rev. Immunol. 2016, 34, 31–64. [Google Scholar] [CrossRef]
- Liu, T.C.; Stappenbeck, T.S. Genetics and pathogenesis of inflammatory bowel disease. Annu. Rev. Pathol. 2016, 11, 127–148. [Google Scholar] [CrossRef] [Green Version]
- Torres, J.; Mehandru, S.; Colombel, J.F.; Peyrin-Biroulet, L. Crohn’s disease. Lancet 2017, 389, 1741–1755. [Google Scholar] [CrossRef]
- Ungaro, R.; Mehandru, S.; Allen, P.B.; Peyrin-Biroulet, L.; Colombel, J.F. Ulcerative colitis. Lancet 2017, 389, 1756–1770. [Google Scholar] [CrossRef]
- Cleynen, I.; Gonzalez, J.R.; Figueroa, C.; Franke, A.; McGovern, D.; Bortlik, M.; Crusius, B.J.; Vecchi, M.; Artieda, M.; Szczypiorska, M.; et al. Genetic factors conferring an increased susceptibility to develop Crohn’s disease also influence disease phenotype: Results from the IBDchip European Project. Gut 2013, 62, 1556–1565. [Google Scholar] [CrossRef] [Green Version]
- Louis, E.; van Kemseke, C.; Reenaers, C. Necessity of phenotypic classification of inflammatory bowel disease. Best. Pract. Res. Clin. Gastroenterol. 2011, 25, S2–S7. [Google Scholar] [CrossRef]
- Levine, A.; Griffiths, A.; Markowitz, J.; Wilson, D.C.; Turner, D.; Russell, R.K.; Fell, J.; Ruemmele, F.M.; Walters, T.; Sherlock, M.; et al. Pediatric modification of the Montreal classification for inflammatory bowel disease: The Paris classification. Inflamm. Bowel Dis. 2011, 17, 1314–1321. [Google Scholar] [CrossRef]
- North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition; Colitis Foundation of America; Bousvaros, A.; Antonioli, D.A.; Colletti, R.B.; Dubinsky, M.C.; Glickman, J.N.; Gold, B.D.; Griffiths, A.M.; Jevon, G.P.; et al. Differentiating ulcerative colitis from Crohn disease in children and young adults: Report of a working group of the North American Society for Pediatric Gastroenterology, Hepatology, and Nutrition and the Crohn’s and Colitis Foundation of America. J. Pediatr. Gastroenterol. Nutr. 2007, 44, 653–674. [Google Scholar]
- Choy, M.C.; Visvanathan, K.; de Cruz, P. An Overview of the innate and adaptive immune system in inflammatory bowel disease. Inflamm. Bowel Dis. 2017, 23, 2–13. [Google Scholar] [CrossRef]
- De Souza, H.S.P.; Fiocchi, C.; Iliopoulos, D. The IBD interactome: An integrated view of aetiology, pathogenesis and therapy. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 739–749. [Google Scholar] [CrossRef]
- Parkes, M.; Jewell, D. Ulcerative colitis and Crohns disease: Molecular genetics and clinical implications. Expert Rev. Mol. Med. 2001, 2001, 1–18. [Google Scholar] [CrossRef]
- Farmer, R.G.; Michener, W.M.; Mortimer, E.A. Studies of family history among patients with inflammatory bowel disease. Clin. Gastroenterol. 1980, 9, 271–277. [Google Scholar] [CrossRef]
- Zhang, H.; Massey, D.; Tremelling, M.; Parkes, M. Genetics of inflammatory bowel disease: Clues to pathogenesis. Br. Med. Bull. 2008, 87, 17–30. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.Z.; van Sommeren, S.; Huang, H.; Ng, S.C.; Alberts, R.; Takahashi, A.; Ripke, S.; Lee, J.C.; Jostins, L.; Shah, T.; et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 2015, 47, 979–986. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.H. The Nod2 gene in Crohn’s disease: Implications for future research into the genetics and immunology of Crohn’s disease. Inflamm. Bowel Dis. 2001, 7, 271–275. [Google Scholar] [CrossRef] [PubMed]
- Bonen, D.K.; Ogura, Y.; Nicolae, D.L.; Inohara, N.; Saab, L.; Tanabe, T.; Chen, F.F.; Foster, S.J.; Duerr, R.H.; Brant, S.R.; et al. Crohn’s disease-associated NOD2 variants share a signaling defect in response to lipopolysaccharide and peptidoglycan. Gastroenterology 2003, 124, 140–146. [Google Scholar] [CrossRef] [PubMed]
- Economou, M.; Trikalinos, T.A.; Loizou, K.T.; Tsianos, E.V.; Ioannidis, J.P. Differential effects of NOD2 variants on Crohn’s disease risk and phenotype in diverse populations: A meta-analysis. Am. J. Gastroenterol. 2004, 99, 2393–2404. [Google Scholar] [CrossRef] [PubMed]
- Leong, R.W.; Armuzzi, A.; Ahmad, T.; Wong, M.L.; Tse, P.; Jewell, D.P.; Sung, J.J. NOD2/CARD15 gene polymorphisms and Crohn’s disease in the Chinese population. Aliment. Pharmacol. Ther. 2003, 17, 1465–1470. [Google Scholar] [CrossRef] [PubMed]
- Yamazaki, K.; Takazoe, M.; Tanaka, T.; Kazumori, T.; Nakamura, Y. Absence of mutation in the NOD2/CARD15 gene among 483 Japanese patients with Crohn’s disease. J. Hum. Genet. 2002, 47, 469–472. [Google Scholar] [CrossRef]
- Hugot, J.P.; Chamaillard, M.; Zouali, H.; Lesage, S.; Cezard, J.P.; Belaiche, J.; Almer, S.; Tysk, C.; O’Morain, C.A.; Gassull, M.; et al. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature 2001, 411, 599–603. [Google Scholar] [CrossRef]
- Rogler, G.; Hausmann, M. The long and winding road: From genetic risk factors to the understanding of disease-pathogenesis in Crohn’s disease. Genes. Immun. 2019, 20, 607–608. [Google Scholar] [CrossRef] [Green Version]
- Gajendran, M.; Loganathan, P.; Catinella, A.P.; Hashash, J.G. A comprehensive review and update on Crohn’s disease. Dis. Mon. 2018, 64, 20–57. [Google Scholar] [CrossRef] [PubMed]
- Plantinga, T.S.; Crisan, T.O.; Oosting, M.; van de Veerdonk, F.L.; de Jong, D.J.; Philpott, D.J.; van der Meer, J.W.; Girardin, S.E.; Joosten, L.A.; Netea, M.G. Crohn’s disease-associated ATG16L1 polymorphism modulates pro-inflammatory cytokine responses selectively upon activation of NOD2. Gut 2011, 60, 1229–1235. [Google Scholar] [CrossRef] [PubMed]
- Coccia, M.; Harrison, O.J.; Schiering, C.; Asquith, M.J.; Becher, B.; Powrie, F.; Maloy, K.J. IL-1beta mediates chronic intestinal inflammation by promoting the accumulation of IL-17A secreting innate lymphoid cells and CD4(+) Th17 cells. J. Exp. Med. 2012, 209, 1595–1609. [Google Scholar] [CrossRef]
- Preza, G.C.; Yang, O.O.; Elliott, J.; Anton, P.A.; Ochoa, M.T. T lymphocyte density and distribution in human colorectal mucosa, and inefficiency of current cell isolation protocols. PLoS ONE 2015, 10, e0122723. [Google Scholar] [CrossRef] [Green Version]
- Tindemans, I.; Joosse, M.E.; Samsom, J.N. Dissecting the heterogeneity in T-cell mediated inflammation in IBD. Cells 2020, 9, 110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nemeth, Z.H.; Bogdanovski, D.A.; Barratt-Stopper, P.; Paglinco, S.R.; Antonioli, L.; Rolandelli, R.H. Crohn’s disease and ulcerative colitis show unique cytokine profiles. Cureus 2017, 9, e1177. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Dasgupta, S.; Fu, Y.; Bailey, B.; Roy, C.; Lightcap, E.; Faustin, B. Subsets of mononuclear phagocytes are enriched in the inflamed colons of patients with IBD. BMC Immunol. 2019, 20, 42. [Google Scholar] [CrossRef] [Green Version]
- Rovedatti, L.; Kudo, T.; Biancheri, P.; Sarra, M.; Knowles, C.H.; Rampton, D.S.; Corazza, G.R.; Monteleone, G.; di Sabatino, A.; Macdonald, T.T. Differential regulation of interleukin 17 and interferon gamma production in inflammatory bowel disease. Gut 2009, 58, 1629–1636. [Google Scholar] [CrossRef]
- Breese, E.; Braegger, C.P.; Corrigan, C.J.; Walker-Smith, J.A.; MacDonald, T.T. Interleukin-2- and interferon-gamma-secreting T cells in normal and diseased human intestinal mucosa. Immunology 1993, 78, 127–131. [Google Scholar]
- Fuss, I.J.; Neurath, M.; Boirivant, M.; Klein, J.S.; de la Motte, C.; Strong, S.A.; Fiocchi, C.; Strober, W. Disparate CD4+ lamina propria (LP) lymphokine secretion profiles in inflammatory bowel disease. Crohn’s disease LP cells manifest increased secretion of IFN-gamma, whereas ulcerative colitis LP cells manifest increased secretion of IL-5. J. Immunol. 1996, 157, 1261–1270. [Google Scholar]
- Verdier, J.; Begue, B.; Cerf-Bensussan, N.; Ruemmele, F.M. Compartmentalized expression of Th1 and Th17 cytokines in pediatric inflammatory bowel diseases. Inflamm. Bowel Dis. 2012, 18, 1260–1266. [Google Scholar] [CrossRef]
- Park, J.H.; Jeong, D.Y.; Peyrin-Biroulet, L.; Eisenhut, M.; Shin, J.I. Insight into the role of TSLP in inflammatory bowel diseases. Autoimmun. Rev. 2017, 16, 55–63. [Google Scholar] [CrossRef] [PubMed]
- Giuffrida, P.; Caprioli, F.; Facciotti, F.; di Sabatino, A. The role of interleukin-13 in chronic inflammatory intestinal disorders. Autoimmun. Rev. 2019, 18, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Maynard, C.L.; Weaver, C.T. Intestinal effector T cells in health and disease. Immunity 2009, 31, 389–400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hansen, J.J. Immune responses to intestinal microbes in inflammatory bowel diseases. Curr. Allergy Asthma Rep. 2015, 15, 61. [Google Scholar] [CrossRef] [PubMed]
- Maloy, K.J.; Powrie, F. Intestinal homeostasis and its breakdown in inflammatory bowel disease. Nature 2011, 474, 298–306. [Google Scholar] [CrossRef]
- Duchmann, R.; Kaiser, I.; Hermann, E.; Mayet, W.; Ewe, K.; Meyer zum Buschenfelde, K.H. Tolerance exists towards resident intestinal flora but is broken in active inflammatory bowel disease (IBD). Clin. Exp. Immunol. 1995, 102, 448–455. [Google Scholar] [CrossRef]
- Liu, S.; Zhao, W.; Lan, P.; Mou, X. The microbiome in inflammatory bowel diseases: From pathogenesis to therapy. Protein Cell 2021, 12, 331–345. [Google Scholar] [CrossRef]
- Sartor, R.B. Mechanisms of disease: Pathogenesis of Crohn’s disease and ulcerative colitis. Nat. Clin. Pract. Gastroenterol. Hepatol. 2006, 3, 390–407. [Google Scholar] [CrossRef]
- Giuffrida, P.; Corazza, G.R.; di Sabatino, A. Old and new lymphocyte players in inflammatory bowel disease. Dig. Dis. Sci. 2018, 63, 277–288. [Google Scholar] [CrossRef] [PubMed]
- Sepahi, A.; Liu, Q.; Friesen, L.; Kim, C.H. Dietary fiber metabolites regulate innate lymphoid cell responses. Mucosal Immunol. 2021, 14, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Luo, W.; Tian, L.; Tan, B.; Shen, Z.; Xiao, M.; Wu, S.; Meng, X.; Wu, X.; Wang, X. Update: Innate lymphoid cells in inflammatory bowel disease. Dig. Dis. Sci. 2021. [Google Scholar] [CrossRef]
- Zhou, W.; Sonnenberg, G.F. Activation and suppression of group 3 innate lymphoid cells in the gut. Trends Immunol. 2020, 41, 721–733. [Google Scholar] [CrossRef]
- Wu, Y.; Shen, J. Innate lymphoid cells in Crohn’s disease. Front. Immunol. 2020, 11, 554880. [Google Scholar] [CrossRef] [PubMed]
- Diefenbach, A.; Gnafakis, S.; Shomrat, O. Innate lymphoid cell-epithelial cell modules sustain intestinal homeostasis. Immunity 2020, 52, 452–463. [Google Scholar] [CrossRef] [PubMed]
- Cupedo, T.; Crellin, N.K.; Papazian, N.; Rombouts, E.J.; Weijer, K.; Grogan, J.L.; Fibbe, W.E.; Cornelissen, J.J.; Spits, H. Human fetal lymphoid tissue-inducer cells are interleukin 17-producing precursors to RORC+ CD127+ natural killer-like cells. Nat. Immunol. 2009, 10, 66–74. [Google Scholar] [CrossRef]
- Cella, M.; Fuchs, A.; Vermi, W.; Facchetti, F.; Otero, K.; Lennerz, J.K.; Doherty, J.M.; Mills, J.C.; Colonna, M. A human natural killer cell subset provides an innate source of IL-22 for mucosal immunity. Nature 2009, 457, 722–725. [Google Scholar] [CrossRef]
- Spits, H.; Artis, D.; Colonna, M.; Diefenbach, A.; di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.; Mebius, R.E.; et al. Innate lymphoid cells—A proposal for uniform nomenclature. Nat. Rev. Immunol. 2013, 13, 145–149. [Google Scholar] [CrossRef]
- Ignacio, A.; Breda, C.N.S.; Camara, N.O.S. Innate lymphoid cells in tissue homeostasis and diseases. World J. Hepatol. 2017, 9, 979–989. [Google Scholar] [CrossRef] [PubMed]
- Eberl, G.; Colonna, M.; di Santo, J.P.; McKenzie, A.N. Innate lymphoid cells. Innate lymphoid cells: A new paradigm in immunology. Science 2015, 348, aaa6566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Artis, D.; Spits, H. The biology of innate lymphoid cells. Nature 2015, 517, 293–301. [Google Scholar] [CrossRef]
- Huang, Y.; Mao, K.; Germain, R.N. Thinking differently about ILCs-Not just tissue resident and not just the same as CD4(+) T-cell effectors. Immunol. Rev. 2018, 286, 160–171. [Google Scholar] [CrossRef] [PubMed]
- Trabanelli, S.; Gomez-Cadena, A.; Salome, B.; Michaud, K.; Mavilio, D.; Landis, B.N.; Jandus, P.; Jandus, C. Human innate lymphoid cells (ILCs): Toward a uniform immune-phenotyping. Cytom. Part B Clin. Cytom. 2018, 94, 392–399. [Google Scholar] [CrossRef]
- Panda, S.K.; Colonna, M. Innate lymphoid cells in mucosal immunity. Front. Immunol. 2019, 10, 861. [Google Scholar] [CrossRef] [Green Version]
- Gasteiger, G.; Fan, X.; Dikiy, S.; Lee, S.Y.; Rudensky, A.Y. Tissue residency of innate lymphoid cells in lymphoid and nonlymphoid organs. Science 2015, 350, 981–985. [Google Scholar] [CrossRef] [Green Version]
- Savage, A.K.; Liang, H.E.; Locksley, R.M. The development of steady-state activation hubs between adult LTi ILC3s and primed macrophages in small intestine. J. Immunol. 2017, 199, 1912–1922. [Google Scholar] [CrossRef]
- Sanos, S.L.; Bui, V.L.; Mortha, A.; Oberle, K.; Heners, C.; Johner, C.; Diefenbach, A. RORgammat and commensal microflora are required for the differentiation of mucosal interleukin 22-producing NKp46+ cells. Nat. Immunol. 2009, 10, 83–91. [Google Scholar] [CrossRef] [Green Version]
- Zook, E.C.; Kee, B.L. Development of innate lymphoid cells. Nat. Immunol. 2016, 17, 775–782. [Google Scholar] [CrossRef] [PubMed]
- Lim, A.I.; Verrier, T.; Vosshenrich, C.A.; di Santo, J.P. Developmental options and functional plasticity of innate lymphoid cells. Curr. Opin. Immunol. 2017, 44, 61–68. [Google Scholar] [CrossRef]
- Mjosberg, J.; Bernink, J.; Peters, C.; Spits, H. Transcriptional control of innate lymphoid cells. Eur. J. Immunol. 2012, 42, 1916–1923. [Google Scholar] [CrossRef] [PubMed]
- Poggi, A.; Benelli, R.; Vene, R.; Costa, D.; Ferrari, N.; Tosetti, F.; Zocchi, M.R. Human gut-associated natural killer cells in health and disease. Front. Immunol. 2019, 10, 961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Simoni, Y.; Newell, E.W. Dissecting human ILC heterogeneity: More than just three subsets. Immunology 2018, 153, 297–303. [Google Scholar] [CrossRef] [Green Version]
- Valle-Noguera, A.; Gomez-Sanchez, M.J.; Girard-Madoux, M.J.H.; Cruz-Adalia, A. Optimized protocol for characterization of mouse gut innate lymphoid cells. Front. Immunol. 2020, 11, 563414. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Marotel, M.; Fauteux-Daniel, S.; Mathieu, A.L.; Viel, S.; Marcais, A.; Walzer, T. T-bet and Eomes govern differentiation and function of mouse and human NK cells and ILC1. Eur. J. Immunol. 2018, 48, 738–750. [Google Scholar] [CrossRef] [Green Version]
- Serafini, N.; Vosshenrich, C.A.; di Santo, J.P. Transcriptional regulation of innate lymphoid cell fate. Nat. Rev. Immunol. 2015, 15, 415–428. [Google Scholar] [CrossRef] [Green Version]
- Vivier, E.; Artis, D.; Colonna, M.; Diefenbach, A.; di Santo, J.P.; Eberl, G.; Koyasu, S.; Locksley, R.M.; McKenzie, A.N.J.; Mebius, R.E.; et al. Innate lymphoid cells: 10 years on. Cell 2018, 174, 1054–1066. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.H.; Hashimoto-Hill, S.; Kim, M. Migration and tissue tropism of innate lymphoid cells. Trends. Immunol. 2016, 37, 68–79. [Google Scholar] [CrossRef] [Green Version]
- Meininger, I.; Carrasco, A.; Rao, A.; Soini, T.; Kokkinou, E.; Mjosberg, J. Tissue-specific features of innate lymphoid cells. Trends Immunol. 2020, 41, 902–917. [Google Scholar] [CrossRef]
- Jiao, Y.; Huntington, N.D.; Belz, G.T.; Seillet, C. Type 1 innate lymphoid cell biology: Lessons learnt from natural killer cells. Front. Immunol. 2016, 7, 426. [Google Scholar] [CrossRef]
- Bonne-Annee, S.; Bush, M.C.; Nutman, T.B. Differential modulation of human innate lymphoid cell (ILC) subsets by IL-10 and TGF-beta. Sci. Rep. 2019, 9, 14305. [Google Scholar] [CrossRef]
- Hildreth, A.D.; O’Sullivan, T.E. Tissue-resident innate and innate-like lymphocyte responses to viral infection. Viruses 2019, 11, 272. [Google Scholar] [CrossRef] [Green Version]
- Adams, N.M.; Sun, J.C. Spatial and temporal coordination of antiviral responses by group 1 ILCs. Immunol. Rev. 2018, 286, 23–36. [Google Scholar] [CrossRef]
- Apraiz, A.; Benedicto, A.; Marquez, J.; Aguera-Lorente, A.; Asumendi, A.; Olaso, E.; Arteta, B. Innate lymphoid cells in the malignant melanoma microenvironment. Cancers 2020, 12, 3177. [Google Scholar] [CrossRef]
- Molgora, M.; Cortez, V.S.; Colonna, M. Killing the invaders: NK cell impact in tumors and anti-tumor therapy. Cancers 2021, 13, 595. [Google Scholar] [CrossRef] [PubMed]
- Sivori, S.; Pende, D.; Quatrini, L.; Pietra, G.; della Chiesa, M.; Vacca, P.; Tumino, N.; Moretta, F.; Mingari, M.C.; Locatelli, F.; et al. NK cells and ILCs in tumor immunotherapy. Mol. Aspects Med. 2020, 100870. [Google Scholar] [CrossRef] [PubMed]
- Zitti, B.; Bryceson, Y.T. Natural killer cells in inflammation and autoimmunity. Cytokine Growth Factor Rev. 2018, 42, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Marcais, A.; Viel, S.; Grau, M.; Henry, T.; Marvel, J.; Walzer, T. Regulation of mouse NK cell development and function by cytokines. Front. Immunol. 2013, 4, 450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kucuksezer, U.C.; Aktas Cetin, E.; Esen, F.; Tahrali, I.; Akdeniz, N.; Gelmez, M.Y.; Deniz, G. The role of natural killer cells in autoimmune diseases. Front. Immunol. 2021, 12, 622306. [Google Scholar] [CrossRef] [PubMed]
- Elemam, N.M.; Hannawi, S.; Maghazachi, A.A. Innate Lymphoid Cells (ILCs) as mediators of inflammation, release of cytokines and lytic molecules. Toxins 2017, 9, 398. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peters, C.P.; Mjosberg, J.M.; Bernink, J.H.; Spits, H. Innate lymphoid cells in inflammatory bowel diseases. Immunol. Lett. 2016, 172, 124–131. [Google Scholar] [CrossRef] [PubMed]
- Bernink, J.H.; Peters, C.P.; Munneke, M.; te Velde, A.A.; Meijer, S.L.; Weijer, K.; Hreggvidsdottir, H.S.; Heinsbroek, S.E.; Legrand, N.; Buskens, C.J.; et al. Human type 1 innate lymphoid cells accumulate in inflamed mucosal tissues. Nat. Immunol. 2013, 14, 221–229. [Google Scholar] [CrossRef]
- Klose, C.S.N.; Flach, M.; Mohle, L.; Rogell, L.; Hoyler, T.; Ebert, K.; Fabiunke, C.; Pfeifer, D.; Sexl, V.; Fonseca-Pereira, D.; et al. Differentiation of type 1 ILCs from a common progenitor to all helper-like innate lymphoid cell lineages. Cell 2014, 157, 340–356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McKenzie, A.N.J.; Spits, H.; Eberl, G. Innate lymphoid cells in inflammation and immunity. Immunity 2014, 41, 366–374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reynders, A.; Yessaad, N.; Vu Manh, T.P.; Dalod, M.; Fenis, A.; Aubry, C.; Nikitas, G.; Escaliere, B.; Renauld, J.C.; Dussurget, O.; et al. Identity, regulation and in vivo function of gut NKp46+RORgammat+ and NKp46+RORgammat− lymphoid cells. EMBO J. 2011, 30, 2934–2947. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weizman, O.E.; Adams, N.M.; Schuster, I.S.; Krishna, C.; Pritykin, Y.; Lau, C.; Degli-Esposti, M.A.; Leslie, C.S.; Sun, J.C.; O’Sullivan, T.E. ILC1 confer early host protection at initial sites of viral infection. Cell 2017, 171, 795–808.e12. [Google Scholar] [CrossRef] [PubMed]
- Helfrich, S.; Mindt, B.C.; Fritz, J.H.; Duerr, C.U. Group 2 innate lymphoid cells in respiratory allergic inflammation. Front. Immunol. 2019, 10, 930. [Google Scholar] [CrossRef]
- Herbert, D.R.; Douglas, B.; Zullo, K. Group 2 innate lymphoid cells (ILC2): Type 2 immunity and Helminth immunity. Int. J. Mol. Sci. 2019, 20, 2276. [Google Scholar] [CrossRef] [Green Version]
- Rafei-Shamsabadi, D.A.; Klose, C.S.N.; Halim, T.Y.F.; Tanriver, Y.; Jakob, T. Context dependent role of type 2 innate lymphoid cells in allergic skin inflammation. Front. Immunol. 2019, 10, 2591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hwang, Y.Y.; McKenzie, A.N. Innate lymphoid cells in immunity and disease. Adv. Exp. Med. Biol. 2013, 785, 9–26. [Google Scholar]
- Placek, K.; Schultze, J.L.; Netea, M.G. Immune memory characteristics of innate lymphoid cells. Curr. Opin. Infect. Dis. 2019, 32, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Gonzalez, I.; Matha, L.; Steer, C.A.; Ghaedi, M.; Poon, G.F.; Takei, F. Allergen-experienced group 2 innate lymphoid cells acquire memory-like properties and enhance allergic lung inflammation. Immunity 2016, 45, 198–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eljaszewicz, A.; Ruchti, F.; Radzikowska, U.; Globinska, A.; Boonpiyathad, T.; Gschwend, A.; Morita, H.; Helbling, A.; Arasi, S.; Kahlert, H.; et al. Trained immunity and tolerance in innate lymphoid cells, monocytes, and dendritic cells during allergen-specific immunotherapy. J. Allergy Clin. Immunol. 2021, 147, 1865–1877. [Google Scholar] [CrossRef] [PubMed]
- Kim, B.S.; Wojno, E.D.; Artis, D. Innate lymphoid cells and allergic inflammation. Curr. Opin. Immunol. 2013, 25, 738–744. [Google Scholar] [CrossRef] [Green Version]
- Jiang, M.; Tao, S.; Zhang, S.; Wang, J.; Zhang, F.; Li, F.; Ding, J. Type 2 innate lymphoid cells participate in IL-33-stimulated Th2-associated immune response in chronic obstructive pulmonary disease. Exp. Ther. Med. 2019, 18, 3109–3116. [Google Scholar] [CrossRef] [Green Version]
- Moro, K.; Yamada, T.; Tanabe, M.; Takeuchi, T.; Ikawa, T.; Kawamoto, H.; Furusawa, J.; Ohtani, M.; Fujii, H.; Koyasu, S. Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature 2010, 463, 540–544. [Google Scholar] [CrossRef]
- Neill, D.R.; Wong, S.H.; Bellosi, A.; Flynn, R.J.; Daly, M.; Langford, T.K.; Bucks, C.; Kane, C.M.; Fallon, P.G.; Pannell, R.; et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature 2010, 464, 1367–1370. [Google Scholar] [CrossRef] [Green Version]
- Price, A.E.; Liang, H.E.; Sullivan, B.M.; Reinhardt, R.L.; Eisley, C.J.; Erle, D.J.; Locksley, R.M. Systemically dispersed innate IL-13-expressing cells in type 2 immunity. Proc. Natl. Acad. Sci. USA 2010, 107, 11489–11494. [Google Scholar] [CrossRef] [Green Version]
- Klein Wolterink, R.G.; Serafini, N.; van Nimwegen, M.; Vosshenrich, C.A.; de Bruijn, M.J.; Fonseca Pereira, D.; Veiga Fernandes, H.; Hendriks, R.W.; di Santo, J.P. Essential, dose-dependent role for the transcription factor Gata3 in the development of IL-5+ and IL-13+ type 2 innate lymphoid cells. Proc. Natl. Acad. Sci. USA 2013, 110, 10240–10245. [Google Scholar] [CrossRef] [Green Version]
- Hoyler, T.; Klose, C.S.; Souabni, A.; Turqueti-Neves, A.; Pfeifer, D.; Rawlins, E.L.; Voehringer, D.; Busslinger, M.; Diefenbach, A. The transcription factor GATA-3 controls cell fate and maintenance of type 2 innate lymphoid cells. Immunity 2012, 37, 634–648. [Google Scholar] [CrossRef] [Green Version]
- Spooner, C.J.; Lesch, J.; Yan, D.; Khan, A.A.; Abbas, A.; Ramirez-Carrozzi, V.; Zhou, M.; Soriano, R.; Eastham-Anderson, J.; Diehl, L.; et al. Specification of type 2 innate lymphocytes by the transcriptional determinant Gfi1. Nat. Immunol. 2013, 14, 1229–1236. [Google Scholar] [CrossRef]
- Yu, Y.; Wang, C.; Clare, S.; Wang, J.; Lee, S.C.; Brandt, C.; Burke, S.; Lu, L.; He, D.; Jenkins, N.A.; et al. The transcription factor Bcl11b is specifically expressed in group 2 innate lymphoid cells and is essential for their development. J. Exp. Med. 2015, 212, 865–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, J.; Heller, J.J.; Guo, X.; Chen, Z.M.; Fish, K.; Fu, Y.X.; Zhou, L. The aryl hydrocarbon receptor regulates gut immunity through modulation of innate lymphoid cells. Immunity 2012, 36, 92–104. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Britanova, L.; Diefenbach, A. Interplay of innate lymphoid cells and the microbiota. Immunol. Rev. 2017, 279, 36–51. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Koroleva, E.P.; Kruglov, A.A.; Kuprash, D.V.; Nedospasov, S.A.; Fu, Y.X.; Tumanov, A.V. Lymphotoxin beta receptor signaling in intestinal epithelial cells orchestrates innate immune responses against mucosal bacterial infection. Immunity 2010, 32, 403–413. [Google Scholar] [CrossRef] [Green Version]
- Tumanov, A.V.; Koroleva, E.P.; Guo, X.; Wang, Y.; Kruglov, A.; Nedospasov, S.; Fu, Y.X. Lymphotoxin controls the IL-22 protection pathway in gut innate lymphoid cells during mucosal pathogen challenge. Cell Host Microbe 2011, 10, 44–53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sonnenberg, G.F.; Monticelli, L.A.; Alenghat, T.; Fung, T.C.; Hutnick, N.A.; Kunisawa, J.; Shibata, N.; Grunberg, S.; Sinha, R.; Zahm, A.M.; et al. Innate lymphoid cells promote anatomical containment of lymphoid-resident commensal bacteria. Science 2012, 336, 1321–1325. [Google Scholar] [CrossRef] [Green Version]
- Luci, C.; Reynders, A.; Ivanov, I.I.; Cognet, C.; Chiche, L.; Chasson, L.; Hardwigsen, J.; Anguiano, E.; Banchereau, J.; Chaussabel, D.; et al. Influence of the transcription factor RORgammat on the development of NKp46+ cell populations in gut and skin. Nat. Immunol. 2009, 10, 75–82. [Google Scholar] [CrossRef]
- Eberl, G.; Marmon, S.; Sunshine, M.J.; Rennert, P.D.; Choi, Y.; Littman, D.R. An essential function for the nuclear receptor RORgamma(t) in the generation of fetal lymphoid tissue inducer cells. Nat. Immunol. 2004, 5, 64–73. [Google Scholar] [CrossRef]
- Mebius, R.E.; Rennert, P.; Weissman, I.L. Developing lymph nodes collect CD4+CD3− LTbeta+ cells that can differentiate to APC, NK cells, and follicular cells but not T or B cells. Immunity 1997, 7, 493–504. [Google Scholar] [CrossRef] [Green Version]
- Klose, C.S.; Kiss, E.A.; Schwierzeck, V.; Ebert, K.; Hoyler, T.; d’Hargues, Y.; Goppert, N.; Croxford, A.L.; Waisman, A.; Tanriver, Y.; et al. A T-bet gradient controls the fate and function of CCR6-RORgammat+ innate lymphoid cells. Nature 2013, 494, 261–265. [Google Scholar] [CrossRef]
- Reboldi, A.; Arnon, T.I.; Rodda, L.B.; Atakilit, A.; Sheppard, D.; Cyster, J.G. IgA production requires B cell interaction with subepithelial dendritic cells in Peyer’s patches. Science 2016, 352, aaf4822. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kruglov, A.A.; Grivennikov, S.I.; Kuprash, D.V.; Winsauer, C.; Prepens, S.; Seleznik, G.M.; Eberl, G.; Littman, D.R.; Heikenwalder, M.; Tumanov, A.V.; et al. Nonredundant function of soluble LTalpha3 produced by innate lymphoid cells in intestinal homeostasis. Science 2013, 342, 1243–1246. [Google Scholar] [CrossRef] [Green Version]
- Takatori, H.; Kanno, Y.; Watford, W.T.; Tato, C.M.; Weiss, G.; Ivanov, I.I.; Littman, D.R.; O’Shea, J.J. Lymphoid tissue inducer-like cells are an innate source of IL-17 and IL-22. J. Exp. Med. 2009, 206, 35–41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, C.; Lee, J.S.; Gilfillan, S.; Robinette, M.L.; Newberry, R.D.; Stappenbeck, T.S.; Mack, M.; Cella, M.; Colonna, M. Unique and redundant functions of NKp46+ ILC3s in models of intestinal inflammation. J. Exp. Med. 2015, 212, 1869–1882. [Google Scholar] [CrossRef] [PubMed]
- Zeng, B.; Shi, S.; Ashworth, G.; Dong, C.; Liu, J.; Xing, F. ILC3 function as a double-edged sword in inflammatory bowel diseases. Cell Death Dis. 2019, 10, 315. [Google Scholar] [CrossRef] [Green Version]
- Powell, N.; Lo, J.W.; Biancheri, P.; Vossenkamper, A.; Pantazi, E.; Walker, A.W.; Stolarczyk, E.; Ammoscato, F.; Goldberg, R.; Scott, P.; et al. Interleukin 6 increases production of cytokines by colonic innate lymphoid cells in mice and patients with chronic intestinal inflammation. Gastroenterology 2015, 149, 456–467.e15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearson, C.; Thornton, E.E.; McKenzie, B.; Schaupp, A.L.; Huskens, N.; Griseri, T.; West, N.; Tung, S.; Seddon, B.P.; Uhlig, H.H.; et al. ILC3 GM-CSF production and mobilisation orchestrate acute intestinal inflammation. eLife 2016, 5, e10066. [Google Scholar] [CrossRef]
- Kramer, B.; Goeser, F.; Lutz, P.; Glassner, A.; Boesecke, C.; Schwarze-Zander, C.; Kaczmarek, D.; Nischalke, H.D.; Branchi, V.; Manekeller, S.; et al. Compartment-specific distribution of human intestinal innate lymphoid cells is altered in HIV patients under effective therapy. PLoS Pathog. 2017, 13, e1006373. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, A.; Vermi, W.; Lee, J.S.; Lonardi, S.; Gilfillan, S.; Newberry, R.D.; Cella, M.; Colonna, M. Intraepithelial type 1 innate lymphoid cells are a unique subset of IL-12- and IL-15-responsive IFN-gamma-producing cells. Immunity 2013, 38, 769–781. [Google Scholar] [CrossRef] [Green Version]
- Ochel, A.; Tiegs, G.; Neumann, K. Type 2 innate lymphoid cells in liver and gut: From current knowledge to future perspectives. Int. J. Mol. Sci. 2019, 20, 1896. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mackley, E.C.; Houston, S.; Marriott, C.L.; Halford, E.E.; Lucas, B.; Cerovic, V.; Filbey, K.J.; Maizels, R.M.; Hepworth, M.R.; Sonnenberg, G.F.; et al. CCR7-dependent trafficking of RORgamma(+) ILCs creates a unique microenvironment within mucosal draining lymph nodes. Nat. Commun. 2015, 6, 5862. [Google Scholar] [CrossRef] [PubMed]
- Belkaid, Y.; Hand, T.W. Role of the microbiota in immunity and inflammation. Cell 2014, 157, 121–141. [Google Scholar] [CrossRef] [Green Version]
- Constantinides, M.G. Interactions between the microbiota and innate and innate-like lymphocytes. J. Leukoc. Biol. 2018, 103, 409–419. [Google Scholar] [CrossRef] [PubMed]
- Ganal-Vonarburg, S.C.; Duerr, C.U. The interaction of intestinal microbiota and innate lymphoid cells in health and disease throughout life. Immunology 2020, 159, 39–51. [Google Scholar] [CrossRef]
- Blander, J.M.; Longman, R.S.; Iliev, I.D.; Sonnenberg, G.F.; Artis, D. Regulation of inflammation by microbiota interactions with the host. Nat. Immunol. 2017, 18, 851–860. [Google Scholar] [CrossRef]
- Rooks, M.G.; Garrett, W.S. Gut microbiota, metabolites and host immunity. Nat. Rev. Immunol. 2016, 16, 341–352. [Google Scholar] [CrossRef]
- Chun, E.; Lavoie, S.; Fonseca-Pereira, D.; Bae, S.; Michaud, M.; Hoveyda, H.R.; Fraser, G.L.; Gallini Comeau, C.A.; Glickman, J.N.; Fuller, M.H.; et al. Metabolite-sensing receptor Ffar2 regulates colonic group 3 innate lymphoid cells and gut immunity. Immunity 2019, 51, 871–884.e6. [Google Scholar] [CrossRef]
- Sawa, S.; Lochner, M.; Satoh-Takayama, N.; Dulauroy, S.; Berard, M.; Kleinschek, M.; Cua, D.; di Santo, J.P.; Eberl, G. RORgammat+ innate lymphoid cells regulate intestinal homeostasis by integrating negative signals from the symbiotic microbiota. Nat. Immunol. 2011, 12, 320–326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, S.H.; Cho, B.H.; Kiyono, H.; Jang, Y.S. Microbiota-derived butyrate suppresses group 3 innate lymphoid cells in terminal ileal Peyer’s patches. Sci. Rep. 2017, 7, 3980. [Google Scholar] [CrossRef] [PubMed]
- Thio, C.L.; Chi, P.Y.; Lai, A.C.; Chang, Y.J. Regulation of type 2 innate lymphoid cell-dependent airway hyperreactivity by butyrate. J. Allergy Clin. Immunol. 2018, 142, 1867–1883.e12. [Google Scholar] [CrossRef] [Green Version]
- Toki, S.; Goleniewska, K.; Reiss, S.; Zhou, W.; Newcomb, D.C.; Bloodworth, M.H.; Stier, M.T.; Boyd, K.L.; Polosukhin, V.V.; Subramaniam, S.; et al. The histone deacetylase inhibitor trichostatin A suppresses murine innate allergic inflammation by blocking group 2 innate lymphoid cell (ILC2) activation. Thorax 2016, 71, 633–645. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barrow, A.D.; Martin, C.J.; Colonna, M. The natural cytotoxicity receptors in health and disease. Front. Immunol. 2019, 10, 909. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Glatzer, T.; Killig, M.; Meisig, J.; Ommert, I.; Luetke-Eversloh, M.; Babic, M.; Paclik, D.; Bluthgen, N.; Seidl, R.; Seifarth, C.; et al. RORgammat(+) innate lymphoid cells acquire a proinflammatory program upon engagement of the activating receptor NKp44. Immunity 2013, 38, 1223–1235. [Google Scholar] [CrossRef] [Green Version]
- Castleman, M.J.; Dillon, S.M.; Purba, C.M.; Cogswell, A.C.; Kibbie, J.J.; McCarter, M.D.; Santiago, M.L.; Barker, E.; Wilson, C.C. Commensal and pathogenic bacteria indirectly induce IL-22 but not IFNgamma production from human colonic ILC3s via multiple mechanisms. Front. Immunol. 2019, 10, 649. [Google Scholar] [CrossRef] [Green Version]
- Crellin, N.K.; Trifari, S.; Kaplan, C.D.; Satoh-Takayama, N.; di Santo, J.P.; Spits, H. Regulation of cytokine secretion in human CD127(+) LTi-like innate lymphoid cells by Toll-like receptor 2. Immunity 2010, 33, 752–764. [Google Scholar] [CrossRef] [Green Version]
- Mortha, A.; Chudnovskiy, A.; Hashimoto, D.; Bogunovic, M.; Spencer, S.P.; Belkaid, Y.; Merad, M. Microbiota-dependent crosstalk between macrophages and ILC3 promotes intestinal homeostasis. Science 2014, 343, 1249288. [Google Scholar] [CrossRef] [Green Version]
- Zhou, L.; Chu, C.; Teng, F.; Bessman, N.J.; Goc, J.; Santosa, E.K.; Putzel, G.G.; Kabata, H.; Kelsen, J.R.; Baldassano, R.N.; et al. Innate lymphoid cells support regulatory T cells in the intestine through interleukin-2. Nature 2019, 568, 405–409. [Google Scholar] [CrossRef]
- Bauche, D.; Joyce-Shaikh, B.; Jain, R.; Grein, J.; Ku, K.S.; Blumenschein, W.M.; Ganal-Vonarburg, S.C.; Wilson, D.C.; McClanahan, T.K.; Malefyt, R.W.; et al. LAG3(+) regulatory T cells restrain interleukin-23-producing CX3CR1(+) gut-resident macrophages during group 3 innate lymphoid cell-driven colitis. Immunity 2018, 49, 342–352.e5. [Google Scholar] [CrossRef] [Green Version]
- Bartizal, K.F.; Salkowski, C.; Pleasants, J.R.; Balish, E. The effect of microbial flora, diet, and age on the tumoricidal activity of natural killer cells. J. Leukoc. Biol. 1984, 36, 739–750. [Google Scholar] [CrossRef]
- Abt, M.C.; Osborne, L.C.; Monticelli, L.A.; Doering, T.A.; Alenghat, T.; Sonnenberg, G.F.; Paley, M.A.; Antenus, M.; Williams, K.L.; Erikson, J.; et al. Commensal bacteria calibrate the activation threshold of innate antiviral immunity. Immunity 2012, 37, 158–170. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ganal, S.C.; Sanos, S.L.; Kallfass, C.; Oberle, K.; Johner, C.; Kirschning, C.; Lienenklaus, S.; Weiss, S.; Staeheli, P.; Aichele, P.; et al. Priming of natural killer cells by nonmucosal mononuclear phagocytes requires instructive signals from commensal microbiota. Immunity 2012, 37, 171–186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamimura, Y.; Lanier, L.L. Homeostatic control of memory cell progenitors in the natural killer cell lineage. Cell Rep. 2015, 10, 280–291. [Google Scholar] [CrossRef] [Green Version]
- Bouskra, D.; Brezillon, C.; Berard, M.; Werts, C.; Varona, R.; Boneca, I.G.; Eberl, G. Lymphoid tissue genesis induced by commensals through NOD1 regulates intestinal homeostasis. Nature 2008, 456, 507–510. [Google Scholar] [CrossRef]
- Gury-BenAri, M.; Thaiss, C.A.; Serafini, N.; Winter, D.R.; Giladi, A.; Lara-Astiaso, D.; Levy, M.; Salame, T.M.; Weiner, A.; David, E.; et al. The spectrum and regulatory landscape of intestinal innate lymphoid cells are shaped by the microbiome. Cell 2016, 166, 1231–1246.e13. [Google Scholar] [CrossRef] [PubMed]
- Monticelli, L.A.; Sonnenberg, G.F.; Abt, M.C.; Alenghat, T.; Ziegler, C.G.; Doering, T.A.; Angelosanto, J.M.; Laidlaw, B.J.; Yang, C.Y.; Sathaliyawala, T.; et al. Innate lymphoid cells promote lung-tissue homeostasis after infection with influenza virus. Nat. Immunol. 2011, 12, 1045–1054. [Google Scholar] [CrossRef]
- Kernbauer, E.; Ding, Y.; Cadwell, K. An enteric virus can replace the beneficial function of commensal bacteria. Nature 2014, 516, 94–98. [Google Scholar] [CrossRef] [PubMed]
- Maggi, L.; Montaini, G.; Mazzoni, A.; Rossettini, B.; Capone, M.; Rossi, M.C.; Santarlasci, V.; Liotta, F.; Rossi, O.; Gallo, O.; et al. Human circulating group 2 innate lymphoid cells can express CD154 and promote IgE production. J. Allergy Clin. Immunol. 2017, 139, 964–976.e4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Von Moltke, J.; Ji, M.; Liang, H.E.; Locksley, R.M. Tuft-cell-derived IL-25 regulates an intestinal ILC2-epithelial response circuit. Nature 2016, 529, 221–225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frisbee, A.L.; Saleh, M.M.; Young, M.K.; Leslie, J.L.; Simpson, M.E.; Abhyankar, M.M.; Cowardin, C.A.; Ma, J.Z.; Pramoonjago, P.; Turner, S.D.; et al. IL-33 drives group 2 innate lymphoid cell-mediated protection during Clostridium difficile infection. Nat. Commun. 2019, 10, 2712. [Google Scholar] [CrossRef]
- Garrido-Mesa, N.; Schroeder, J.H.; Stolarczyk, E.; Gallagher, A.L.; Lo, J.W.; Bailey, C.; Campbell, L.; Sexl, V.; MacDonald, T.T.; Howard, J.K.; et al. T-bet controls intestinal mucosa immune responses via repression of type 2 innate lymphoid cell function. Mucosal Immunol. 2019, 12, 51–63. [Google Scholar] [CrossRef] [PubMed]
- Zhu, P.; Zhu, X.; Wu, J.; He, L.; Lu, T.; Wang, Y.; Liu, B.; Ye, B.; Sun, L.; Fan, D.; et al. IL-13 secreted by ILC2s promotes the self-renewal of intestinal stem cells through circular RNA circPan3. Nat. Immunol. 2019, 20, 183–194. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.; Salimi, M.; Panse, I.; Mjosberg, J.M.; McKenzie, A.N.; Spits, H.; Klenerman, P.; Ogg, G. Prostaglandin D2 activates group 2 innate lymphoid cells through chemoattractant receptor-homologous molecule expressed on TH2 cells. J. Allergy Clin. Immunol. 2014, 133, 1184–1194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Monticelli, L.A.; Osborne, L.C.; Noti, M.; Tran, S.V.; Zaiss, D.M.; Artis, D. IL-33 promotes an innate immune pathway of intestinal tissue protection dependent on amphiregulin-EGFR interactions. Proc. Natl. Acad. Sci. USA 2015, 112, 10762–10767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wallrapp, A.; Riesenfeld, S.J.; Burkett, P.R.; Kuchroo, V.K. Type 2 innate lymphoid cells in the induction and resolution of tissue inflammation. Immunol. Rev. 2018, 286, 53–73. [Google Scholar] [CrossRef]
- Mullin, J.M.; Snock, K.V. Effect of tumor necrosis factor on epithelial tight junctions and transepithelial permeability. Cancer Res. 1990, 50, 2172–2176. [Google Scholar] [PubMed]
- Clark, E.; Hoare, C.; Tanianis-Hughes, J.; Carlson, G.L.; Warhurst, G. Interferon gamma induces translocation of commensal Escherichia coli across gut epithelial cells via a lipid raft-mediated process. Gastroenterology 2005, 128, 1258–1267. [Google Scholar] [CrossRef] [PubMed]
- Sonnenberg, G.F.; Fouser, L.A.; Artis, D. Border patrol: Regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22. Nat. Immunol. 2011, 12, 383–390. [Google Scholar] [CrossRef] [PubMed]
- Penny, H.A.; Hodge, S.H.; Hepworth, M.R. Orchestration of intestinal homeostasis and tolerance by group 3 innate lymphoid cells. Semin. Immunopathol. 2018, 40, 357–370. [Google Scholar] [CrossRef] [Green Version]
- Kinnebrew, M.A.; Buffie, C.G.; Diehl, G.E.; Zenewicz, L.A.; Leiner, I.; Hohl, T.M.; Flavell, R.A.; Littman, D.R.; Pamer, E.G. Interleukin 23 production by intestinal CD103(+)CD11b(+) dendritic cells in response to bacterial flagellin enhances mucosal innate immune defense. Immunity 2012, 36, 276–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wolk, K.; Kunz, S.; Witte, E.; Friedrich, M.; Asadullah, K.; Sabat, R. IL-22 increases the innate immunity of tissues. Immunity 2004, 21, 241–254. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, Y.; Valdez, P.A.; Danilenko, D.M.; Hu, Y.; Sa, S.M.; Gong, Q.; Abbas, A.R.; Modrusan, Z.; Ghilardi, N.; de Sauvage, F.J.; et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat. Med. 2008, 14, 282–289. [Google Scholar] [CrossRef]
- Gronke, K.; Hernandez, P.P.; Zimmermann, J.; Klose, C.S.N.; Kofoed-Branzk, M.; Guendel, F.; Witkowski, M.; Tizian, C.; Amann, L.; Schumacher, F.; et al. Interleukin-22 protects intestinal stem cells against genotoxic stress. Nature 2019, 566, 249–253. [Google Scholar] [CrossRef] [PubMed]
- Goto, Y.; Obata, T.; Kunisawa, J.; Sato, S.; Ivanov, I.I.; Lamichhane, A.; Takeyama, N.; Kamioka, M.; Sakamoto, M.; Matsuki, T.; et al. Innate lymphoid cells regulate intestinal epithelial cell glycosylation. Science 2014, 345, 1254009. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goto, Y.; Kiyono, H. Epithelial barrier: An interface for the cross-communication between gut flora and immune system. Immunol. Rev. 2012, 245, 147–163. [Google Scholar] [CrossRef] [PubMed]
- Bry, L.; Falk, P.G.; Midtvedt, T.; Gordon, J.I. A model of host-microbial interactions in an open mammalian ecosystem. Science 1996, 273, 1380–1383. [Google Scholar] [CrossRef] [PubMed]
- Hepworth, M.R.; Fung, T.C.; Masur, S.H.; Kelsen, J.R.; McConnell, F.M.; Dubrot, J.; Withers, D.R.; Hugues, S.; Farrar, M.A.; Reith, W.; et al. Immune tolerance. Group 3 innate lymphoid cells mediate intestinal selection of commensal bacteria-specific CD4(+) T cells. Science 2015, 348, 1031–1035. [Google Scholar] [CrossRef] [Green Version]
- Hepworth, M.R.; Monticelli, L.A.; Fung, T.C.; Ziegler, C.G.; Grunberg, S.; Sinha, R.; Mantegazza, A.R.; Ma, H.L.; Crawford, A.; Angelosanto, J.M.; et al. Innate lymphoid cells regulate CD4+ T-cell responses to intestinal commensal bacteria. Nature 2013, 498, 113–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- DeGruttola, A.K.; Low, D.; Mizoguchi, A.; Mizoguchi, E. Current understanding of dysbiosis in disease in human and animal models. Inflamm. Bowel Dis. 2016, 22, 1137–1150. [Google Scholar] [CrossRef] [Green Version]
- Jangid, A.; Fukuda, S.; Seki, M.; Horiuchi, T.; Suzuki, Y.; Taylor, T.D.; Ohno, H.; Prakash, T. Association of colitis with gut-microbiota dysbiosis in clathrin adapter AP-1B knockout mice. PLoS ONE 2020, 15, e0228358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borowczyk, J.; Shutova, M.; Brembilla, N.C.; Boehncke, W.H. IL-25 (IL-17E) in epithelial immunology and pathophysiology. J. Allergy Clin. Immunol. 2021, 148, 40–52. [Google Scholar] [CrossRef] [PubMed]
- Schulz-Kuhnt, A.; Wirtz, S.; Neurath, M.F.; Atreya, I. Regulation of human innate lymphoid cells in the context of mucosal inflammation. Front. Immunol. 2020, 11, 1062. [Google Scholar] [CrossRef] [PubMed]
- Forkel, M.; van Tol, S.; Hoog, C.; Michaelsson, J.; Almer, S.; Mjosberg, J. Distinct Alterations in the composition of mucosal innate lymphoid cells in newly diagnosed and established Crohn’s disease and ulcerative colitis. J. Crohns Colitis 2019, 13, 67–78. [Google Scholar] [CrossRef] [PubMed]
- Geremia, A.; Arancibia-Carcamo, C.V.; Fleming, M.P.; Rust, N.; Singh, B.; Mortensen, N.J.; Travis, S.P.; Powrie, F. IL-23-responsive innate lymphoid cells are increased in inflammatory bowel disease. J. Exp. Med. 2011, 208, 1127–1133. [Google Scholar] [CrossRef] [Green Version]
- Buonocore, S.; Ahern, P.P.; Uhlig, H.H.; Ivanov, I.I.; Littman, D.R.; Maloy, K.J.; Powrie, F. Innate lymphoid cells drive interleukin-23-dependent innate intestinal pathology. Nature 2010, 464, 1371–1375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Powell, N.; Walker, A.W.; Stolarczyk, E.; Canavan, J.B.; Gokmen, M.R.; Marks, E.; Jackson, I.; Hashim, A.; Curtis, M.A.; Jenner, R.G.; et al. The transcription factor T-bet regulates intestinal inflammation mediated by interleukin-7 receptor+ innate lymphoid cells. Immunity 2012, 37, 674–684. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munoz, M.; Eidenschenk, C.; Ota, N.; Wong, K.; Lohmann, U.; Kuhl, A.A.; Wang, X.; Manzanillo, P.; Li, Y.; Rutz, S.; et al. Interleukin-22 induces interleukin-18 expression from epithelial cells during intestinal infection. Immunity 2015, 42, 321–331. [Google Scholar] [CrossRef] [Green Version]
- Satoh-Takayama, N.; Vosshenrich, C.A.; Lesjean-Pottier, S.; Sawa, S.; Lochner, M.; Rattis, F.; Mention, J.J.; Thiam, K.; Cerf-Bensussan, N.; Mandelboim, O.; et al. Microbial flora drives interleukin 22 production in intestinal NKp46+ cells that provide innate mucosal immune defense. Immunity 2008, 29, 958–970. [Google Scholar] [CrossRef] [PubMed]
- Tsai, P.Y.; Zhang, B.; He, W.Q.; Zha, J.M.; Odenwald, M.A.; Singh, G.; Tamura, A.; Shen, L.; Sailer, A.; Yeruva, S.; et al. IL-22 upregulates epithelial claudin-2 to drive diarrhea and enteric pathogen clearance. Cell Host Microbe 2017, 21, 671–681.e4. [Google Scholar] [CrossRef] [Green Version]
- Li, J.; Shi, W.; Sun, H.; Ji, Y.; Chen, Y.; Guo, X.; Sheng, H.; Shu, J.; Zhou, L.; Cai, T.; et al. Activation of DR3 signaling causes loss of ILC3s and exacerbates intestinal inflammation. Nat. Commun. 2019, 10, 3371. [Google Scholar] [CrossRef] [Green Version]
- Gerosa, F.; Baldani-Guerra, B.; Nisii, C.; Marchesini, V.; Carra, G.; Trinchieri, G. Reciprocal activating interaction between natural killer cells and dendritic cells. J. Exp. Med. 2002, 195, 327–333. [Google Scholar] [CrossRef]
- Mocikat, R.; Braumuller, H.; Gumy, A.; Egeter, O.; Ziegler, H.; Reusch, U.; Bubeck, A.; Louis, J.; Mailhammer, R.; Riethmuller, G.; et al. Natural killer cells activated by MHC class I(low) targets prime dendritic cells to induce protective CD8 T cell responses. Immunity 2003, 19, 561–569. [Google Scholar] [CrossRef] [Green Version]
- Hosomi, S.; Grootjans, J.; Tschurtschenthaler, M.; Krupka, N.; Matute, J.D.; Flak, M.B.; Martinez-Naves, E.; Gomez del Moral, M.; Glickman, J.N.; Ohira, M.; et al. Intestinal epithelial cell endoplasmic reticulum stress promotes MULT1 up-regulation and NKG2D-mediated inflammation. J. Exp. Med. 2017, 214, 2985–2997. [Google Scholar] [CrossRef] [PubMed]
- Ng, S.C.; Plamondon, S.; Al-Hassi, H.O.; English, N.; Gellatly, N.; Kamm, M.A.; Knight, S.C.; Stagg, A.J. A novel population of human CD56+ human leucocyte antigen D-related (HLA-DR+) colonic lamina propria cells is associated with inflammation in ulcerative colitis. Clin. Exp. Immunol. 2009, 158, 205–218. [Google Scholar] [CrossRef] [PubMed]
- Lin, L.; Ma, C.; Wei, B.; Aziz, N.; Rajalingam, R.; Yusung, S.; Erlich, H.A.; Trachtenberg, E.A.; Targan, S.R.; McGovern, D.P.; et al. Human NK cells licensed by killer Ig receptor genes have an altered cytokine program that modifies CD4+ T cell function. J. Immunol. 2014, 193, 940–949. [Google Scholar] [CrossRef] [Green Version]
- Takayama, T.; Kamada, N.; Chinen, H.; Okamoto, S.; Kitazume, M.T.; Chang, J.; Matuzaki, Y.; Suzuki, S.; Sugita, A.; Koganei, K.; et al. Imbalance of NKp44(+)NKp46(-) and NKp44(-)NKp46(+) natural killer cells in the intestinal mucosa of patients with Crohn’s disease. Gastroenterology 2010, 139, 882–892.e3. [Google Scholar] [CrossRef] [PubMed]
- Gwela, A.; Siddhanathi, P.; Chapman, R.W.; Travis, S.; Powrie, F.; Arancibia-Carcamo, C.V.; Geremia, A. Th1 and innate lymphoid cells accumulate in primary sclerosing cholangitis-associated inflammatory bowel disease. J. Crohns Colitis 2017, 11, 1124–1134. [Google Scholar] [CrossRef] [PubMed]
- Kobori, A.; Yagi, Y.; Imaeda, H.; Ban, H.; Bamba, S.; Tsujikawa, T.; Saito, Y.; Fujiyama, Y.; Andoh, A. Interleukin-33 expression is specifically enhanced in inflamed mucosa of ulcerative colitis. J. Gastroenterol. 2010, 45, 999–1007. [Google Scholar] [CrossRef] [PubMed]
- Schiering, C.; Krausgruber, T.; Chomka, A.; Frohlich, A.; Adelmann, K.; Wohlfert, E.A.; Pott, J.; Griseri, T.; Bollrath, J.; Hegazy, A.N.; et al. The alarmin IL-33 promotes regulatory T-cell function in the intestine. Nature 2014, 513, 564–568. [Google Scholar] [CrossRef] [Green Version]
- Camelo, A.; Barlow, J.L.; Drynan, L.F.; Neill, D.R.; Ballantyne, S.J.; Wong, S.H.; Pannell, R.; Gao, W.; Wrigley, K.; Sprenkle, J.; et al. Blocking IL-25 signalling protects against gut inflammation in a type-2 model of colitis by suppressing nuocyte and NKT derived IL-13. J. Gastroenterol. 2012, 47, 1198–1211. [Google Scholar] [CrossRef] [Green Version]
- Jostins, L.; Ripke, S.; Weersma, R.K.; Duerr, R.H.; McGovern, D.P.; Hui, K.Y.; Lee, J.C.; Schumm, L.P.; Sharma, Y.; Anderson, C.A.; et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012, 491, 119–124. [Google Scholar] [CrossRef] [Green Version]
- Mazzurana, L.; Bonfiglio, F.; Forkel, M.; d’Amato, M.; Halfvarson, J.; Mjosberg, J. Crohn’s disease is associated with activation of circulating innate lymphoid cells. Inflamm. Bowel Dis. 2021, 27, 1128–1138. [Google Scholar] [CrossRef]
- De Salvo, C.; Buela, K.A.; Creyns, B.; Corridoni, D.; Rana, N.; Wargo, H.L.; Cominelli, C.L.; Delaney, P.G.; Rodriguez-Palacios, A.; Cominelli, F.; et al. NOD2 drives early IL-33-dependent expansion of group 2 innate lymphoid cells during Crohn’s disease-like ileitis. J. Clin. Investig. 2021, 131. [Google Scholar] [CrossRef]
- Pizarro, T.T.; Pastorelli, L.; Bamias, G.; Garg, R.R.; Reuter, B.K.; Mercado, J.R.; Chieppa, M.; Arseneau, K.O.; Ley, K.; Cominelli, F. SAMP1/YitFc mouse strain: A spontaneous model of Crohn’s disease-like ileitis. Inflamm. Bowel Dis. 2011, 17, 2566–2584. [Google Scholar] [CrossRef] [Green Version]
- Knipfer, L.; Schulz-Kuhnt, A.; Kindermann, M.; Greif, V.; Symowski, C.; Voehringer, D.; Neurath, M.F.; Atreya, I.; Wirtz, S. A CCL1/CCR8-dependent feed-forward mechanism drives ILC2 functions in type 2-mediated inflammation. J. Exp. Med. 2019, 216, 2763–2777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Puttur, F.; Denney, L.; Gregory, L.G.; Vuononvirta, J.; Oliver, R.; Entwistle, L.J.; Walker, S.A.; Headley, M.B.; McGhee, E.J.; Pease, J.E.; et al. Pulmonary environmental cues drive group 2 innate lymphoid cell dynamics in mice and humans. Sci. Immunol. 2019, 4, eaav7638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caballer, M.A.V.; Gonzalez-Granado, J.M.; Zorita, V.; Abu Nabah, Y.N.; Silvestre-Roig, C.; del Monte-Monge, A.; Molina-Sanchez, P.; Ait-Oufella, H.; Andres-Manzano, M.J.; Sanz, M.J.; et al. Disruption of the CCL1-CCR8 axis inhibits vascular Treg recruitment and function and promotes atherosclerosis in mice. J. Mol. Cell. Cardiol. 2019, 132, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Fernández, B.H.; Gomez-Bris, R.; Somovilla-Crespo, B.; Gonzalez-Granado, J.M. Immunobiology of atherosclerosis: A complex net of interactions. Int. J. Mol. Sci. 2019, 20, 5293. [Google Scholar] [CrossRef] [Green Version]
- Panina-Bordignon, P.; Papi, A.; Mariani, M.; di Lucia, P.; Casoni, G.; Bellettato, C.; Buonsanti, C.; Miotto, D.; Mapp, C.; Villa, A.; et al. The C-C chemokine receptors CCR4 and CCR8 identify airway T cells of allergen-challenged atopic asthmatics. J. Clin. Investig. 2001, 107, 1357–1364. [Google Scholar] [CrossRef] [Green Version]
- Mikhak, Z.; Fukui, M.; Farsidjani, A.; Medoff, B.D.; Tager, A.M.; Luster, A.D. Contribution of CCR4 and CCR8 to antigen-specific T(H)2 cell trafficking in allergic pulmonary inflammation. J. Allergy Clin. Immunol. 2009, 123, 67–73.e3. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barsheshet, Y.; Wildbaum, G.; Levy, E.; Vitenshtein, A.; Akinseye, C.; Griggs, J.; Lira, S.A.; Karin, N. CCR8(+)FOXp3(+) Treg cells as master drivers of immune regulation. Proc. Natl. Acad. Sci. USA 2017, 114, 6086–6091. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kang, L.; Schmalzl, A.; Leupold, T.; Gonzalez-Acera, M.; Atreya, R.; Neurath, M.F.; Becker, C.; Wirtz, S. CCR8 Signaling via CCL1 regulates responses of intestinal IFN-gamma producing innate lymphoid celis and protects from experimental colitis. Front. Immunol. 2020, 11, 609400. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Liu, D.; Tan, Y.; Deng, F.; Li, R. M1 Macrophage exosomes MiR-21a-5p aggravates inflammatory bowel disease through decreasing E-cadherin and subsequent ILC2 activation. J. Cell. Mol. Med. 2021, 25, 3041–3050. [Google Scholar] [CrossRef] [PubMed]
- Schnoor, M. E-cadherin is important for the maintenance of intestinal epithelial homeostasis under basal and inflammatory conditions. Dig. Dis. Sci. 2015, 60, 816–818. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiong, X.; Cheng, Z.; Wu, F.; Hu, M.; Liu, Z.; Dong, R.; Chen, G. Berberine in the treatment of ulcerative colitis: A possible pathway through Tuft cells. Biomed. Pharmacother. 2021, 134, 111129. [Google Scholar] [CrossRef]
- Bernink, J.H.; Krabbendam, L.; Germar, K.; de Jong, E.; Gronke, K.; Kofoed-Nielsen, M.; Munneke, J.M.; Hazenberg, M.D.; Villaudy, J.; Buskens, C.J.; et al. Interleukin-12 and -23 control plasticity of CD127(+) group 1 and group 3 innate lymphoid cells in the intestinal lamina propria. Immunity 2015, 43, 146–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vonarbourg, C.; Mortha, A.; Bui, V.L.; Hernandez, P.P.; Kiss, E.A.; Hoyler, T.; Flach, M.; Bengsch, B.; Thimme, R.; Holscher, C.; et al. Regulated expression of nuclear receptor RORgammat confers distinct functional fates to NK cell receptor-expressing RORgammat(+) innate lymphocytes. Immunity 2010, 33, 736–751. [Google Scholar] [CrossRef] [Green Version]
- Tang, Y.; Tan, S.A.; Iqbal, A.; Li, J.; Glover, S.C. STAT3 genotypic variant rs744166 and increased tyrosine phosphorylation of STAT3 in IL-23 responsive innate lymphoid cells during pathogenesis of Crohn’s disease. J. Immunol. Res. 2019, 2019, 9406146. [Google Scholar] [CrossRef] [Green Version]
- Guo, X.; Qiu, J.; Tu, T.; Yang, X.; Deng, L.; Anders, R.A.; Zhou, L.; Fu, Y.X. Induction of innate lymphoid cell-derived interleukin-22 by the transcription factor STAT3 mediates protection against intestinal infection. Immunity 2014, 40, 25–39. [Google Scholar] [CrossRef] [Green Version]
- Rankin, L.C.; Girard-Madoux, M.J.; Seillet, C.; Mielke, L.A.; Kerdiles, Y.; Fenis, A.; Wieduwild, E.; Putoczki, T.; Mondot, S.; Lantz, O.; et al. Complementarity and redundancy of IL-22-producing innate lymphoid cells. Nat. Immunol. 2016, 17, 179–186. [Google Scholar] [CrossRef]
- Teunissen, M.B.M.; Munneke, J.M.; Bernink, J.H.; Spuls, P.I.; Res, P.C.M.; te Velde, A.; Cheuk, S.; Brouwer, M.W.D.; Menting, S.P.; Eidsmo, L.; et al. Composition of innate lymphoid cell subsets in the human skin: Enrichment of NCR(+) ILC3 in lesional skin and blood of psoriasis patients. J. Investig. Dermatol. 2014, 134, 2351–2360. [Google Scholar] [CrossRef] [Green Version]
- Chea, S.; Perchet, T.; Petit, M.; Verrier, T.; Guy-Grand, D.; Banchi, E.G.; Vosshenrich, C.A.; di Santo, J.P.; Cumano, A.; Golub, R. Notch signaling in group 3 innate lymphoid cells modulates their plasticity. Sci. Signal. 2016, 9, ra45. [Google Scholar] [CrossRef] [Green Version]
- Viant, C.; Rankin, L.C.; Girard-Madoux, M.J.; Seillet, C.; Shi, W.; Smyth, M.J.; Bartholin, L.; Walzer, T.; Huntington, N.D.; Vivier, E.; et al. Transforming growth factor-beta and Notch ligands act as opposing environmental cues in regulating the plasticity of type 3 innate lymphoid cells. Sci. Signal. 2016, 9, ra46. [Google Scholar] [CrossRef] [PubMed]
- Ebbo, M.; Crinier, A.; Vely, F.; Vivier, E. Innate lymphoid cells: Major players in inflammatory diseases. Nat. Rev. Immunol. 2017, 17, 665–678. [Google Scholar] [CrossRef] [PubMed]
- Travis, S.P.; Schnell, D.; Krzeski, P.; Abreu, M.T.; Altman, D.G.; Colombel, J.F.; Feagan, B.G.; Hanauer, S.B.; Lichtenstein, G.R.; Marteau, P.R.; et al. Reliability and initial validation of the ulcerative colitis endoscopic index of severity. Gastroenterology 2013, 145, 987–995. [Google Scholar] [CrossRef] [Green Version]
- Lim, A.I.; Menegatti, S.; Bustamante, J.; le Bourhis, L.; Allez, M.; Rogge, L.; Casanova, J.L.; Yssel, H.; di Santo, J.P. IL-12 drives functional plasticity of human group 2 innate lymphoid cells. J. Exp. Med. 2016, 213, 569–583. [Google Scholar] [CrossRef] [Green Version]
- Li, S.; Bostick, J.W.; Ye, J.; Qiu, J.; Zhang, B.; Urban, J.F., Jr.; Avram, D.; Zhou, L. Aryl hydrocarbon receptor signaling cell intrinsically inhibits intestinal group 2 innate lymphoid cell function. Immunity 2018, 49, 915–928.e5. [Google Scholar] [CrossRef] [Green Version]
- Bal, S.M.; Bernink, J.H.; Nagasawa, M.; Groot, J.; Shikhagaie, M.M.; Golebski, K.; van Drunen, C.M.; Lutter, R.; Jonkers, R.E.; Hombrink, P.; et al. IL-1beta, IL-4 and IL-12 control the fate of group 2 innate lymphoid cells in human airway inflammation in the lungs. Nat. Immunol. 2016, 17, 636–645. [Google Scholar] [CrossRef] [PubMed]
- De Salvo, C.; Buela, K.A.; Pizarro, T.T. Cytokine-mediated regulation of innate lymphoid cell plasticity in gut mucosal immunity. Front. Immunol. 2020, 11, 585319. [Google Scholar] [CrossRef] [PubMed]
- Gao, Y.; Souza-Fonseca-Guimaraes, F.; Bald, T.; Ng, S.S.; Young, A.; Ngiow, S.F.; Rautela, J.; Straube, J.; Waddell, N.; Blake, S.J.; et al. Tumor immunoevasion by the conversion of effector NK cells into type 1 innate lymphoid cells. Nat. Immunol. 2017, 18, 1004–1015. [Google Scholar] [CrossRef] [PubMed]
- Cortez, V.S.; Ulland, T.K.; Cervantes-Barragan, L.; Bando, J.K.; Robinette, M.L.; Wang, Q.; White, A.J.; Gilfillan, S.; Cella, M.; Colonna, M. SMAD4 impedes the conversion of NK cells into ILC1-like cells by curtailing non-canonical TGF-beta signaling. Nat. Immunol. 2017, 18, 995–1003. [Google Scholar] [CrossRef] [PubMed] [Green Version]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Saez, A.; Gomez-Bris, R.; Herrero-Fernandez, B.; Mingorance, C.; Rius, C.; Gonzalez-Granado, J.M. Innate Lymphoid Cells in Intestinal Homeostasis and Inflammatory Bowel Disease. Int. J. Mol. Sci. 2021, 22, 7618. https://doi.org/10.3390/ijms22147618
Saez A, Gomez-Bris R, Herrero-Fernandez B, Mingorance C, Rius C, Gonzalez-Granado JM. Innate Lymphoid Cells in Intestinal Homeostasis and Inflammatory Bowel Disease. International Journal of Molecular Sciences. 2021; 22(14):7618. https://doi.org/10.3390/ijms22147618
Chicago/Turabian StyleSaez, Angela, Raquel Gomez-Bris, Beatriz Herrero-Fernandez, Claudia Mingorance, Cristina Rius, and Jose M. Gonzalez-Granado. 2021. "Innate Lymphoid Cells in Intestinal Homeostasis and Inflammatory Bowel Disease" International Journal of Molecular Sciences 22, no. 14: 7618. https://doi.org/10.3390/ijms22147618
APA StyleSaez, A., Gomez-Bris, R., Herrero-Fernandez, B., Mingorance, C., Rius, C., & Gonzalez-Granado, J. M. (2021). Innate Lymphoid Cells in Intestinal Homeostasis and Inflammatory Bowel Disease. International Journal of Molecular Sciences, 22(14), 7618. https://doi.org/10.3390/ijms22147618