
Mucus Release and Airway Constriction by TMEM16A May Worsen Pathology in Inflammatory Lung Disease

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
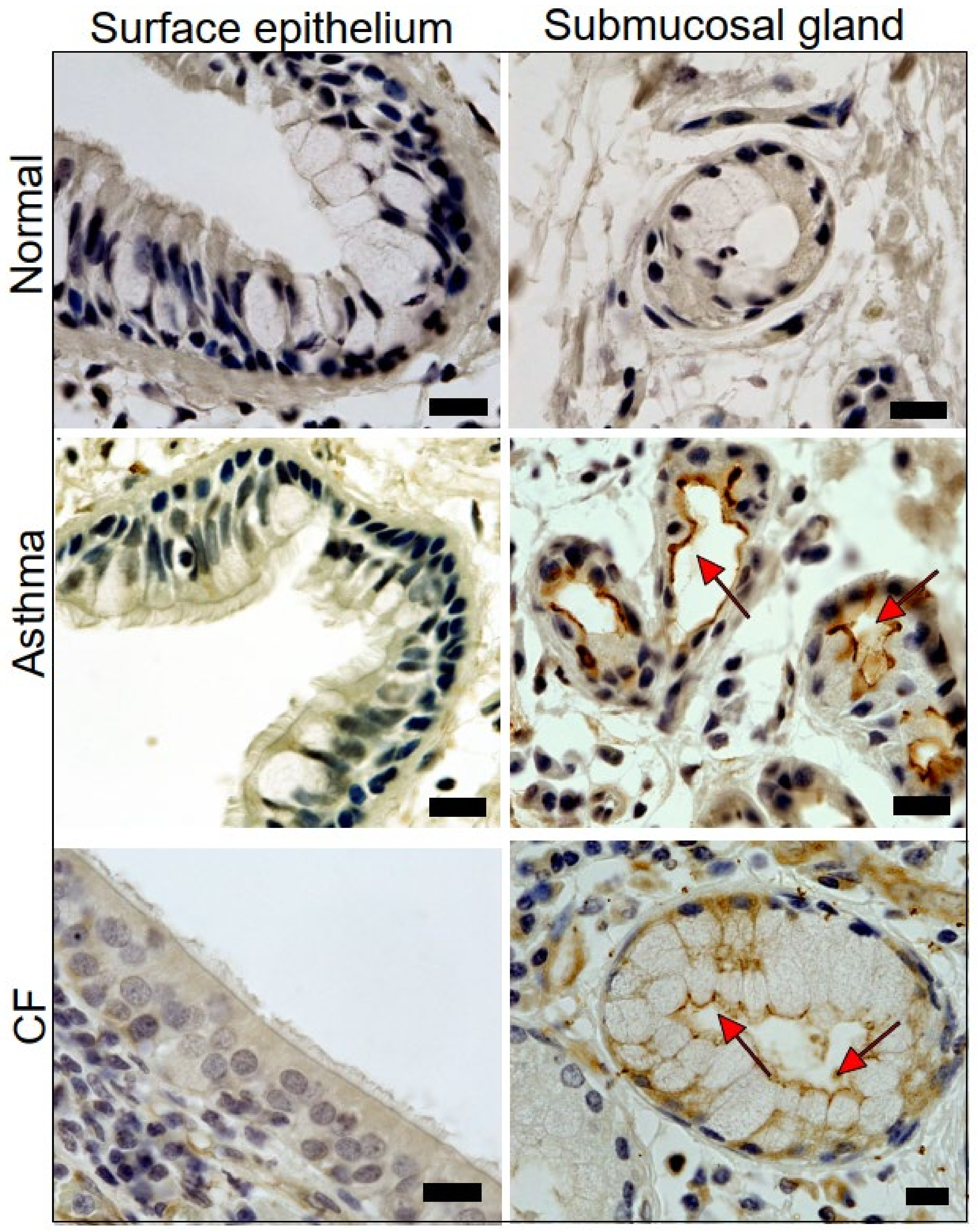
2.1. Expression of TMEM16A in Healthy, Asthmatic, and CF Lungs
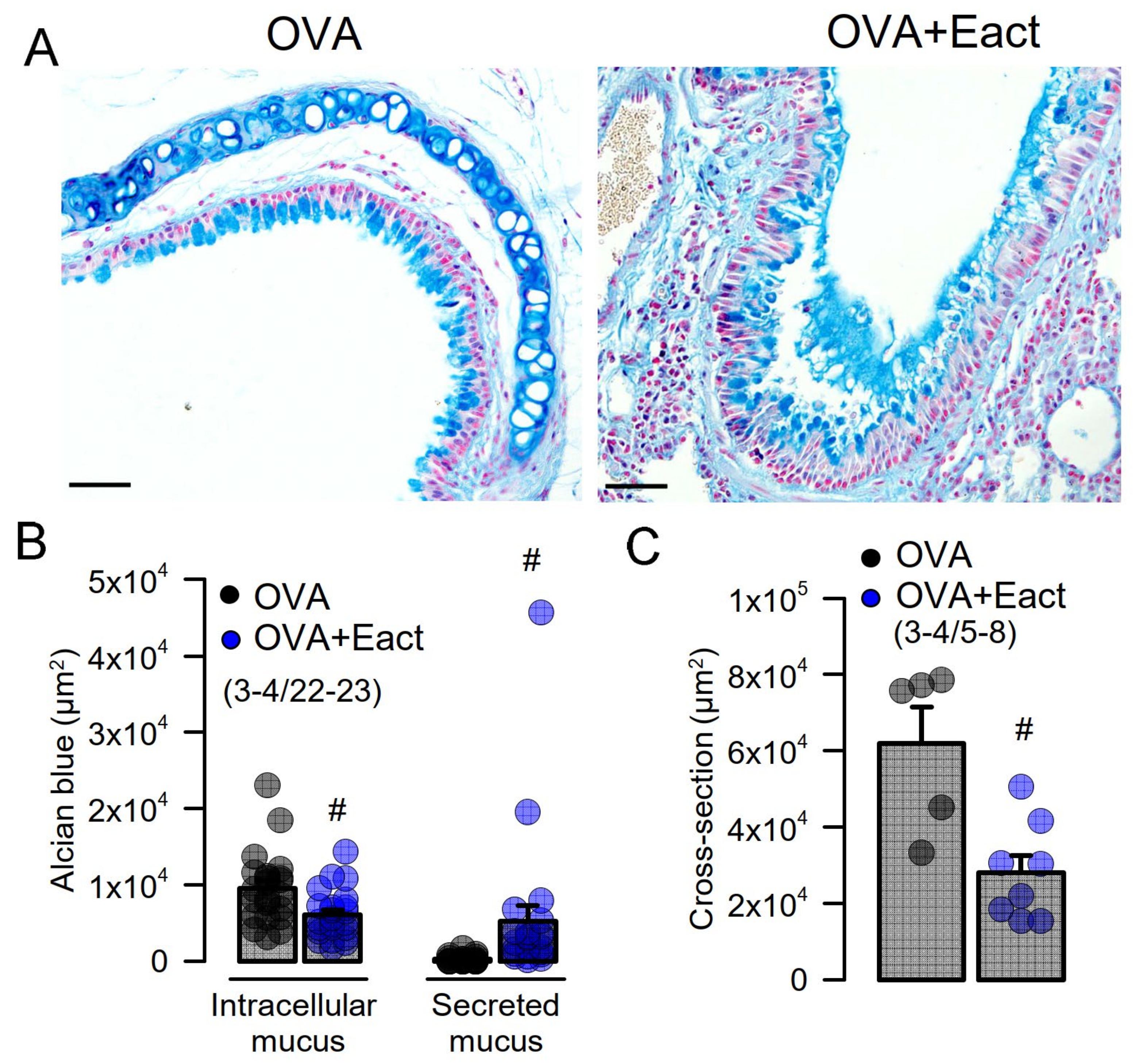
2.2. Acute Activation of TMEM16A by Eact Induces Mucus Release and Airway Constriction
2.3. Mucus Production and Mucus Secretion Is Strongly Inhibited by Niclosamide
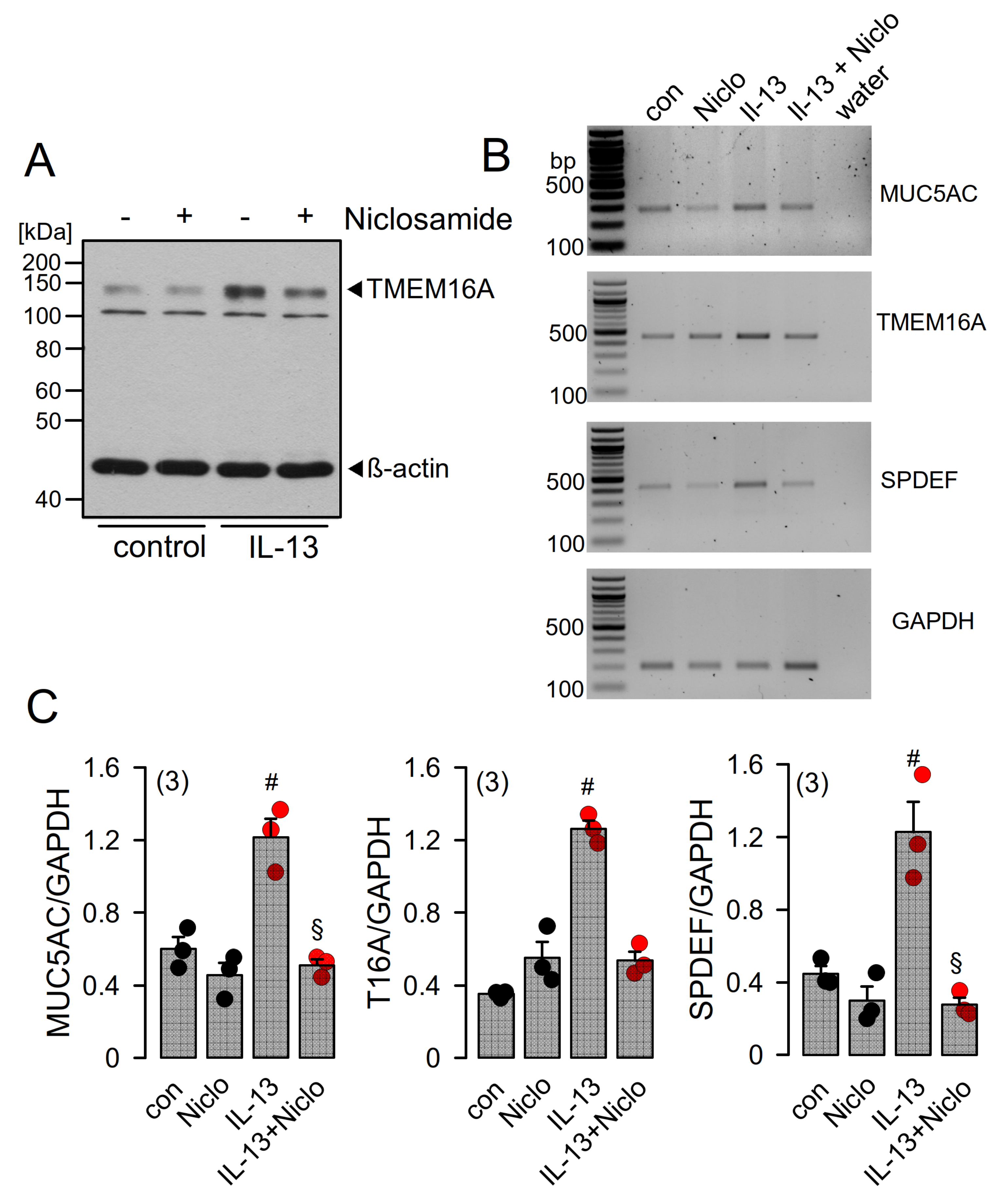
2.4. Expression of MUC5AC and Activation of TMEM16A in Calu-3 Cells Are Inhibited by Niclosamide
2.5. Niclosamide Inhibits Expression of TMEM16A, MUC5AC, and SPDEF in Calu-3 Cells
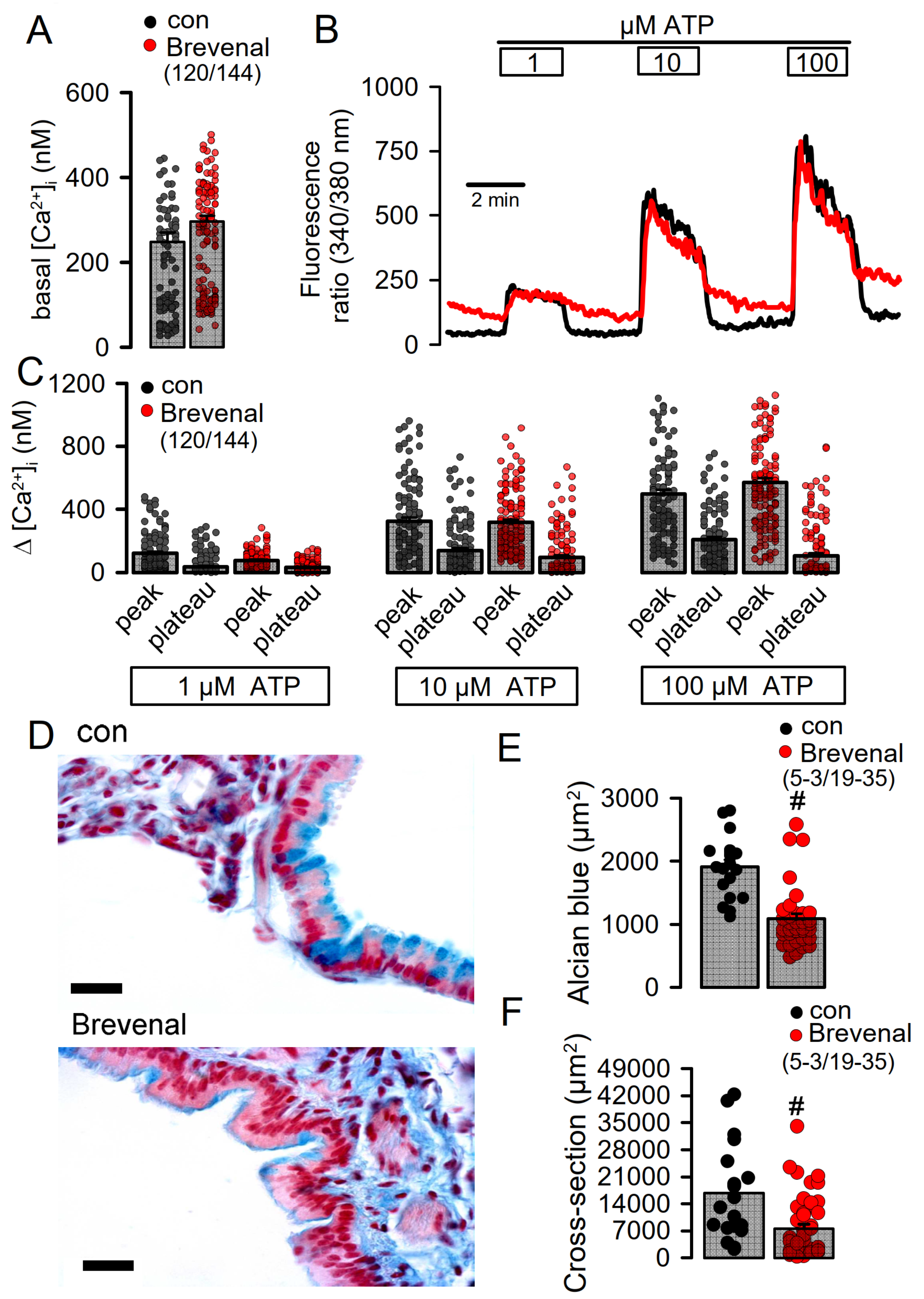
2.6. Potentiation of TMEM16A Whole-Cell Currents by Brevenal Released Airway Mucus and Caused Bronchoconstriction
3. Discussion
4. Materials and Methods
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Danahay, H.L.; Lilley, S.; Fox, R.; Charlton, H.; Sabater, J.; Button, B.; McCarthy, C.; Collingwood, S.P.; Gosling, M. TMEM16A potentiation: A novel therapeutic approach for the treatment of cystic fibrosis. Am. J. Respir. Crit. Care Med. 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knowles, M.R.; Clarke, L.L.; Boucher, R.C. Activation by extracellular nucleotides of chloride secretion in the airway epithelia of patients with cystic fibrosis. N. Engl. J. Med. 1991, 325, 533–538. [Google Scholar] [CrossRef] [PubMed]
- Mall, M.; Gonska, T.; Thomas, J.; Schreiber, R.; Seydewitz, H.H.; Kuehr, J.; Brandis, M.; Kunzelmann, K. Modulation of Ca2+ activated Cl− secretion by basolateral K+ channels in human normal and cystic fibrosis airway epithelia. Pediatr. Res. 2003, 53, 608–618. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Son, M.; Ito, Y.; Sato, S.; Ishikawa, T.; Kondo, M.; Nakayama, S.; Shimokata, K.; Kume, H. Apical and basolateral ATP-induced anion secretion in polarized human airway epithelia. Am. J. Respir. Cell Mol. Biol. 2004, 30, 411–419. [Google Scholar] [CrossRef] [PubMed]
- Kunzelmann, K.; Mall, M. Pharmacotherapy of the ion transport defect in cystic fibrosis: Role of purinergic receptor agonists and other potential therapeutics. Am. J. Respir. Med. 2003, 2, 299–309. [Google Scholar] [CrossRef]
- Ratjen, F.; Durham, T.; Navratil, T.; Schaberg, A.; Accurso, F.J.; Wainwright, C.; Barnes, M.; Moss, R.B. Long term effects of denufosol tetrasodium in patients with cystic fibrosis. J. Cyst. Fibros. 2012, 11, 539–549. [Google Scholar] [CrossRef] [Green Version]
- Eber, E.; Trawinska-Bartnicka, M.; Sands, D.; Bellon, G.; Mellies, U.; Bolbás, K.; Quattrucci, S.; Mazurek, H.; Widmann, R.; Schoergenhofer, C.; et al. Aerosolized lancovutide in adolescents (≥12 years) and adults with cystic fibrosis—A randomized trial. J. Cyst. Fibros. 2020. [Google Scholar] [CrossRef]
- Caci, E.; Scudieri, P.; Di Carlo, E.; Morelli, P.; Bruno, S.; De Fino, I.; Bragonzi, A.; Gianotti, A.; Sondo, E.; Ferrera, L.; et al. Upregulation of TMEM16A protein in bronchial epithelial cells by bacterial pyocyanin. PLoS ONE 2015, 10, e0131775. [Google Scholar] [CrossRef]
- Miner, K.; Labitzke, K.; Liu, B.; Elliot, R.; Wang, P.; Henckels, K.; Gaida, K.; Elliot, R.; Chen, J.J.; Liu, L.; et al. Drug repurposing: The anthelmintics niclosamide and nitazoxanide are potent TMEM16A antagonists that fully bronchodilate airways. Front. Pharmacol. 2019, 10, 51. [Google Scholar] [CrossRef] [Green Version]
- Benedetto, R.; Ousingsawat, J.; Wanitchakool, P.; Zhang, Y.; Holtzman, M.J.; Amaral, M.; Rock, J.R.; Schreiber, R.; Kunzelmann, K. Epithelial chloride transport by CFTR requires TMEM16A. Sci. Rep. 2017, 7, 12397. [Google Scholar] [CrossRef] [Green Version]
- Benedetto, R.; Centeio, R.; Ousingsawat, J.; Schreiber, R.; Janda, M.; Kunzelmann, K. Transport properties in CFTR-/- knockout piglets suggest normal airway surface liquid pH and enhanced amiloride-sensitive Na+ absorption. Pflugers Arch. Eur. J. Physiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Kondo, M.; Tsuji, M.; Hara, K.; Arimura, K.; Yagi, O.; Tagaya, E.; Takeyama, K.; Tamaoki, J. Chloride ion transport and overexpression of TMEM16A in a guinea pig asthma model. Clin. Exp. Allergy 2017, 47, 795–804. [Google Scholar] [CrossRef] [PubMed]
- Okada, S.F.; Zhang, L.; Kreda, S.M.; Abdullah, L.H.; Davis, C.W.; Pickles, R.J.; Lazarowski, E.R.; Boucher, R.C. Coupled nucleotide and mucin hypersecretion from goblet-cell metaplastic human airway epithelium. Am. J. Respir. Cell Mol. Biol. 2011, 45, 253–260. [Google Scholar] [CrossRef] [PubMed]
- Davis, C.W.; Dickey, B.F. Regulated airway goblet cell mucin secretion. Annu. Rev. Physiol. 2008, 70, 487–512. [Google Scholar] [CrossRef]
- Olivier, K.N.; Bennett, W.D.; Hohneker, K.W.; Zeman, K.L.; Edwards, L.J.; Boucher, R.C.; Knowles, M.R. Acute safety and effects on mucociliary clearance of aerosolized uridine 5′-triphosphate +/− amiloride in normal human adults. Am. J. Respir. Crit. Care Med. 1996, 154, 217–223. [Google Scholar] [CrossRef]
- Bennett, W.D.; Olivier, K.N.; Zeman, K.L.; Hohneker, K.W.; Boucher, R.C.; Knowles, M.R. Effect of uridine 5′-triphosphate plus amiloride on mucociliary clearance in adult cystic fibrosis. Am. J. Respir. Crit. Care Med. 1996, 153, 1796–1801. [Google Scholar] [CrossRef]
- Danahay, H.; Fox, R.; Lilley, S.; Charlton, H.; Adley, K.; Christie, L.; Ansari, E.; Ehre, C.; Flen, A.; Tuvim, M.J.; et al. Potentiating TMEM16A does not stimulate airway mucus secretion or bronchial and pulmonary arterial smooth muscle contraction. FASEB BioAdv. 2020, 2, 464–477. [Google Scholar] [CrossRef]
- Scudieri, P.; Caci, E.; Bruno, S.; Ferrera, L.; Schiavon, M.; Sondo, E.; Tomati, V.; Gianotti, A.; Zegarra-Moran, O.; Pedemonte, N.; et al. Association of TMEM16A chloride channel overexpression with airway goblet cells metaplasia. J. Physiol. 2012, 590, 6141–6155. [Google Scholar] [CrossRef]
- Huang, F.; Zhang, H.; Wu, M.; Yang, H.; Kudo, M.; Peters, C.J.; Woodruff, P.G.; Solberg, O.D.; Donne, M.L.; Huang, X.; et al. Calcium-activated chloride channel TMEM16A modulates mucin secretion and airway smooth muscle contraction. Proc. Natl. Acad. Sci. USA 2012, 109, 16354–16359. [Google Scholar] [CrossRef] [Green Version]
- Lin, J.; Jiang, Y.; Li, L.; Liu, Y.; Tang, H.; Jiang, D. TMEM16A mediates the hypersecretion of mucus induced by Interleukin-13. Exp. Cell Res. 2015, 334, 260–269. [Google Scholar] [CrossRef]
- Benedetto, R.; Cabrita, I.; Schreiber, R.; Kunzelmann, K. TMEM16A is indispensable for basal mucus secretion in airways and intestine. FASEB J. 2019, 33, 4502–4512. [Google Scholar] [CrossRef] [PubMed]
- Zhang, C.H.; Li, Y.; Zhao, W.; Lifshitz, L.M.; Li, H.; Harfe, B.D.; Zhu, M.S.; ZhuGe, R. The transmembrane protein 16A Ca2+-activated Cl− channel in airway smooth muscle contributes to airway hyperresponsiveness. Am. J. Respir. Crit. Care Med. 2013, 187, 374–381. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danielsson, J.; Perez-Zoghbi, J.; Bernstein, K.; Barajas, M.B.; Zhang, Y.; Kumar, S.; Sharma, P.K.; Gallos, G.; Emala, C.W. Antagonists of the TMEM16A calcium-activated chloride channel modulate airway smooth muscle tone and intracellular calcium. Anesthesiology 2015, 123, 569–581. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danielsson, J.; Kuforiji, A.S.; Yocum, G.T.; Zhang, Y.; Xu, D.; Gallos, G.; Emala, C.W.S. Agonism of the TMEM16A calcium-activated chloride channel modulates airway smooth muscle tone. Am. J. Physiol. Lung Cell. Mol. Physiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Kent, B.D.; Lane, S.J.; van Beek, E.J.; Dodd, J.D.; Costello, R.W.; Tiddens, H.A. Asthma and cystic fibrosis: A tangled web. Pediatr. Pulmonol. 2014, 49, 205–213. [Google Scholar] [CrossRef] [PubMed]
- Sun, H.; Xia, Y.; Paudel, O.; Yang, X.R.; Sham, J.S. Chronic hypoxia-induced upregulation of Ca2+-activated Cl− channel in pulmonary arterial myocytes: A mechanism contributing to enhanced vasoreactivity. J. Physiol. 2012, 590, 3507–3521. [Google Scholar] [CrossRef]
- Forrest, A.S.; Joyce, T.C.; Huebner, M.L.; Ayon, R.J.; Wiwchar, M.; Joyce, J.; Freitas, N.; Davis, A.J.; Ye, L.; Duan, D.D.; et al. Increased TMEM16A-encoded calcium-activated chloride channel activity is associated with pulmonary hypertension. Am. J. Physiol. Cell Physiol. 2012, 303, C1229–C1243. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allawzi, A.M.; Vang, A.; Clements, R.T.; Jhun, B.S.; Kue, N.R.; Mancini, T.J.; Landi, A.K.; Terentyev, D.; O-Uchi, J.; Comhair, S.A.; et al. Activation of anoctamin-1 limits pulmonary endothelial cell proliferation via p38-MAPK-dependent apoptosis. Am. J. Respir. Cell Mol. Biol. 2017. [Google Scholar] [CrossRef]
- Papp, R.; Nagaraj, C.; Zabini, D.; Nagy, B.M.; Lengyel, M.; Maurer, D.S.; Sharma, N.; Egemnazarov, B.; Kovacs, G.; Kwapiszewska, G.; et al. Targeting TMEM16A to reverse vasoconstriction and remodelling in idiopathic PAH. Eur. Respir. J. 2019. [Google Scholar] [CrossRef] [PubMed]
- Wells, J.M.; Farris, R.F.; Gosdin, T.A.; Dransfield, M.T.; Wood, M.E.; Bell, S.C.; Rowe, S.M. Pulmonary artery enlargement and cystic fibrosis pulmonary exacerbations: A cohort study. Lancet Respir. Med. 2016, 4, 636–645. [Google Scholar] [CrossRef] [Green Version]
- Zouk, A.N.; Gulati, S.; Xing, D.; Wille, K.M.; Rowe, S.M.; Wells, J.M. Pulmonary artery enlargement is associated with pulmonary hypertension and decreased survival in severe cystic fibrosis: A cohort study. PLoS ONE 2020, 15, e0229173. [Google Scholar] [CrossRef] [Green Version]
- Namkung, W.; Yao, Z.; Finkbeiner, W.E.; Verkman, A.S. Small-molecule activators of TMEM16A, a calcium-activated chloride channel, stimulate epithelial chloride secretion and intestinal contraction. FASEB J. 2011, 25, 4048–4062. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Centeio, R.; Cabrita, I.; Benedetto, R.; Talbi, K.; Ousingsawat, J.; Schreiber, R.; Sullivan, J.K.; Kunzelmann, K. Pharmacological inhibition and activation of the Ca2+ activated Cl− channel TMEM16A. Int. J. Mol. Sci. 2020, 21, 2557. [Google Scholar] [CrossRef] [Green Version]
- Genovese, M.; Borrelli, A.; Venturini, A.; Guidone, D.; Caci, E.; Viscido, G.; Gambardella, G.; di Bernardo, D.; Scudieri, P.; Galietta, L.J.V. TRPV4 and purinergic receptor signalling pathways are separately linked in airway epithelia to CFTR and TMEM16A chloride channels. J. Physiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Cabrita, I.; Benedetto, R.; Schreiber, R.; Kunzelmann, K. Niclosamide repurposed for the treatment of inflammatory airway disease. JCI Insight 2019, 8, 128414. [Google Scholar] [CrossRef]
- Chua, R.L.; Lukassen, S.; Trump, S.; Hennig, B.P.; Wendisch, D.; Pott, F.; Debnath, O.; Thürmann, L.; Kurth, F.; Völker, M.T.; et al. COVID-19 severity correlates with airway epithelium-immune cell interactions identified by single-cell analysis. Nat. Bitechnol. 2020, 38, 970–979. [Google Scholar] [CrossRef] [PubMed]
- Cabrita, I.; Benedetto, R.; Wanitchakool, P.; Lerias, J.; Centeio, R.; Ousingsawat, J.; Schreiber, R.; Kunzelmann, K. TMEM16A mediated mucus production in human airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Cabrita, I.; Kraus, A.; Scholz, J.K.; Skoczynski, K.; Schreiber, R.; Kunzelmann, K.; Buchholz, B. Cyst growth in ADPKD is prevented by pharmacological and genetic inhibition of TMEM16A In Vivo. Nat. Commun. 2020, 11, 4320. [Google Scholar] [CrossRef]
- Rajavelu, P.; Chen, G.; Xu, Y.; Kitzmiller, J.A.; Korfhagen, T.R.; Whitsett, J.A. Airway epithelial SPDEF integrates goblet cell differentiation and pulmonary Th2 inflammation. J. Clin. Investig. 2015, 125, 2021–2031. [Google Scholar] [CrossRef] [Green Version]
- Chen, G.; Korfhagen, T.R.; Xu, Y.; Kitzmiller, J.; Wert, S.E.; Maeda, Y.; Gregorieff, A.; Clevers, H.; Whitsett, J.A. SPDEF is required for mouse pulmonary goblet cell differentiation and regulates a network of genes associated with mucus production. J. Clin. Investig. 2009, 119, 2914–2924. [Google Scholar] [CrossRef] [Green Version]
- Park, K.S.; Korfhagen, T.R.; Bruno, M.D.; Kitzmiller, J.A.; Wan, H.; Wert, S.E.; Khurana Hershey, G.K.; Chen, G.; Whitsett, J.A. SPDEF regulates goblet cell hyperplasia in the airway epithelium. J. Clin. Investig. 2007, 117, 978–988. [Google Scholar] [CrossRef]
- Abraham, W.M.; Bourdelais, A.J.; Sabater, J.R.; Ahmed, A.; Lee, T.A.; Serebriakov, I.; Baden, D.G. Airway responses to aerosolized brevetoxins in an animal model of asthma. Am. J. Respir. Crit. Care Med. 2005, 171, 26–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fatima, N.; Cohen, C.; Siddiqui, M.T. DOG1 utility in diagnosing gastrointestinal stromal tumors on fine-needle aspiration. Cancer Cytopathol. 2011, 119, 202–208. [Google Scholar] [CrossRef]
- Ballard, S.T.; Fountain, J.D.; Inglis, S.K.; Corboz, M.R.; Taylor, A.E. Chloride secretion across distal airway epithelium: Relationship to submucosal gland distribution. Am. J. Physiol. 1995, 268, L526–L531. [Google Scholar] [CrossRef] [PubMed]
- Finkbeiner, W.E.; Zlock, L.T.; Morikawa, M.; Lao, A.Y.; Dasari, V.; Widdicombe, J.H. Cystic fibrosis and the relationship between mucin and chloride secretion by cultures of human airway gland mucous cells. Am. J. Physiol. Lung Cell. Mol. Physiol. 2011, 301, L402–L414. [Google Scholar] [CrossRef] [Green Version]
- Lerias, J.; Pinto, M.; Benedetto, R.; Schreiber, R.; Amaral, M.; Aureli, M.; Kunzelmann, K. Compartmentalized crosstalk of CFTR and TMEM16A (ANO1) through EPAC1 and ADCY1. Cell. Signal. 2018, 44, 10–19. [Google Scholar] [CrossRef]
- Matusovsky, O.S.; Kachmar, L.; Ijpma, G.; Panariti, A.; Benedetti, A.; Martin, J.G.; Lauzon, A.M. Contractile properties of intrapulmonary airway smooth muscle in cystic fibrosis. Am. J. Respir. Cell Mol. Biol. 2019, 60, 434–444. [Google Scholar] [CrossRef] [PubMed]
- McCuaig, S.; Martin, J.G. How the airway smooth muscle in cystic fibrosis reacts in proinflammatory conditions: Implications for airway hyper-responsiveness and asthma in cystic fibrosis. Lancet Respir. Med. 2013, 1, 137–147. [Google Scholar] [CrossRef]
- Kunzelmann, K.; Ousingsawat, J.; Cabrita, I.; Doušová, T.; Bähr, A.; Janda, M.; Schreiber, R.; Benedetto, R. TMEM16A in cystic fibrosis: Activating or inhibiting? Front. Pharmacol. 2019, 10, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duran, C.; Thompson, C.H.; Xiao, Q.; Hartzell, H.C. Chloride channels: Often enigmatic, rarely predictable. Annu. Rev. Physiol. 2010, 17, 95–121. [Google Scholar] [CrossRef] [Green Version]
- Keeler, D.M.; Grandal, M.K.; McCall, J.R. Brevenal, a marine natural product, is anti-inflammatory and an immunomodulator of macrophage and lung epithelial cells. Mar. Drugs 2019, 17, 184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hammer, C.; Wanitchakool, P.; Sirianant, L.; Papiol, S.; Monnheimer, M.; Faria, D.; Ousingsawat, J.; Schramek, N.; Schmitt, C.; Margos, G.; et al. A coding variant of ANO10, affecting volume regulation of macrophages, is associated with Borrelia seropositivity. Mol. Med. 2015, 21, 26–37. [Google Scholar] [CrossRef] [PubMed]
- Perez, F.J.; Iturra, P.A.; Ponce, C.A.; Magne, F.; Garcia-Angulo, V.; Vargas, S.L. Niflumic acid reverses airway mucus excess and improves survival in the rat model of steroid-induced Pneumocystis pneumonia. Front. Microbiol. 2019, 10, 1522. [Google Scholar] [CrossRef] [PubMed]
- Kunzelmann, K. Getting hands on a drug for Covid-19: Inhaled and intranasal Niclosamide. Lancet Reg. Health Eur. 2021. [Google Scholar] [CrossRef]
- Schreiber, R.; Castrop, H.; Kunzelmann, K. Allergen induced airway hyperresponsiveness is absent in ecto-5′-nucleotidase (CD73) deficient mice. Pflugers Arch. Eur. J. Physiol. 2008, 457, 431–440. [Google Scholar] [CrossRef]
- Martins, J.R.; Faria, D.; Kongsuphol, P.; Reisch, B.; Schreiber, R.; Kunzelmann, K. Anoctamin 6 is an essential component of the outwardly rectifying chloride channel. Proc. Natl. Acad. Sci. USA 2011, 108, 18168–18172. [Google Scholar] [CrossRef] [Green Version]
- Grynkiewicz, G.; Poenie, M.; Tsien, R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985, 260, 3440–3450. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TMEM16A | forward: 5′-CGACTACGTGTACATTTTCCG reverse: 5′-GATTCCGATGTCTTTGGCTC | 445 bp |
| MUC5AC | forward: 5′-GCTCAGCTGTTCTCTGGACG reverse: 5′-GTCACATTCCTCAGCGAGGTC | 279 bp |
| SPDF | forward: 5′-GTGCTCAAGGACATCGAGAC reverse: 5′-CCTAATGAAGCGGCCATAGC | 423 bp |
| GAPDH | forward: 5′-GTATTGGGCGCCTGGTCAC reverse: 5′-CTCCTGGAAGATGGTGATGG | 200 bp |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Centeio, R.; Ousingsawat, J.; Cabrita, I.; Schreiber, R.; Talbi, K.; Benedetto, R.; Doušová, T.; Verbeken, E.K.; De Boeck, K.; Cohen, I.; et al. Mucus Release and Airway Constriction by TMEM16A May Worsen Pathology in Inflammatory Lung Disease. Int. J. Mol. Sci. 2021, 22, 7852. https://doi.org/10.3390/ijms22157852
Centeio R, Ousingsawat J, Cabrita I, Schreiber R, Talbi K, Benedetto R, Doušová T, Verbeken EK, De Boeck K, Cohen I, et al. Mucus Release and Airway Constriction by TMEM16A May Worsen Pathology in Inflammatory Lung Disease. International Journal of Molecular Sciences. 2021; 22(15):7852. https://doi.org/10.3390/ijms22157852
Chicago/Turabian StyleCenteio, Raquel, Jiraporn Ousingsawat, Inês Cabrita, Rainer Schreiber, Khaoula Talbi, Roberta Benedetto, Tereza Doušová, Eric K. Verbeken, Kris De Boeck, Isaac Cohen, and et al. 2021. "Mucus Release and Airway Constriction by TMEM16A May Worsen Pathology in Inflammatory Lung Disease" International Journal of Molecular Sciences 22, no. 15: 7852. https://doi.org/10.3390/ijms22157852
APA StyleCenteio, R., Ousingsawat, J., Cabrita, I., Schreiber, R., Talbi, K., Benedetto, R., Doušová, T., Verbeken, E. K., De Boeck, K., Cohen, I., & Kunzelmann, K. (2021). Mucus Release and Airway Constriction by TMEM16A May Worsen Pathology in Inflammatory Lung Disease. International Journal of Molecular Sciences, 22(15), 7852. https://doi.org/10.3390/ijms22157852