1. Introduction
Second-generation sequencing (SGS) platforms, such as Illumina, have significant limitations, although they are widely used in bacterial-genome research [
1]. First, for a single laboratory, an SGS device requires significant capital investments (about USD 980,000), and the operation of the instrument has strict requirements in terms of the laboratory environment and the operators’ skills. Second, the process of SGS sample preparation and library construction is cumbersome. In addition, the sequencing process is time-consuming, and the output lags. Third, SGS platforms are based on PCR amplification for carrying out DNA molecule sequencing. Thus, they are limited by amplification and sequencing bias complications, and the read lengths are restricted to a few hundred bases. Short-read lengths make it difficult to achieve complete de novo assembly for genomes that include longer repetitive elements. The structures of repetitive regions include, for example, resistance gene cassettes, insertion sequences, and transposons.
The first established long-read technology was that of Pacific Biosciences (PacBio), which uses a sequencing-by-synthesis approach [
2]. For the PacBio Sequel machine, the average length of reads is in the range of 10–14 kb. The error rate for raw reads is about 13–15%. The throughput can reach up to about 30 Gb per run. However, the cost of genome sequencing with the PacBio technology is still relatively high, as it involves a high initial cost for the platform (about USD 350,000) and about USD 100 per Gb.
The recent platform of Oxford Nanopore Technologies (ONT) delivers the real-time long-read sequencing of individual molecules [
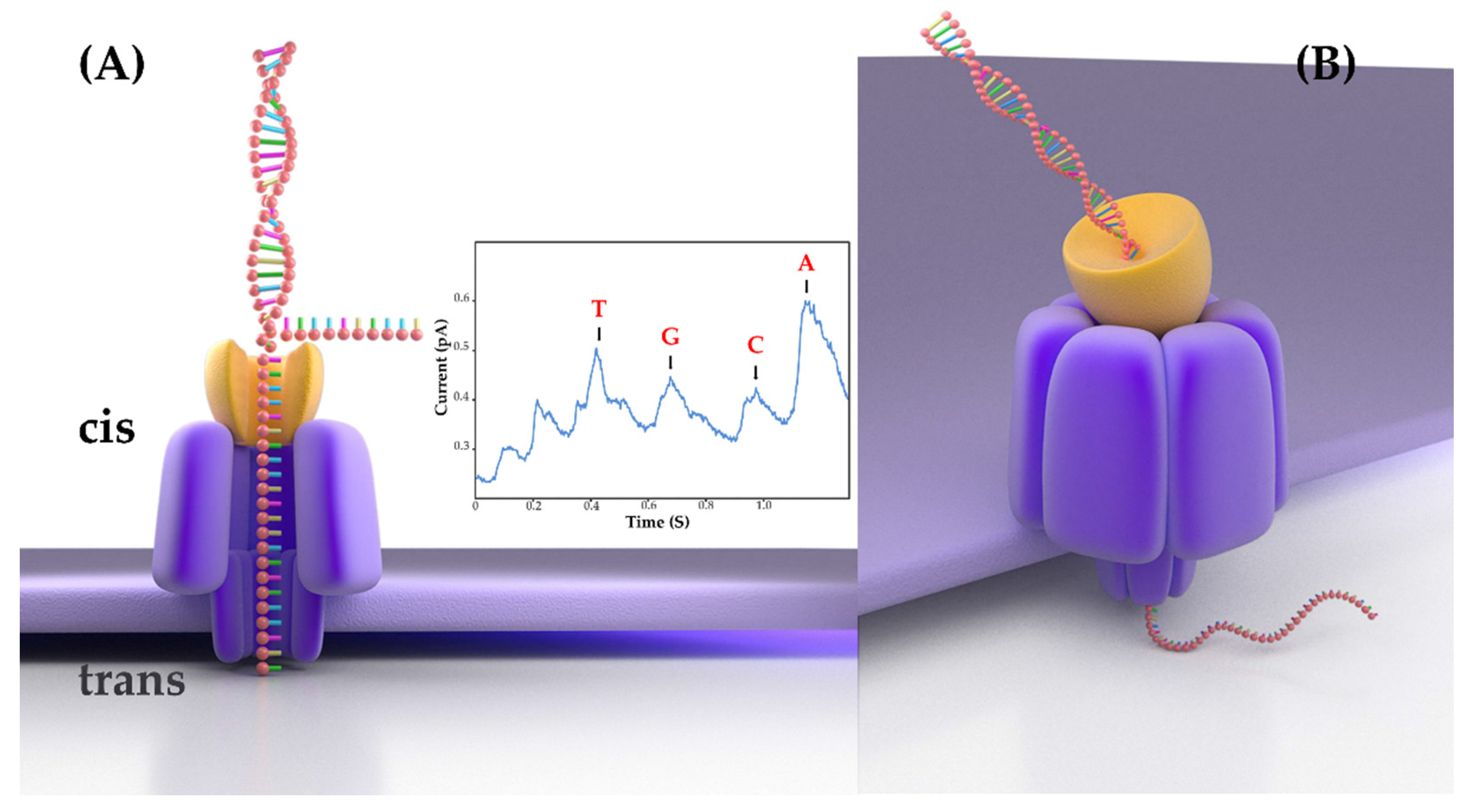
3]. It has a distinctive principle: ONT sequencing measures the disruption in ionic current when a single-stranded DNA molecule passes through a nanopore of an electrically resistant yet voltage-applied membrane. The change in ionic current is translated into sequence information in real time by base-calling software. The process of ONT sequencing adopts a PCR-free method to directly sequence genomic DNA molecules (
Figure 1). Therefore, as long as the DNA strand remains intact during sample preparation, there is no upper limit to the read length. Compared with SGS, ONT has many advantages. The MinION sequencer, one of the ONT platforms, is the first handheld sequencer; it measures only 2 cm × 4 cm × 9 cm and weighs about 100 g. It can typically work in a variety of environments, even in space and polar regions. In addition, the construction process for ONT sequencing libraries is relatively brief and straightforward, requiring only 1 h. Hence, because of its portable size, low price, and low computing requirements, MinION could revolutionize the discipline of genomics. In recent years, ONT has been greatly developed, but it is still used less in veterinary microbiology research and clinical diagnosis.
Here, we report a comparative study on the genome assemblies of
Haemophilus parasuis using different strategies from three different sequencing platforms: NovaSeq from Illumina (NGS), and long-read platforms PacBio Sequel and ONT MinION.
Haemophilus parasuis causes Glässer’s disease, which is characterized by fibrinous polyserositis and arthritis in swine [
4]. The strains are classified into 15 serovars, and nontypeable isolates represent a high percentage (about 25%). Isolates of different serovars, and even within the same serovar, are heterogeneous in their genomic traits and virulence. There is a lack of clear genetic markers for the virulence that can be used to distinguish highly pathogenic strains, which brings challenges for clinical diagnosis and vaccine development. To solve this problem, it is necessary to analyze genomic differences among a large number of virulent strains by relying on efficient and low-cost sequencing techniques. We compared the assembled genomes in terms of the accuracy, quality, and completeness of the long-read-only assembly, hybrid assembly, and assembly with polishing. Lastly, we aligned the transcripts obtained through RNA-seq with the genome assemblies to explore the effects of sequencing errors on protein prediction.
3. Discussion
The
H. parasuis genome could be assembled de novo with a read depth of about 50× by using PacBio or ONT platforms for the independent assembly strategy. Moreover, the complete circular genome can be directly assembled by using ONT reads. However, a continuous genomic sequence was not obtained by using Illumina short reads alone. On the SGS platform, the continuous assembled genome could be improved by increasing the length of the inserted fragments, such as mate-pair libraries, the insert sizes of which could range from 8 to 40 kb [
5]. However, compared with the ONT platform (>100 kb for prokaryotes and >1 Mb for eukaryotes), the library-construction process in the SGS platform for the use of the long-read method was complicated and costly, and the return on investment was relatively low. Long-read sequencing has transformed genome assembly. This should be the starting point for all new genome-assembly projects. Compared with PacBio, the ONT platform allows for researchers to sequence microbial genomes more quickly and at a lower cost [
6,
7]. It can be used in various genome-sequencing projects due to its unique technical principles, ultralong reads, and portability, although its accuracy is slightly worse than that of the PacBio platform.
Second, for the hybrid assembly strategy, more continuous assemblies can be achieved when using long reads in conjunction with Illumina reads. This strategy initiated the hybrid assembly with high-quality Illumina short reads and filled the gaps with ONT or PacBio long reads. We assembled the complete genome with Unicycler and MaSuRCA instead of SPAdes. Moreover, hybrid assembly using Illumina reads with ONT reads was superior to using Illumina reads with PacBio reads. This may be because the ultralong reads of ONT provide more adequate information about the genome arrangement. Similarly, Zhao Chen et al. reported a genome assembly project of 12 strains with a hybrid assembly strategy (
Escherichia coli,
Klebsiella variicola,
Klebsiella pneumonia,
Enterobacter cancerogenus,
Salmonella,
Citrobacter braakii,
Cronobacter sakazakii,
Listeria monocytogenes,
Staphylococcus aureus,
Campylobacter jejuni,
Campylobacter coli). The results showed that SPAdes failed to completely assemble any of the genomes. Unicycler completed the genomes of 10 of the 12 strains, and MaSuRCA produced complete assemblies of seven strains. However, SPAdes and Unicycler produced more accurate assemblies and performed better in genomic analyses of AMR, virulence potential, and pangenome compared to MaSuRCA [
8]. Unicycler exhibited improved assemblies, suggesting algorithmic approaches following that model may be the most fruitful in the future.
Third, for the strategy of assembly followed by polishing, the genomes assembled with ONT or PacBio long reads and polished with Illumina short reads were optimal in terms of accuracy, continuity, and completeness. An equally excellent method was the correction with Homopolish after the ONT-only assembly, which corrected systematic ONT errors by using homologous sequences and had the advantage of avoiding hybrid sequencing with Illumina. The developers of Homopolish tested the software’s polishing ability against bacterial genomes (
Enterococcus faecalis,
Pseudomonas aeruginosa,
Salmonella enterica, et al.), a viral genome (
Lambda phage), and a fungal genome (
Saccharomyces cerevisiae). When combined with Medaka/HELEN, genome quality can exceed Q50 on R9.4 flow cells, achieving similar precision to that of hybrid assemblies with Unicycler [
9]. Results showed that ONT-only sequencing could produce sufficiently high-quality microbial genomes for downstream analysis. Hence, considering the costs and efficiencies of these experiments, we recommend the use of ONT assembly followed by Homopolish correction in microbial-genome sequencing. However, the efficiency of Homopolish is related to the abundance of related genomes in NCBI, and it has not been tested on noncoding regions, which represent a large proportion of eukaryotic genomes. Therefore, for all eukaryotes and for prokaryotes that lack related genomes in NCBI, the strategy of assembly with long-read technology combined with accurate short-read-technology error correction is more feasible. For example, Miten Jain reported the sequencing and assembly of a reference genome for the human GM12878 Utah/Ceph cell line using the MinION sequencer. The final assembled genome was 2867 million bases in size, covering 85.8% of the reference. Assembly accuracy, after incorporating complementary short-read sequencing data, exceeded 99.8% [
10]. In addition, long-read sequencing is a great tool to overcome the low resolution of reconstructing the repetitive regions and polyploidy of plant genomes. In combination with the SGS technology, the genomes of various plants were completely assembled [
11,
12,
13]. This leads to a deeper understanding of plants’ genomic diversity, evolution, and gene function, in turn accelerating the process of plant breeding and the production of improved varieties. In summary, there is no perfect sequencing technology at present, and the advantages and disadvantages of different mainstream sequencing platforms are shown in
Table 8. For actual genomic-sequencing projects, we need to select the appropriate sequencing platform according to the genomic characteristics of different species, and take advantage of the benefits of different platforms, such as the ultralong reads of ONT, and the high-accuracy reads and high throughput of Illumina to solve complex genomic-sequencing challenges. Moreover, the low-cost, scalable, and easy-to-operate ONT platform offers researchers worldwide the opportunity to independently resolve the genomes of most organisms at any location, unlike the previous need to rely on third-party sequencing facilities, which will increase the efficiency of genome sequencing. At the same time, the large number of front-line users will ensure the sustainability of ONT sequencing technology.
By aligning the 16S rDNA sequences from different assemblies, most regions of the ONT-read-only assembly were highly accurate, and the predominant errors were indels that were concentrated in the homopolymer region. For the R 9.4.1 flow cells, the raw signal was mainly influenced by three central nucleotides (k-mers) that occupied the pores. By introducing frameshifts and premature stop codons, these errors could potentially critically affect the interpretation of the translated regions [
14,
15,
16]. In order to improve signal robustness, ONT chemistry involves the attachment of a motor protein to the DNA, which slows down the translocation and allows for the k-mers to reside within the pore for long enough to differentiate the signal from noise. Nevertheless, despite the reduced translocation speed, it is difficult to detect the transition between two identical k-mers, which complicates the detection of homopolymers that are longer than the k-mer. Because the translocation speed for nucleotides is generally nonuniform, it is not accurate to infer the homopolymer length from the duration of the measured signal, leading to the generation of indels [
17].
As we aligned the transcripts with assemblies, indels were present in many aligned genes (664 of 2116) in the unpolished ONT-long-read assembly genome. Correction using ONT reads was able to reduce the number of genes with indels from 664 to 278; this number was further reduced to about 180 after another round of correction using Homopolish or Pilon. Results showed that using only ONT reads for polishing could achieve the purpose of filling indels. However, further polishing is necessary in order to obtain a higher-quality genome. Then, we aligned the transcripts with the predicted CDSs of the different assemblies and obtained the same conclusions as above. Errors in long-read assemblies can critically affect protein prediction. One way to tackle this problem is to polish the genome assembly with long reads by using Medaka or with short reads by using Pilon. Another option is the use of Homopolish for polishing, as it corrects sequencing errors by retrieving homologs from closely related genomes and a trained ML model. The polished genomes were reported to achieve an accuracy of Q40–90 (>99.99%) [
12]. From the perspective of the nature of the data, most of the related genomes utilized by Homopolish were sequenced with the Illumina platform in its early stages. Therefore, this is equivalent to using multiple ultralong Illumina reads to polish a genome assembly. Recently, ONT has started offering novel reagents that allow for the continuous sequencing of the forward and reverse strands of a single DNA molecule. With these reagents, mode raw-read-length accuracy of greater than 99% is expected to be generated. Other updates include ultrahigh-precision base-calling options, a new generation of sequencing instruments, and a new control method for the sequencing process. These important updates make it possible to directly generate ultrahigh-precision genomes by using ONT sequencing.
The H. parasuis disease occurs in swine populations around the world, irrespective of health status. Considering the increasing pressure to reduce reliance on antibiotics, vaccination strategies for preventing systemic infection and mortality are more emphasized. However, cross-protection between different serovars and even within the same serovar is variable and difficult predict. To design effective new universal vaccines, genomic methods are needed in order to screen for antigens with protective potential. Compared with the SGS technology, long-read ONT sequencing can provide more abundant genetic information for solving this challenge.
In conclusion, the assembly of bacterial genomes can be directly achieved by using long-read sequencing techniques, such as PacBio and ONT. Compared with PacBio, the ONT platform has the advantages of lower cost, faster sequencing speed, longer read length, and greater ease of operation. Its current accuracy is comparable to that of PacBio, but with updates to ONT sequencing, its accuracy could reach or even exceed that of SGS platforms. Moreover, in current research, to maximize assembly accuracy, it is essential to polish the assembly with homologous sequences of related genomes or sequencing data from short-read technology. When necessary, indels and errors can be checked by aligning known proteins and cDNA or mRNA sequences against the genome and fixing them manually. Furthermore, we found discrepancies between the published reference genome and our assemblies. This suggests that the genomes of different isolates are highly variable and that the genomes of individuals are not representative of the species as a whole. Therefore, for the foreseeable future, to better explain the process of genetic evolution and variation in bacteria, the large-scale genome sequencing and construction of pangenomes for specific species of bacteria are needed. At the same time, no sequencing-assembly pipelines should be set in stone, but they need to be continuously updated and optimized for the organism to be sequenced.


 ,
, 





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}