
Protective Role of Glutathione in the Hippocampus after Brain Ischemia


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Protective Roles of Glutathione (GSH) in Ischemia-Induced Hippocampal Injury
2.1. Anti-Oxidative Role of GSH in Ischemia-Induced Oxidative Stress
2.2. GSH Protects Neurons from Ischemia-Induced Disruption of Intracellular Zn2+ Homeostasis
3. Neuroprotective Effects of EAAC1
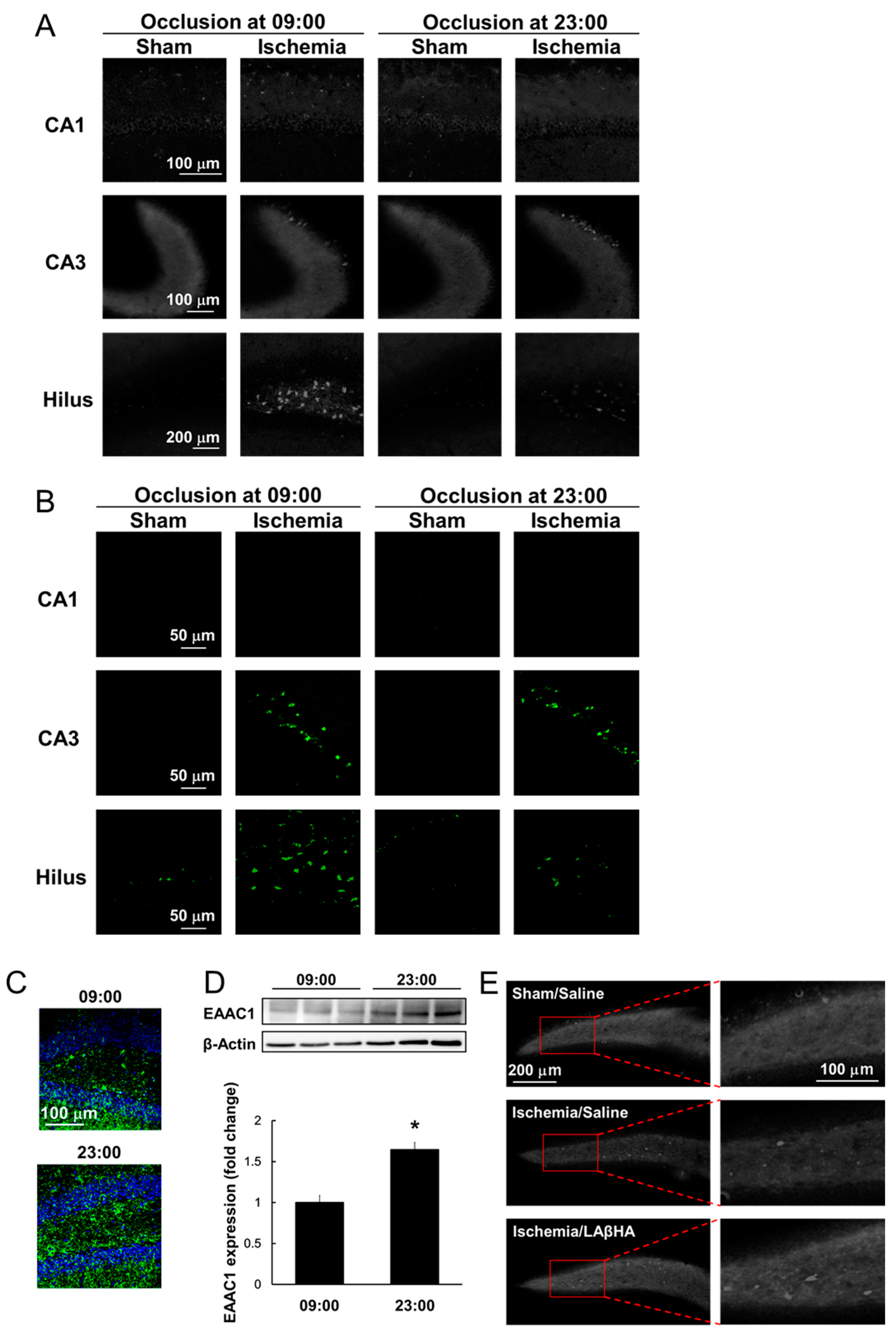
4. Neuroprotective Role of GSH and Time-of-Day Variations in Ischemic Injury
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Conflicts of Interest
References
- GBD 2016 Neurology Collaborators. Global, regional, and national burden of neurological disorders, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2019, 18, 459–480. [Google Scholar] [CrossRef] [Green Version]
- Feigin, V.L.; Krishnamurthi, R. Public health strategies could reduce the global stroke epidemic. Lancet Neurol. 2010, 9, 847–848. [Google Scholar] [CrossRef]
- Izquierdo, I.; Medina, J.H. Memory formation: The sequence of biochemical events in the hippocampus and its connection to activity in other brain structures. Neurobiol. Learn. Mem. 1997, 68, 285–316. [Google Scholar] [CrossRef] [Green Version]
- Schaapsmeerders, P.; van Uden, I.W.M.; Tuladhar, A.M.; Maaijwee, N.A.M.; van Dijk, E.J.; Rutten-Jacobs, L.C.A.; Arntz, R.M.; Schoonderwaldt, H.C.; Dorresteijn, L.D.A.; de Leeuw, F.E.; et al. Ipsilateral hippocampal atrophy is associated with long-term memory dysfunction after ischemic stroke in young adults. Hum. Brain Mapp. 2015, 36, 2432–2442. [Google Scholar] [CrossRef]
- Oliver, C.N.; Starke-Reed, P.E.; Stadtman, E.R.; Liu, G.J.; Carney, J.M.; Floyd, R.A. Oxidative damage to brain proteins, loss of glutamine synthetase activity, and production of free radicals during ischemia/reperfusion-induced injury to gerbil brain. Proc. Natl. Acad. Sci. USA 1990, 87, 5144–5147. [Google Scholar] [CrossRef] [Green Version]
- Rodrigo, R.; Fernández-Gajardo, R.; Gutiérrez, R.; Matamala, J.M.; Carrasco, R.; Miranda-Merchak, A.; Feuerhake, W. Oxidative stress and pathophysiology of ischemic stroke: Novel therapeutic opportunities. CNS Neurol. Disord. Drug Targets 2013, 12, 698–714. [Google Scholar] [CrossRef]
- Takeda, A. Zinc homeostasis and functions of zinc in the brain. Biometals 2001, 14, 343–351. [Google Scholar] [CrossRef] [PubMed]
- Koh, J.Y.; Suh, S.W.; Gwag, B.J.; He, Y.Y.; Hsu, C.Y.; Choi, D.W. The role of zinc in selective neuronal death after transient global cerebral ischemia. Science 1996, 272, 1013–1016. [Google Scholar] [CrossRef] [PubMed]
- Won, S.J.; Yoo, B.H.; Brennan, A.M.; Shin, B.S.; Kauppinen, T.M.; Berman, A.E.; Swanson, R.A.; Suh, S.W. EAAC1 gene deletion alters zinc homeostasis and exacerbates neuronal injury after transient cerebral ischemia. J. Neurosci. 2010, 30, 15409–15418. [Google Scholar] [CrossRef] [PubMed]
- Aratake, T.; Higashi, Y.; Hamada, T.; Ueba, Y.; Shimizu, T.; Shimizu, S.; Yawata, T.; Ueba, T.; Saito, M. The role of diurnal fluctuations in excitatory amino acid carrier 1 levels in post-ischemic hippocampal Zn2+ accumulation. Exp. Neurol. 2021, 336, 113538. [Google Scholar] [CrossRef]
- Kapoor, M.; Sharma, S.; Sandhir, R.; Nehru, B. Temporal changes in physiological and molecular markers in various brain regions following transient global ischemia in rats. Mol. Biol. Rep. 2019, 46, 6215–6230. [Google Scholar] [CrossRef]
- Kho, A.R.; Choi, B.Y.; Lee, S.H.; Hong, D.K.; Lee, S.H.; Jeong, J.H.; Park, K.H.; Song, H.K.; Choi, H.C.; Suh, S.W. Effects of protocatechuic acid (PCA) on global cerebral ischemia-induced hippocampal neuronal death. Int. J. Mol. Sci. 2018, 19, 1420. [Google Scholar] [CrossRef] [Green Version]
- Love, S. Oxidative stress in brain ischemia. Brain Pathol. 1999, 9, 119–131. [Google Scholar] [CrossRef]
- Nikonenko, A.G.; Radenovic, L.; Andjus, P.R.; Skibo, G.G. Structural features of ischemic damage in the hippocampus. Anat. Rec. 2009, 292, 1914–1921. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Tompkins, K.D.; Simonyi, A.; Korthuis, R.J.; Sun, A.Y.; Sun, G.Y. Apocynin protects against global cerebral ischemia-reperfusion-induced oxidative stress and injury in the gerbil hippocampus. Brain Res. 2006, 1090, 182–189. [Google Scholar] [CrossRef] [PubMed]
- Galano, A.; Alvarez-Idaboy, J.R. Glutathione: Mechanism and kinetics of its non-enzymatic defense action against free radicals. RSC Adv. 2011, 1, 1763–1771. [Google Scholar] [CrossRef]
- Orian, L.; Cozza, G.; Maiorino, M.; Toppo, S.; Ursini, F. The catalytic mechanism of glutathione peroxidases (Chapter 3). In Glutathione, 1st ed.; Flohé, L., Ed.; CRC Press: Boca Raton, FL, USA, 2018; pp. 35–52. [Google Scholar]
- Couto, N.; Wood, J.; Barber, J. The role of glutathione reductase and related enzymes on cellular redox homoeostasis network. Free Radic. Biol. Med. 2016, 95, 27–42. [Google Scholar] [CrossRef] [PubMed]
- Hirrlinger, J.; Dringen, R. Multidrug resistance protein 1-mediated export of glutathione and glutathione disulfide from brain astrocytes. Methods Enzymol. 2005, 400, 395–409. [Google Scholar] [CrossRef]
- Homolya, L.; Váradi, A.; Sarkadi, B. Multidrug resistance-associated proteins: Export pumps for conjugates with glutathione, glucuronate or sulfate. BioFactors 2003, 17, 103–114. [Google Scholar] [CrossRef]
- Wu, T.; Yin, F.; Kong, H.; Peng, J. Germacrone attenuates cerebral ischemia/reperfusion injury in rats via antioxidative and antiapoptotic mechanisms. J. Cell. Biochem. 2019, 120, 18901–18909. [Google Scholar] [CrossRef]
- Li, Z.; Yulei, J.; Yaqing, J.; Jinmin, Z.; Xinyong, L.; Jing, G.; Min, L. Protective effects of tetramethylpyrazine analogue Z-11 on cerebral ischemia reperfusion injury. Eur. J. Pharmacol. 2019, 844, 156–164. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Fattah, M.M.; Messiha, B.A.S.; Mansour, A.M. Modulation of brain ACE and ACE2 may be a promising protective strategy against cerebral ischemia/reperfusion injury: An experimental trial in rats. Naunyn-Schmiedebergs Arch. Pharmacol. 2018, 391, 1003–1020. [Google Scholar] [CrossRef]
- Yabuki, Y.; Fukunaga, K. Oral administration of glutathione improves memory deficits following transient brain ischemia by reducing brain oxidative stress. Neuroscience 2013, 250, 394–407. [Google Scholar] [CrossRef] [PubMed]
- Anderson, M.F.; Nilsson, M.; Eriksson, P.S.; Sims, N.R. Glutathione monoethyl ester provides neuroprotection in a rat model of stroke. Neurosci. Lett. 2004, 354, 163–165. [Google Scholar] [CrossRef]
- Won, S.J.; Kim, J.E.; Cittolin-Santos, G.F.; Swanson, R.A. Assessment at the single-cell level identifies neuronal glutathione depletion as both a cause and effect of ischemia-reperfusion oxidative stress. J. Neurosci. 2015, 35, 7143–7152. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dringen, R.; Pfeiffer, B.; Hamprecht, B. Synthesis of the antioxidant glutathione in neurons: Supply by astrocytes of CysGly as precursor for neuronal glutathione. J. Neurosci. 1999, 19, 562–569. [Google Scholar] [CrossRef] [PubMed]
- Frederickson, C.J. Neurobiology of zinc and zinc-containing neurons. Int. Rev. Neurobiol. 1989, 31, 145–238. [Google Scholar] [CrossRef]
- Cole, T.B.; Wenzel, H.J.; Kafer, K.E.; Schwartzkroin, P.A.; Palmiter, R.D. Elimination of zinc from synaptic vesicles in the intact mouse brain by disruption of the ZnT3 gene. Proc. Natl. Acad. Sci. USA 1999, 96, 1716–1721. [Google Scholar] [CrossRef] [Green Version]
- Danscher, G.; Howell, G.; Pérez-Clausell, J.; Hertel, N. The dithizone, Timm’s sulphide silver and the selenium methods demonstrate a chelatable pool of zinc in CNS. A proton activation (PIXE) analysis of carbon tetrachloride extracts from rat brains and spinal cords intravitally treated with dithizone. Histochemistry 1985, 83, 419–422. [Google Scholar] [CrossRef]
- Sensi, S.L.; Paoletti, P.; Bush, A.I.; Sekler, I. Zinc in the physiology and pathology of the CNS. Nat. Rev. Neurosci. 2009, 10, 780–791. [Google Scholar] [CrossRef]
- Frederickson, C.J.; Koh, J.Y.; Bush, A.I. The neurobiology of zinc in health and disease. Nat. Rev. Neurosci. 2005, 6, 449–462. [Google Scholar] [CrossRef] [PubMed]
- Frederickson, C.J.; Giblin, L.J.; Krezel, A.; McAdoo, D.J.; Mueller, R.N.; Zeng, Y.; Balaji, R.V.; Masalha, R.; Thompson, R.B.; Fierke, C.A.; et al. Concentrations of extracellular free zinc (pZn)e in the central nervous system during simple anesthetization, ischemia and reperfusion. Exp. Neurol. 2006, 198, 285–293. [Google Scholar] [CrossRef]
- Lee, J.Y.; Kim, J.H.; Palmiter, R.D.; Koh, J.Y. Zinc released from metallothionein-III may contribute to hippocampal CA1 and thalamic neuronal death following acute brain injury. Exp. Neurol. 2003, 184, 337–347. [Google Scholar] [CrossRef]
- Koh, J.Y.; Lee, S.J. Metallothionein-3 as a multifunctional player in the control of cellular processes and diseases. Mol. Brain 2020, 13, 116. [Google Scholar] [CrossRef] [PubMed]
- Krężel, A.; Maret, W. The functions of metamorphic metallothioneins in zinc and copper metabolism. Int. J. Mol. Sci. 2017, 18, 1237. [Google Scholar] [CrossRef] [Green Version]
- Medvedeva, Y.V.; Ji, S.G.; Yin, H.Z.; Weiss, J.H. Differential vulnerability of CA1 versus CA3 pyramidal neurons after ischemia: Possible relationship to sources of Zn2+ accumulation and its entry into and prolonged effects on mitochondria. J. Neurosci. 2017, 37, 726–737. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maret, W. Oxidative metal release from metallothionein via zinc-thiol/disulfide interchange. Proc. Natl. Acad. Sci. USA 1994, 91, 237–241. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, T.; Zheng, W.; Xu, H.; Zhou, J.M.; Wang, Z.Y. Clioquinol inhibits zinc-triggered caspase activation in the hippocampal CA1 region of a global ischemic gerbil model. PLoS ONE 2010, 5, e11888. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.J.; Liao, S.L. Zinc toxicity on neonatal cortical neurons: Involvement of glutathione chelation. J. Neurochem. 2003, 85, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Hong, D.K.; Kho, A.R.; Lee, S.H.; Jeong, J.H.; Kang, B.S.; Kang, D.H.; Park, M.K.; Park, K.H.; Lim, M.S.; Choi, B.Y.; et al. Transient receptor potential melastatin 2 (TRPM2) inhibition by antioxidant, n-acetyl-l-cysteine, reduces global cerebral ischemia-induced neuronal death. Int. J. Mol. Sci. 2020, 21, 6026. [Google Scholar] [CrossRef]
- Mize, C.E.; Langdon, R.G. Hepatic glutathione reductase. I. Purification and general kinetic properties. J. Biol. Chem. 1962, 237, 1589–1595. [Google Scholar] [CrossRef]
- Splittgerber, A.G.; Tappel, A.L. Inhibition of glutathione peroxidase by cadmium and other metal ions. Arch. Biochem. Biophys. 1979, 197, 534–542. [Google Scholar] [CrossRef]
- Coco, S.; Verderio, C.; Trotti, D.; Rothstein, J.D.; Volterra, A.; Matteoli, M. Non-synaptic localization of the glutamate transporter EAAC1 in cultured hippocampal neurons. Eur. J. Neurosci. 1997, 9, 1902–1910. [Google Scholar] [CrossRef]
- Shashidharan, P.; Huntley, G.W.; Murray, J.M.; Buku, A.; Moran, T.; Walsh, M.J.; Morrison, J.H.; Plaitakis, A. Immunohistochemical localization of the neuron-specific glutamate transporter EAAC1 (EAAT3) in rat brain and spinal cord revealed by a novel monoclonal antibody. Brain Res. 1997, 773, 139–148. [Google Scholar] [CrossRef]
- Rothstein, J.D.; Martin, L.; Levey, A.I.; Dykes-Hoberg, M.; Jin, L.; Wu, D.; Nash, N.; Kuncl, R.W. Localization of neuronal and glial glutamate transporters. Neuron 1994, 13, 713–725. [Google Scholar] [CrossRef]
- Kanai, Y.; Hediger, M.A. Primary structure and functional characterization of a high-affinity glutamate transporter. Nature 1992, 360, 467–471. [Google Scholar] [CrossRef]
- Faden, A.I.; Demediuk, P.; Panter, S.S.; Vink, R. The role of excitatory amino acids and NMDA receptors in traumatic brain injury. Science 1989, 244, 798–800. [Google Scholar] [CrossRef]
- Watase, K.; Hashimoto, K.; Kano, M.; Yamada, K.; Watanabe, M.; Inoue, Y.; Okuyama, S.; Sakagawa, T.; Ogawa, S.I.; Kawashima, N.; et al. Motor discoordination and increased susceptibility to cerebellar injury in GLAST mutant mice. Eur. J. Neurosci. 1998, 10, 976–988. [Google Scholar] [CrossRef] [PubMed]
- Bendahan, A.; Armon, A.; Madani, N.; Kavanaugh, M.P.; Kanner, B.I. Arginine 447 plays a pivotal role in substrate interactions in a neuronal glutamate transporter. J. Biol. Chem. 2000, 275, 37436–37442. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.; Swanson, R.A. The glutamate transporters EAAT2 and EAAT3 mediate cysteine uptake in cortical neuron cultures. J. Neurochem. 2003, 84, 1332–1339. [Google Scholar] [CrossRef]
- Himi, T.; Ikeda, M.; Yasuhara, T.; Nishida, M.; Morita, I. Role of neuronal glutamate transporter in the cysteine uptake and intracellular glutathione levels in cultured cortical neurons. J. Neural Transm. 2003, 110, 1337–1348. [Google Scholar] [CrossRef]
- Watts, S.D.; Torres-Salazar, D.; Divito, C.B.; Amara, S.G. Cysteine transport through excitatory amino acid transporter 3 (EAAT3). PLoS ONE 2014, 9, e109245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zerangue, N.; Kavanaugh, M.P. Interaction of L-cysteine with a human excitatory amino acid transporter. J. Physiol. 1996, 493, 419–423. [Google Scholar] [CrossRef] [PubMed]
- Aoyama, K.; Suh, S.W.; Hamby, A.M.; Liu, J.; Chan, W.Y.; Chen, Y.; Swanson, R.A. Neuronal glutathione deficiency and age-dependent neurodegeneration in the EAAC1 deficient mouse. Nat. Neurosci. 2006, 9, 119–126. [Google Scholar] [CrossRef]
- Jang, B.G.; Won, S.J.; Kim, J.H.; Choi, B.Y.; Lee, M.W.; Sohn, M.; Song, H.K.; Suh, S.W. EAAC1 gene deletion alters zinc homeostasis and enhances cortical neuronal injury after transient cerebral ischemia in mice. J. Trace Elem. Med. Biol. 2012, 26, 85–88. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.Y.; Kim, J.H.; Kim, H.J.; Lee, B.E.; Kim, I.Y.; Sohn, M.; Suh, S.W. EAAC1 gene deletion increases neuronal death and blood brain barrier disruption after transient cerebral ischemia in female mice. Int. J. Mol. Sci. 2014, 15, 19444–19457. [Google Scholar] [CrossRef] [Green Version]
- Gottlieb, M.; Domercq, M.; Matute, C. Altered expression of the glutamate transporter EAAC1 in neurons and immature oligodendrocytes after transient forebrain ischemia. J. Cereb. Blood Flow Metab. 2000, 20, 678–687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Praag, H.; Schinder, A.F.; Christie, B.R.; Toni, N.; Palmer, T.D.; Gage, F.H. Functional neurogenesis in the adult hippocampus. Nature 2002, 415, 1030–1034. [Google Scholar] [CrossRef]
- Zhao, C.; Teng, E.M.; Summers, R.G.; Ming, G.L.; Gage, F.H. Distinct morphological stages of dentate granule neuron maturation in the adult mouse hippocampus. J. Neurosci. 2006, 26, 3–11. [Google Scholar] [CrossRef]
- Takagi, Y.; Nozaki, K.; Takahashi, J.; Yodoi, J.; Ishikawa, M.; Hashimoto, N. Proliferation of neuronal precursor cells in the dentate gyrus is accelerated after transient forebrain ischemia in mice. Brain Res. 1999, 831, 283–287. [Google Scholar] [CrossRef]
- Türeyen, K.; Vemuganti, R.; Sailor, K.A.; Bowen, K.K.; Dempsey, R.J. Transient focal cerebral ischemia-induced neurogenesis in the dentate gyrus of the adult mouse. J. Neurosurg. 2004, 101, 799–805. [Google Scholar] [CrossRef]
- Tobin, M.K.; Bonds, J.A.; Minshall, R.D.; Pelligrino, D.A.; Testai, F.D.; Lazarov, O. Neurogenesis and inflammation after ischemic stroke: What is known and where we go from here. J. Cereb. Blood Flow Metab. 2014, 34, 1573–1584. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, B.Y.; Won, S.J.; Kim, J.H.; Sohn, M.; Song, H.K.; Chung, T.N.; Kim, T.Y.; Suh, S.W. EAAC1 gene deletion reduces adult hippocampal neurogenesis after transient cerebral ischemia. Sci. Rep. 2018, 8, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Argentino, C.; Toni, D.; Rasura, M.; Violi, F.; Sacchetti, M.L.; Allegretta, A.; Balsano, F.; Fieschi, C. Circadian variation in the frequency of ischemic stroke. Stroke 1990, 21, 387–389. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gallerani, M.; Manfredini, R.; Ricci, L.; Goldoni, C.; Cocurullo, A.; Pareschi, P.L. Circadian variation in the onset of acute myocardial infarction: Lack of an effect due to age and sex. J. Int. Med. Res. 1993, 21, 158–160. [Google Scholar] [CrossRef] [PubMed]
- Gallerani, M.; Manfredini, R.; Ricci, L.; Cocurullo, A.; Goldoni, C.; Bigoni, M.; Fersini, C. Chronobiological aspects of acute cerebrovascular diseases. Acta Neurol. Scand. 1993, 87, 482–487. [Google Scholar] [CrossRef] [PubMed]
- Marshall, J. Diurnal variation in occurrence of strokes. Stroke 1977, 8, 230–231. [Google Scholar] [CrossRef] [Green Version]
- Tsementzis, S.A.; Gill, J.S.; Hitchcock, E.R.; Gill, S.K.; Beevers, D.G. Diurnal variation of and activity during the onset of stroke. Neurosurgery 1985, 17, 901–904. [Google Scholar] [CrossRef]
- Elliott, W.J. Circadian variation in the timing of stroke onset: A meta-analysis. Stroke 1998, 29, 992–996. [Google Scholar] [CrossRef]
- Jiménez-Conde, J.; Ois, A.; Rodríguez-Campello, A.; Gomis, M.; Roquer, J. Does sleep protect against ischemic stroke? Less frequent ischemic strokes but more severe ones. J. Neurol. 2007, 254, 782–788. [Google Scholar] [CrossRef]
- Ripamonti, L.; Riva, R.; Maioli, F.; Zenesini, C.; Procaccianti, G. Daily variation in the occurrence of different subtypes of stroke. Stroke Res. Treat. 2017, 2017, 9091250. [Google Scholar] [CrossRef] [PubMed]
- Beker, M.C.; Caglayan, B.; Yalcin, E.; Caglayan, A.B.; Turkseven, S.; Gurel, B.; Kelestemur, T.; Sertel, E.; Sahin, Z.; Kutlu, S.; et al. Time-of-day dependent neuronal injury After ischemic stroke: Implication of circadian clock transcriptional factor Bmal1 and survival kinase AKT. Mol. Neurobiol. 2018, 55, 2565–2576. [Google Scholar] [CrossRef]
- Weil, Z.M.; Karelina, K.; Su, A.J.; Barker, J.M.; Norman, G.J.; Zhang, N.; DeVries, A.C.; Nelson, R.J. Time-of-day determines neuronal damage and mortality after cardiac arrest. Neurobiol. Dis. 2009, 36, 352–360. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.H.; Kim, E.Y.; Gwag, B.J.; Sohn, S.; Koh, J.Y. Zinc-induced cortical neuronal death with features of apoptosis and necrosis: Mediation by free radicals. Neuroscience 1999, 89, 175–182. [Google Scholar] [CrossRef]
- Zhang, C.; Wang, Y.; Zhao, X.; Wang, D.; Liu, L.; Wang, C.; Pu, Y.; Zou, X.; Du, W.; Jing, J.; et al. Distal single subcortical infarction had a better clinical outcome compared with proximal single subcortical infarction. Stroke 2014, 45, 2613–2619. [Google Scholar] [CrossRef] [Green Version]
- Kinoshita, C.; Aoyama, K.; Matsumura, N.; Kikuchi-Utsumi, K.; Watabe, M.; Nakaki, T. Rhythmic oscillations of the microRNA mir-96-5p play a neuroprotective role by indirectly regulating glutathione levels. Nat. Commun. 2014, 5, 3823. [Google Scholar] [CrossRef] [Green Version]
- Aoyama, K.; Nakaki, T. Glutathione in Cellular Redox Homeostasis: Association with the Excitatory Amino Acid Carrier 1 (EAAC1). Molecules 2015, 20, 8742–8758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hodgson, N.; Trivedi, M.; Muratore, C.; Li, S.; Deth, R. Soluble oligomers of amyloid-β cause changes in redox state, DNA methylation, and gene transcription by inhibiting EAAT3 mediated cysteine uptake. J. Alzheimer’s Dis. 2013, 36, 197–209. [Google Scholar] [CrossRef] [PubMed]
- Duerson, K.; Woltjer, R.L.; Mookherjee, P.; Leverenz, J.B.; Montine, T.J.; Bird, T.D.; Pow, D.V.; Rauen, T.; Cook, D.G. Detergent-insoluble EAAC1/EAAT3 aberrantly accumulates in hippocampal neurons of Alzheimer’s disease patients. Brain Pathol. 2009, 19, 267–278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacoste, M.G.; Ponce, I.T.; Golini, R.L.; Delgado, S.M.; Anzulovich, A.C. Aging modifies daily variation of antioxidant enzymes and oxidative status in the hippocampus. Exp. Gerontol. 2017, 88, 42–50. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Higashi, Y.; Aratake, T.; Shimizu, T.; Shimizu, S.; Saito, M. Protective Role of Glutathione in the Hippocampus after Brain Ischemia. Int. J. Mol. Sci. 2021, 22, 7765. https://doi.org/10.3390/ijms22157765
Higashi Y, Aratake T, Shimizu T, Shimizu S, Saito M. Protective Role of Glutathione in the Hippocampus after Brain Ischemia. International Journal of Molecular Sciences. 2021; 22(15):7765. https://doi.org/10.3390/ijms22157765
Chicago/Turabian StyleHigashi, Youichirou, Takaaki Aratake, Takahiro Shimizu, Shogo Shimizu, and Motoaki Saito. 2021. "Protective Role of Glutathione in the Hippocampus after Brain Ischemia" International Journal of Molecular Sciences 22, no. 15: 7765. https://doi.org/10.3390/ijms22157765
APA StyleHigashi, Y., Aratake, T., Shimizu, T., Shimizu, S., & Saito, M. (2021). Protective Role of Glutathione in the Hippocampus after Brain Ischemia. International Journal of Molecular Sciences, 22(15), 7765. https://doi.org/10.3390/ijms22157765