
Global Deletion of 11β-HSD1 Prevents Muscle Wasting Associated with Glucocorticoid Therapy in Polyarthritis

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
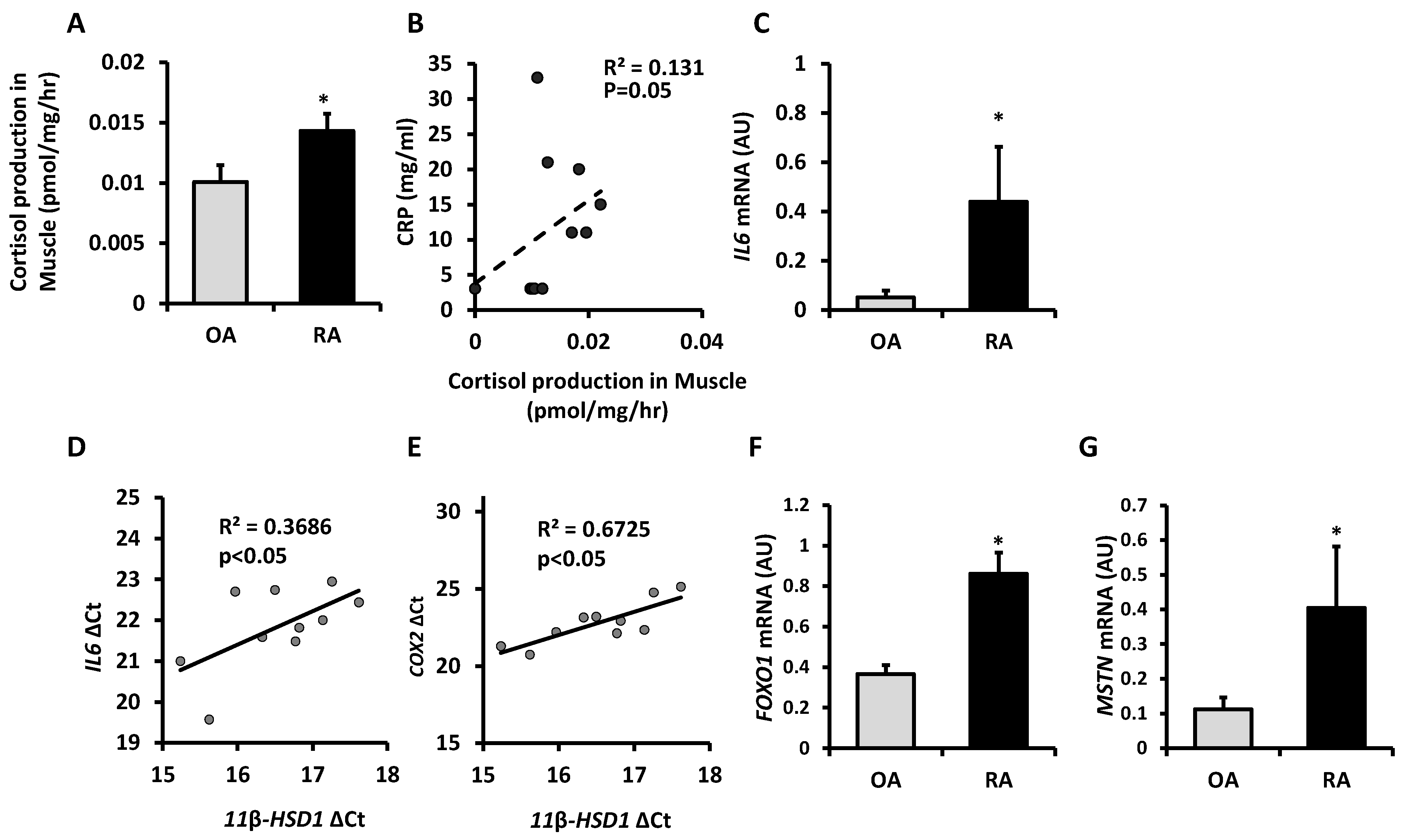
2.1. Cortisol Activation by 11β-HSD1 Is Increased in Skeletal Muscle in Rheumatoid Arthritis and Correlates with Inflammation
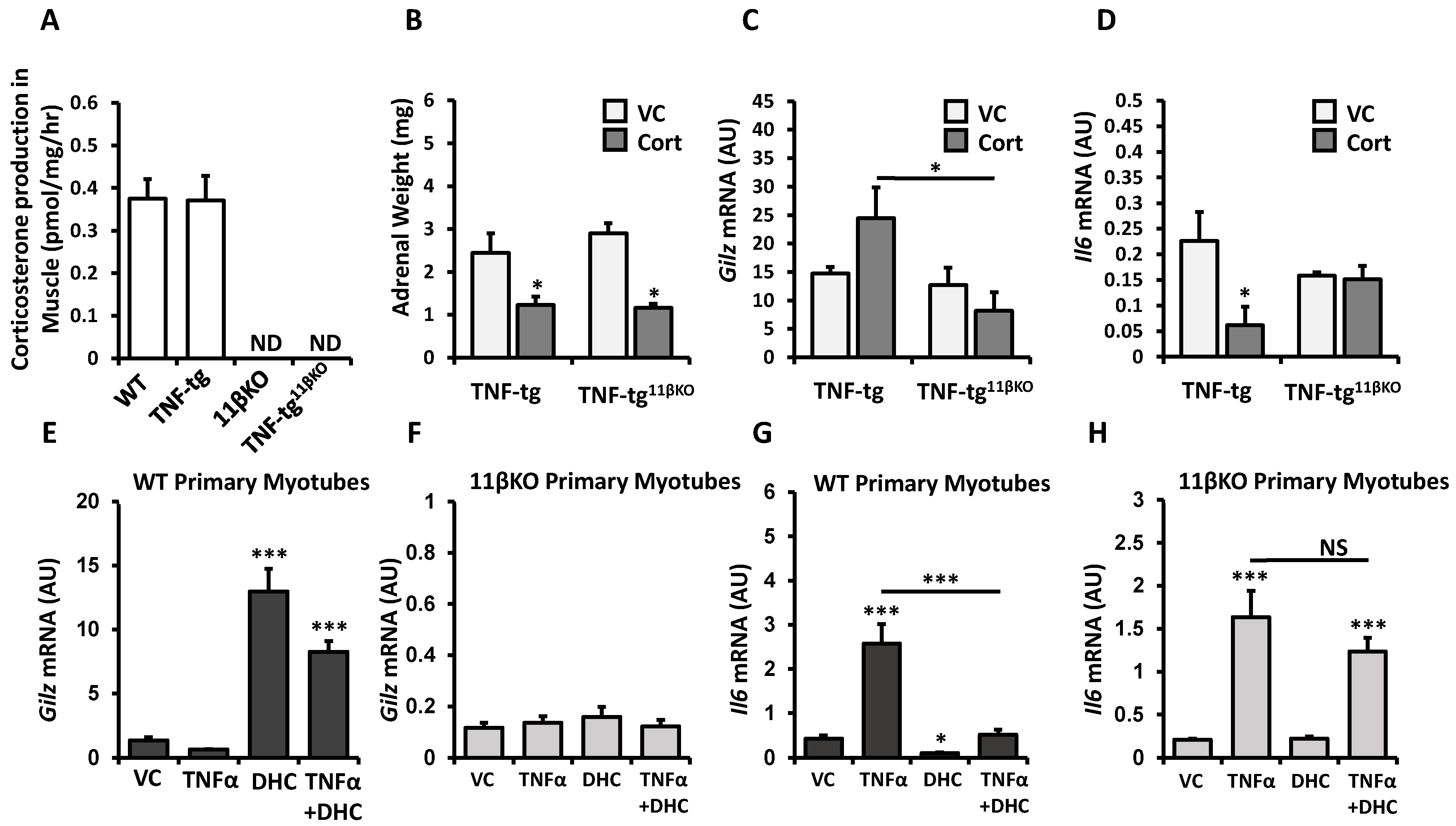
2.2. Deletion of 11β-HSD1 in a Model of Chronic Inflammation Causes Resistance to the Anti-Inflammatory Responses of Therapeutic Glucocorticoid in Muscle
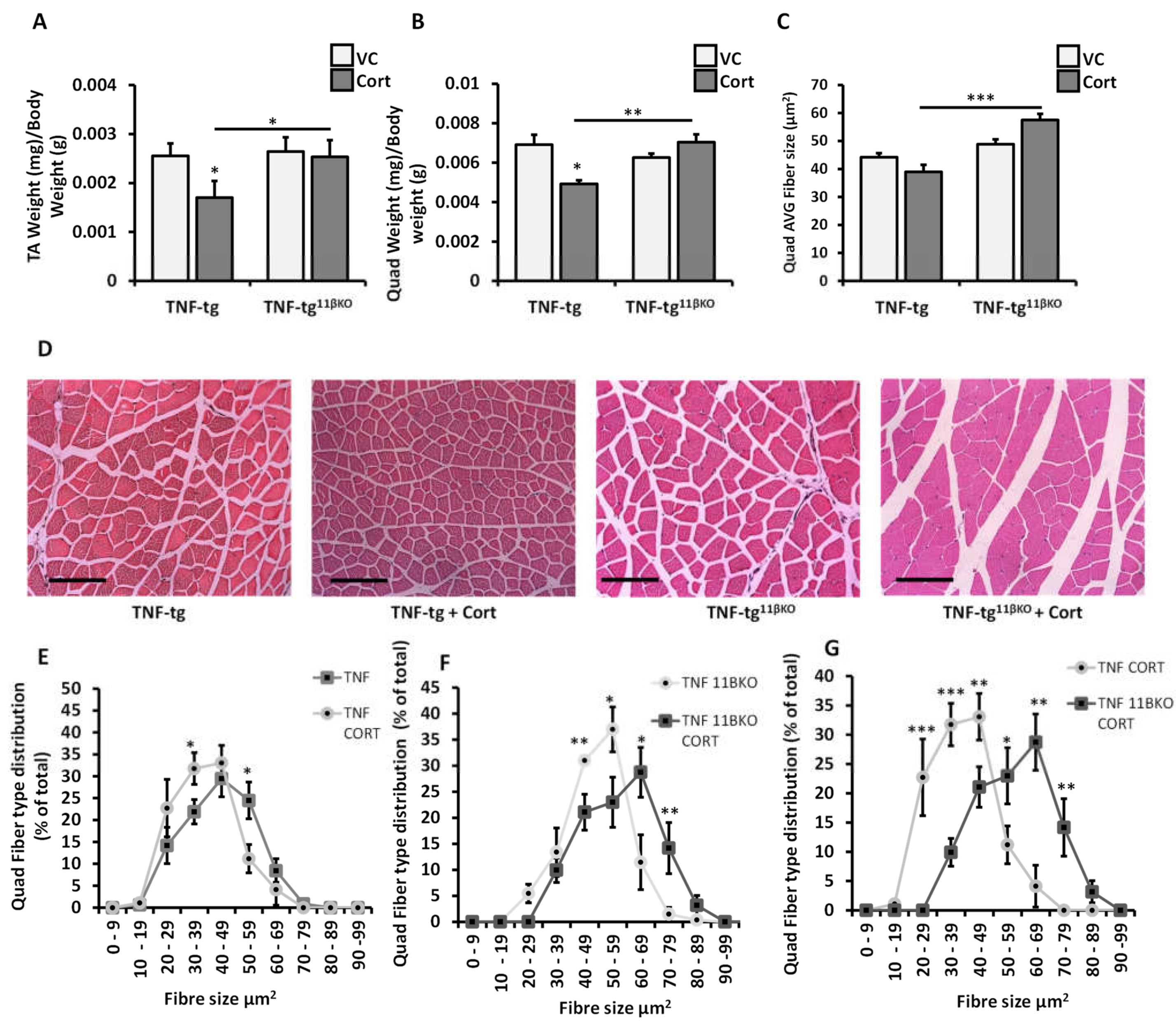
2.3. 11β-HSD1 Mediates Muscle Wasting in Response to Therapeutic Glucocorticoids in Chronic Inflammation
2.4. 11β-HSD1 KO Diminishes Activation of Muscle Catabolic Pathways in Response to Therapeutic Glucocorticoid
3. Discussion
4. Materials and Methods
4.1. Human Skeletal Muscle Biopsies
4.2. Animal Models
4.3. 11β-HSD1 Enzymatic Activity Assay
4.4. Primary Murine Muscle Cell Culture
4.5. RNA Isolation and Analysis of Gene Expression
4.6. Histological Analysis of Muscle
4.7. Western Blots
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 11β-HSD1 | 11beta-hydroxysteroid dehydrogenase type 1 |
| DHC | dehydrocorticosterone |
| HPA | hypothalamic-pituitary-adrenal |
| OA | osteoarthritis |
| RA | rheumatoid arthritis |
| TNF-tg | transgenic expression of human TNF-α |
| 11βKO | global genetic deletion of 11β-HSD1 |
| WT | wild-type genotype without genetic modifications |
| TA | tibialis anterior |
| NS | not significant |
| ND | not detectable |
| ESR | erythrocyte sedimentation rate |
| CRP | C-reactive protein |
References
- Coutinho, A.E.; Chapman, K.E. The anti-inflammatory and immunosuppressive effects of glucocorticoids, recent developments and mechanistic insights. Mol. Cell. Endocrinol. 2011, 335, 2–13. [Google Scholar] [CrossRef]
- Rhen, T.; Cidlowski, J.A. Antiinflammatory action of glucocorticoids—New mechanisms for old drugs. N. Engl. J. Med. 2005, 353, 1711–1723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oray, M.; Abu Samra, K.; Ebrahimiadib, N.; Meese, H.; Foster, C.S. Long-term side effects of glucocorticoids. Expert Opin. Drug Saf. 2016, 15, 457–465. [Google Scholar] [CrossRef]
- Schakman, O.; Kalista, S.; Barbé, C.; Loumaye, A.; Thissen, J.P. Glucocorticoid-induced skeletal muscle atrophy. Int. J. Biochem. Cell Biol. 2013, 45, 2163–2172. [Google Scholar] [CrossRef]
- Beaudart, C.; Zaaria, M.; Pasleau, F.; Reginster, J.Y.; Bruyère, O. Health Outcomes of Sarcopenia: A Systematic Review and Meta-Analysis. PLoS ONE 2017, 12, e0169548. [Google Scholar] [CrossRef] [Green Version]
- Gathercole, L.L.; Lavery, G.G.; Morgan, S.A.; Cooper, M.S.; Sinclair, A.J.; Tomlinson, J.W.; Stewart, P.M. 11β-Hydroxysteroid dehydrogenase 1: Translational and therapeutic aspects. Endocr. Rev. 2013, 34, 525–555. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Morgan, S.A.; Hassan-Smith, Z.K.; Doig, C.L.; Sherlock, M.; Stewart, P.M.; Lavery, G.G. Glucocorticoids and 11β-HSD1 are major regulators of intramyocellular protein metabolism. J. Endocrinol. 2016, 229, 277–286. [Google Scholar] [CrossRef] [PubMed]
- Morgan, S.A.; McCabe, E.L.; Gathercole, L.L.; Hassan-Smith, Z.K.; Larner, D.P.; Bujalska, I.J.; Stewart, P.M.; Tomlinson, J.W.; Lavery, G.G. 11β-HSD1 is the major regulator of the tissue-specific effects of circulating glucocorticoid excess. Proc. Natl. Acad. Sci. USA 2014, 111, E2482–E2491. [Google Scholar] [CrossRef] [Green Version]
- Feig, P.U.; Shah, S.; Hermanowski-Vosatka, A.; Plotkin, D.; Springer, M.S.; Donahue, S.; Thach, C.; Klein, E.J.; Lai, E.; Kaufman, K.D. Effects of an 11beta-hydroxysteroid dehydrogenase type 1 inhibitor, MK-0916, in patients with type 2 diabetes mellitus and metabolic syndrome. Diabetes Obes. Metab. 2011, 13, 498–504. [Google Scholar] [CrossRef] [PubMed]
- Stefan, N.; Ramsauer, M.; Jordan, P.; Nowotny, B.; Kantartzis, K.; Machann, J.; Hwang, J.H.; Nowotny, P.; Kahl, S.; Harreiter, J.; et al. Inhibition of 11beta-HSD1 with RO5093151 for non-alcoholic fatty liver disease: A multicentre, randomised, double-blind, placebo-controlled trial. Lancet Diabetes Endocrinol. 2014, 2, 406–416. [Google Scholar] [CrossRef]
- Hardy, R.S.; Botfield, H.; Markey, K.; Mitchell, J.L.; Alimajstorovic, Z.; Westgate, C.S.J.; Sagmeister, M.; Fairclough, R.J.; Ottridge, R.S.; Yiangou, A.; et al. 11βHSD1 Inhibition with AZD4017 Improves Lipid Profiles and Lean Muscle Mass in Idiopathic Intracranial Hypertension. J. Clin. Endocrinol. Metab. 2021, 106, 174–187. [Google Scholar] [CrossRef]
- Ahasan, M.M.; Hardy, R.; Jones, C.; Kaur, K.; Nanus, D.; Juarez, M.; Morgan, S.A.; Hassan-Smith, Z.; Benezech, C.; Caamano, J.H.; et al. Inflammatory regulation of glucocorticoid metabolism in mesenchymal stromal cells. Arthritis Rheum. 2012, 64, 2404–2413. [Google Scholar] [CrossRef]
- Hardy, R.S.; Doig, C.L.; Hussain, Z.; O’Leary, M.; Morgan, S.A.; Pearson, M.J.; Naylor, A.; Jones, S.W.; Filer, A.; Stewart, P.M.; et al. 11β-Hydroxysteroid dehydrogenase type 1 within muscle protects against the adverse effects of local inflammation. J. Pathol. 2016, 240, 472–483. [Google Scholar] [CrossRef]
- Webster, J.M.; Kempen, L.; Hardy, R.S.; Langen, R.C.J. Inflammation and Skeletal Muscle Wasting During Cachexia. Front. Physiol. 2020, 11, 597675. [Google Scholar] [CrossRef] [PubMed]
- Fenton, C.G.; Webster, J.M.; Martin, C.S.; Fareed, S.; Wehmeyer, C.; Mackie, H.; Jones, R.; Seabright, A.P.; Lewis, J.W.; Lai, Y.C.; et al. Therapeutic glucocorticoids prevent bone loss but drive muscle wasting when administered in chronic polyarthritis. Arthritis Res. Ther. 2019, 21, 182. [Google Scholar] [CrossRef] [Green Version]
- Fenton, C.; Martin, C.; Jones, R.; Croft, A.; Campos, J.; Naylor, A.J.; Taylor, A.E.; Chimen, M.; Cooper, M.; Lavery, G.G.; et al. Local steroid activation is a critical mediator of the anti-inflammatory actions of therapeutic glucocorticoids. Ann. Rheum. Dis. 2021, 80, 250–260. [Google Scholar] [CrossRef]
- Jang, C.; Obeyesekere, V.R.; Alford, F.P.; Inder, W.J. Skeletal muscle 11β hydroxysteroid dehydrogenase type 1 activity is upregulated following elective abdominal surgery. Eur. J. Endocrinol. 2009, 160, 249–255. [Google Scholar] [CrossRef]
- Hardy, R.; Rabbitt, E.H.; Filer, A.; Emery, P.; Hewison, M.; Stewart, P.M.; Gittoes, N.J.; Buckley, C.D.; Raza, K.; Cooper, M.S. Local and systemic glucocorticoid metabolism in inflammatory arthritis. Ann. Rheum. Dis. 2007, 67, 1204–1210. [Google Scholar] [CrossRef]
- Nanus, D.E.; Filer, A.D.; Yeo, L.; Scheel-Toellner, D.; Hardy, R.; Lavery, G.G.; Stewart, P.M.; Buckley, C.D.; Tomlinson, J.W.; Cooper, M.S.; et al. Differential glucocorticoid metabolism in patients with persistent versus resolving inflammatory arthritis. Arthritis Res. Ther. 2015, 17, 121. [Google Scholar] [CrossRef] [Green Version]
- Sagmeister, M.S.; Taylor, A.E.; Fenton, A.; Wall, N.A.; Chanouzas, D.; Nightingale, P.G.; Ferro, C.J.; Arlt, W.; Cockwell, P.; Hardy, R.S.; et al. Glucocorticoid activation by 11β-hydroxysteroid dehydrogenase enzymes in relation to inflammation and glycaemic control in chronic kidney disease: A cross-sectional study. Clin. Endocrinol. 2019, 90, 241–249. [Google Scholar] [CrossRef]
- Stegk, J.P.; Ebert, B.; Martin, H.J.; Maser, E. Expression profiles of human 11beta-hydroxysteroid dehydrogenases type 1 and type 2 in inflammatory bowel diseases. Mol. Cell. Endocrinol. 2009, 301, 104–108. [Google Scholar] [CrossRef]
- Penner, G.; Gang, G.; Sun, X.; Wray, C.; Hasselgren, P.O. C/EBP DNA-binding activity is upregulated by a glucocorticoid-dependent mechanism in septic muscle. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2002, 282, R439–R444. [Google Scholar] [CrossRef]
- Zhang, G.; Jin, B.; Li, Y.-P. C/EBPβ mediates tumour-induced ubiquitin ligase atrogin1/MAFbx upregulation and muscle wasting. EMBO J. 2011, 30, 4323–4335. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamada, Y.; Tada, M.; Mandai, K.; Hidaka, N.; Inui, K.; Nakamura, H. Glucocorticoid use is an independent risk factor for developing sarcopenia in patients with rheumatoid arthritis: From the CHIKARA study. Clin. Rheumatol. 2020, 39, 1757–1764. [Google Scholar] [CrossRef] [PubMed]
- Dendoncker, K.; Timmermans, S.; Vandewalle, J.; Eggermont, M.; Lempiäinen, J.; Paakinaho, V.; Van Hamme, E.; Dewaele, S.; Vandevyver, S.; Ballegeer, M.; et al. TNF-α inhibits glucocorticoid receptor-induced gene expression by reshaping the GR nuclear cofactor profile. Proc. Natl. Acad. Sci. USA 2019, 116, 12942–12951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hassan-Smith, Z.K.; Morgan, S.A.; Sherlock, M.; Hughes, B.; Taylor, A.E.; Lavery, G.G.; Tomlinson, J.W.; Stewart, P.M. Gender-Specific Differences in Skeletal Muscle 11β-HSD1 Expression Across Healthy Aging. J. Clin. Endocrinol. Metab. 2015, 100, 2673–2681. [Google Scholar] [CrossRef] [Green Version]
- Kilgour, A.H.M.; Gallagher, I.J.; Maclullich, A.M.J.; Andrew, R.; Gray, C.D.; Hyde, P.; Wackerhage, H.; Husi, H.; Ross, J.A.; Starr, J.M.; et al. Increased Skeletal Muscle 11βHSD1 mRNA Is Associated with Lower Muscle Strength in Ageing. PLoS ONE 2013, 8, e84057. [Google Scholar] [CrossRef] [Green Version]
- Keffer, J.; Probert, L.; Cazlaris, H.; Georgopoulos, S.; Kaslaris, E.; Kioussis, D.; Kollias, G. Transgenic mice expressing human tumour necrosis factor: A predictive genetic model of arthritis. EMBO J. 1991, 10, 4025–4031. [Google Scholar] [CrossRef]
- Lewis, M.; Tartaglia, L.A.; Lee, A.; Bennett, G.L.; Rice, G.C.; Wong, G.H.; Chen, E.Y.; Goeddel, D.V. Cloning and expression of cDNAs for two distinct murine tumor necrosis factor receptors demonstrate one receptor is species specific. Proc. Natl. Acad. Sci. USA 1991, 88, 2830–2834. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kontoyiannis, D.; Pasparakis, M.; Pizarro, T.T.; Cominelli, F.; Kollias, G. Impaired On/Off Regulation of TNF Biosynthesis in Mice Lacking TNF AU-Rich Elements. Immunity 1999, 10, 387–398. [Google Scholar] [CrossRef] [Green Version]
- Naylor, A.J.; Desanti, G.; Saghir, A.N.; Hardy, R.S. TNFalpha depleting therapy improves fertility and animal welfare in TNFalpha-driven transgenic models of polyarthritis when administered in their routine breeding. Lab. Anim. 2018, 52, 59–68. [Google Scholar] [CrossRef] [Green Version]
- Rosenblatt, J.D.; Lunt, A.I.; Parry, D.J.; Partridge, T.A. Culturing satellite cells from living single muscle fiber explants. In Vitro Cell. Dev. Biol. Anim. 1995, 31, 773–779. [Google Scholar] [CrossRef] [PubMed]
- O’Leary, M.F.; Wallace, G.R.; Bennett, A.J.; Tsintzas, K.; Jones, S.W. IL-15 promotes human myogenesis and mitigates the detrimental effects of TNFalpha on myotube development. Sci. Rep. 2017, 7, 12997. [Google Scholar] [CrossRef]
- Rasband, W.S. ImageJ; National Institutes of Health: Bethesda, MD, USA, 2018. Available online: https://imagej.nih.gov/ij/ (accessed on 12 July 2021).
- Mutulsky, H. GraphPad Prism; GraphPad Software: San Diego, CA, USA, 2021; Available online: www.graphpad.com (accessed on 12 July 2021).


{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Patient Details | Patients with OA (n = 12) | Patients with RA (n = 10) | Group Comparison (p-Value) |
|---|---|---|---|
| Age (years) | 66.2 + 3.3 | 65.7 + 4.1 | 0.93 |
| CRP (mg/L) | 2.3 + 1.7 | 11.0 + 3.5 | 0.03 |
| ESR (mm/h) | 1.9 + 1.0 | 24.13 + 7.1 | 0.001 |
| Methotrexate (n) | 0 | 5 | na |
| Anti-TNF therapy (n) | 0 | 2 | na |
| Prednisolone | 0 | 0 | na |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Webster, J.M.; Sagmeister, M.S.; Fenton, C.G.; Seabright, A.P.; Lai, Y.-C.; Jones, S.W.; Filer, A.; Cooper, M.S.; Lavery, G.G.; Raza, K.; et al. Global Deletion of 11β-HSD1 Prevents Muscle Wasting Associated with Glucocorticoid Therapy in Polyarthritis. Int. J. Mol. Sci. 2021, 22, 7828. https://doi.org/10.3390/ijms22157828
Webster JM, Sagmeister MS, Fenton CG, Seabright AP, Lai Y-C, Jones SW, Filer A, Cooper MS, Lavery GG, Raza K, et al. Global Deletion of 11β-HSD1 Prevents Muscle Wasting Associated with Glucocorticoid Therapy in Polyarthritis. International Journal of Molecular Sciences. 2021; 22(15):7828. https://doi.org/10.3390/ijms22157828
Chicago/Turabian StyleWebster, Justine M., Michael S. Sagmeister, Chloe G. Fenton, Alex P. Seabright, Yu-Chiang Lai, Simon W. Jones, Andrew Filer, Mark S. Cooper, Gareth G. Lavery, Karim Raza, and et al. 2021. "Global Deletion of 11β-HSD1 Prevents Muscle Wasting Associated with Glucocorticoid Therapy in Polyarthritis" International Journal of Molecular Sciences 22, no. 15: 7828. https://doi.org/10.3390/ijms22157828
APA StyleWebster, J. M., Sagmeister, M. S., Fenton, C. G., Seabright, A. P., Lai, Y.-C., Jones, S. W., Filer, A., Cooper, M. S., Lavery, G. G., Raza, K., Langen, R., & Hardy, R. S. (2021). Global Deletion of 11β-HSD1 Prevents Muscle Wasting Associated with Glucocorticoid Therapy in Polyarthritis. International Journal of Molecular Sciences, 22(15), 7828. https://doi.org/10.3390/ijms22157828