Abstract
Activation of microglia and/or astrocytes often releases proinflammatory molecules as critical pathogenic mediators that can promote neuroinflammation and secondary brain damages in diverse diseases of the central nervous system (CNS). Therefore, controlling the activation of glial cells and their neuroinflammatory responses has been considered as a potential therapeutic strategy for treating neuroinflammatory diseases. Recently, receptor-mediated lysophospholipid signaling, sphingosine 1-phosphate (S1P) receptor- and lysophosphatidic acid (LPA) receptor-mediated signaling in particular, has drawn scientific interest because of its critical roles in pathogenies of diverse neurological diseases such as neuropathic pain, systemic sclerosis, spinal cord injury, multiple sclerosis, cerebral ischemia, traumatic brain injury, hypoxia, hydrocephalus, and neuropsychiatric disorders. Activation of microglia and/or astrocytes is a common pathogenic event shared by most of these CNS disorders, indicating that lysophospholipid receptors could influence glial activation. In fact, many studies have reported that several S1P and LPA receptors can influence glial activation during the pathogenesis of cerebral ischemia and multiple sclerosis. This review aims to provide a comprehensive framework about the roles of S1P and LPA receptors in the activation of microglia and/or astrocytes and their neuroinflammatory responses in CNS diseases.
1. Introduction
Glial cells are non-neuronal central nervous system (CNS)-resident cells that support neurons for CNS homeostasis and normal neuronal functioning in a healthy condition [1]. Abundantly distributed in the CNS, glial cells are in continuous communication with neurons, immune cells, and blood vessels [2,3]. They are primarily divided into four phenotypes, namely, astrocytes, oligodendrocytes, microglia, and ependymal cells [1]. Ependymal cells are present at the lining of the ventricular system. Their roles during CNS injury remain unclear. Oligodendrocytes are myelin-producing cells that play important roles in myelin-degenerating diseases such as multiple sclerosis (MS), neuromyelitis optica, and idiopathic inflammatory demyelinating diseases [4]. Microglia and astrocytes are considered major glial cell types that are critical regulators of brain injury and recovery in diverse neuroinflammatory disorders. Microglia are innate immune cells of the CNS that play important roles in the host defense [5,6]. Astrocytes are in close proximity to neurons and blood vasculatures [7]. Both microglia and astrocytes are the most motile and active glial cells in the CNS. They can sense any changes in the CNS milieu through their processes. Therefore, microglia and astrocytes are the primary cells to be activated upon any hazardous stimuli [8,9]. Activation of microglia and astrocytes in acute CNS injuries is necessary for the host defense [10]. For instance, activated microglia are involved in brain cleaning system as they can remove dead cells and tissue debris [11], whereas activated astrocytes can prevent neurodegeneration through the formation of glial scar [12]. However, chronic activation of microglia and astrocytes are considered as detrimental because these activated cells can promote neuroinflammatory events and ultimately lead to neurodegeneration [13].
Bioactive lysophospholipids and their receptors (lysophospholipid receptors) are believed to be potential targets for drug design to treat various diseases, which ensures the possibility for an entirely new class of lipidomic-based therapeutic agents [14,15]. Originally, lysophospholipids were thought to be precursors and metabolites in the biosynthesis of membrane phospholipids [16,17,18]. Later, two of them, lysophosphatidic acid (LPA) and sphingosine 1-phosphate (S1P), were identified as important extracellular signaling molecules that could participate in various biological functions in organisms, including immunity, inflammation, muscle contraction, development, fibrosis, obesity, cancer, angiogenesis, cellular migration, and neurite extension [17,19,20,21]. Being extracellular signaling molecules, LPA and S1P can signal through binding to and activating at least 11 cognate G protein-coupled receptors (six LPA receptors, LPA1–LPA6 and five S1P receptors, S1P1–S1P5) and mediate a variety of biological functions throughout the body [22,23,24]. LPA and S1P are abundantly present in the CNS, where receptor-mediated LPA and S1P signaling are believed to play crucial roles in neurological disorders involving neuroinflammation, the major cause of neurodegeneration [25,26,27,28]. In fact, LPA and S1P receptors have emerged as novel and fascinating therapeutic targets for several inflammatory CNS diseases, including MS, neuropathic pain, spinal cord injury, cerebral ischemia, traumatic brain injury, hydrocephalus, fetal hypoxia, seizure, hearing loss, Sandhoff disease, and neuropsychiatric disorders [17,20]. In most of these CNS diseases, neuroinflammatory responses initiated by activated microglia and astrocytes are considered major neuroharmful events. In fact, previous studies have revealed that these neuroglial cells express most LPA and S1P receptors [29,30,31,32,33]. Therefore, LPA and S1P receptors can influence glial cell biology, including activation, proliferation, migration, etc. In addition, LPA and S1P receptors can regulate inflammatory responses of microglia and astrocytes, consequently signifying that these receptors are potential drug targets for treating various neuroinflammatory disorders. The aim of this review is to explore the roles of receptor-mediated LPA and S1P signaling in neuroinflammation through the regulation of responses of microglia and astrocytes.
2. S1P Receptors in Activation of Microglia and Their Neuroinflammatory Responses
A growing body of evidence has revealed the involvement of S1P receptors in microglial biology in diverse neuroinflammatory disorders. For example, it has been suggested that S1P might be involved in the migration and morphological alteration of microglia [34,35]. In cultured microglia, S1P can influence ATP release via volume-regulated anion channels [35]. The released ATP is associated with the migration of microglia [35]. S1P lyase-deficient microglia, in which amounts of S1P are found to be increased, show a significant decrease in ramification index of microglia (a morphological phenotype indicating microglial activation), along with increased ionized calcium-binding adapter molecule 1 (Iba1) reactivity [34], suggesting that S1P can promote microglial activation. In addition, mRNA and protein expression levels of proinflammatory mediators (such as tumor necrosis factor-α (TNF-α) and interleukin-6 (IL-6)) and mRNA expression levels of Toll-like receptor 4 (TLR4) are significantly increased in S1P lyase-deficient microglia [34], demonstrating that increased S1P levels are associated with microglial activation and their proinflammatory responses. In mice brains, microinjection of S1P into the corpus callosum can dramatically increase microglial activation, as evidenced by an increased number of Iba1-positive cells [27]. Such an S1P-induced microglial activation is closely associated with increased brain damage upon induction of transient middle cerebral artery occlusion (tMCAO), a type of focal cerebral ischemia [27]. Similarly, in microglia subjected to oxygen–glucose deprivation/re-oxygenation (OGD/R), a popular in vitro model of cerebral ischemia [36,37], S1P exposure can increase the production of IL-17A, which is associated with neuronal apoptosis [38]. Besides S1P itself, sphingosine kinase 1 (SphK1), a principal kinase responsible for S1P production in the brain [39], is also a key regulator of microglial activation and subsequent production of inflammatory mediators [40]. Protein expression levels of SphK1 are upregulated in lipopolysaccharide (LPS)-stimulated BV2 microglia cell line [40]. Suppression of SphK1 activity with either pharmacological or genetic tools can attenuate the upregulation of inflammatory mediators such as TNF-α, IL-1β, and inducible nitric oxide synthase (iNOS) [40], further indicating the role of S1P in neuroinflammatory activation of microglia.
The involvement of S1P receptors in microglial activation and their inflammatory responses has been further validated through experimental studies employing FTY720 (fingolimod, Gilenya®, Novartis, Switzerland), a drug to treat MS. FTY720 is a non-selective modulator of S1P receptors [41,42] with a pharmacological potential as a functional antagonist of S1P1 [43,44,45] and S1P3 [46]. It can decrease microglial activation in diverse CNS diseases, including Alzheimer’s disease (AD), Parkinson’s disease (PD), cerebral ischemia, and MS [47]. FTY720 can significantly attenuate mRNA and protein expression levels of proinflammatory cytokines such as TNF-α, IL-1β, and IL-6 in LPS-stimulated mouse primary microglia [48]. In addition, it can decrease lysophosphatidylcholine-induced production of nitric oxide (NO), TNF-α, and IL-1β from microglia [49]. In a kainic acid-induced neurodegenerative mouse model, FTY720 can significantly decrease the number of Iba1-positive cells in the brain and attenuate JNK phosphorylation in LPS-stimulated microglia [50]. In mice exposed to chronic unpredictable mild stress, FTY720 can decrease depressive-like behaviors by attenuating hippocampal NOD-like receptor pyrin domain-containing protein 3 (NLRP3) inflammasome activation [51], which is believed to occur in microglia because microglia are central regulators of NLRP3 inflammasome activation in the brain [52]. FTY720 decreases mRNA expression levels of iNOS and CD16, but increases mRNA expression levels of arginase 1 (Arg-1) and CD206, suggesting that FTY720 can promote M2 polarization of activated microglia (anti-inflammatory/neuroprotective phenotype) while attenuating M1 polarization (proinflammatory/neuro-harmful phenotype) [51]. FTY720 and SEW2871 (a selective agonist for S1P1 [53]) can significantly attenuate microglial activation in the substantia nigra of 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced PD mouse [54], further indicating the role of S1P receptors in microglial biology. In an ischemic stroke-challenged brain, FTY720 can attenuate microglial activation and M1 polarization as evidenced by attenuated protein expression levels of Iba1, iNOS, and NLRP3 [55]. The effect of FTY720 on microglial polarization in cerebral ischemia is further confirmed in OGD/R-challenged microglia, in which FTY720 treatment decreases mRNA expression levels of M1 polarization markers such as CD86, cyclooxygenase-2 (COX-2), iNOS, IL-1β, IL-6, TNF-α, and interferon-γ (IFN-γ), whereas it increases mRNA expression levels of M2 polarization markers and growth factors such as transforming growth factor-β1 (TGF-β1), TGF-β2, TGF-β3, C-C motif chemokine ligand 2 (CCL2), granulocyte colony-stimulating factor (GCSF), granulocyte-macrophage colony-stimulating factor (GMCSF), and insulin-like growth factor-1α (IGF-1α) [55]. FTY720 can decrease mRNA expression levels of proinflammatory cytokines and chemokines such as IL-1α, IL-1β, TNF-α, IL-6, CCL2, CCL3, CCL4, and CCL9 in LPS-stimulated primary microglial culture [56]. In addition, FTY720 treatment can significantly decrease the number of activated microglia at the proximity of beta-amyloid (Aβ) plaque in AD-prone transgenic mice [57]. Similarly, following a hypoxic/ischemic insult in mice, FTY720 treatment can attenuate microglial activation and upregulation of proinflammatory cytokines such as TNF-α and IL-1β at protein and mRNA expression levels in the white matter of the brain [58]. FTY720 can also attenuate microglial activation in twitcher mice as numbers of Iba1-positive cells and ameboid microglia in the white matter of the cerebellum are decreased after treatment with FTY720 [59,60]. Findings of these in vitro and in vivo studies suggest that FTY720 can promote the anti-inflammatory phenotype and reduce the proinflammatory phenotype of microglia.
Besides FTY720, a few other S1P receptor modulators can also influence microglial activation, further strengthening the notion that receptor-mediated S1P signaling plays an important role in the activation of microglia and their inflammatory responses. In the culture of corticostriatal slices obtained from mice with experimental autoimmune encephalomyelitis (EAE) as a popular animal model of MS [61,62], ozanimod (RPC1063, Zeposia®, Bristol Myers Squibb), a recently developed drug to treat MS by targeting S1P1 and S1P5 [63], can attenuate M1 microglial activation, as evidenced by reduced mRNA expression levels of Iba1, iNOS, IL-1β, and TNF-α, while it induces M2 microglial activation, as evidenced by increased mRNA expression levels of Found in Inflammatory Zone 1 (FIZZ1) [63]. These ex vivo effects of ozanimod on M1 microglial activation were further validated in Th1 cytokines-stimulated BV2 microglia, in which ozanimod decreased mRNA expression levels of IL-6, Regulated upon Activation, Normal T Cell Expressed and Presumably Secreted (RANTES), and TNF-α [64]. Siponimod (BAF312, Mayzent®, Novartis) is also a drug used to treat MS by targeting S1P1 and S1P5 [65]. It can dramatically attenuate microglia activation in the brains of EAE mice [66]. In addition, RP-001, a selective agonist for S1P1 [67], can attenuate microglial activation in mice brains challenged with sub-arachnoid hemorrhage, as evidenced by a significant reduction in Iba1 immunoreactivity [68]. Besides cerebral ischemia and MS, receptor-mediated S1P signaling could also influence other neurodegenerative diseases. FTY720 administration can significantly reduce protein expression levels of proinflammatory mediators (COX-2 and TNF-α) in the hippocampus of Aβ-induced AD-like Wistar rat [69]. FTY720 can also dramatically attenuate microglial activation in 5xFAD AD-like mice [57,70]. In addition, it can significantly decrease the number of ameboid microglia in 5xFAD mice while there is no difference in the number of resting microglia upon FTY720 administration [57], further demonstrating that FTY720 can attenuate microglial activation in AD mice. Ameboid microglia are neurotoxic [71,72]. FTY720 can reduce neurotoxic microglia by attenuating inflammatory microglial activation in an AD mouse brain [57]. Similarly, in APP/PS1 mice, FTY720 administration can attenuate microglial activation, as evidenced by the suppressed Iba1 expression in the hippocampus and neocortex [73,74]. FTY720 can also attenuate microglial activation in experimental models of PD [75,76]. It can attenuate the increase in the number of Iba1-immunopositive cells in the substantia nigra and striatum at 21 days after mice are exposed to 6-hydroxydopamine (6-OHDA) [75]. These studies clearly reflect the involvement of S1P receptors in microglia-mediated neuroinflammatory events during CNS pathogenesis. These aforementioned independent studies clearly indicate that S1P itself and its receptor can influence microglial activation, possibly leading to inflammatory responses in diverse neurodegenerative diseases. In the following sections, we will discuss how each S1P receptor can affect the activation of microglia and their neuroinflammatory responses.
Regulatory roles of S1P1 in the activation of microglia and their neuroinflammatory responses have been well demonstrated in cerebral ischemia and MS. In cerebral ischemia, S1P1 plays a pathogenic role with a close link to neuroinflammation, mainly by influencing microglial activation [77,78]. Suppressing S1P1 activity with the administration of AUY954, one of its functional antagonists [43], can reduce proinflammatory responses and enhance anti-inflammatory responses of activated microglia in post-ischemic brains [78]. Experimental studies employing an S1P1 knockdown approach also support these findings as S1P1 knockdown can reduce mRNA expression levels of proinflammatory cytokines in LPS-stimulated cultured microglia [77]. In fact, suppressing S1P1 activity with either AUY954 administration or S1P1 knockdown can attenuate brain damages after tMCAO challenge [77]. Such neuroprotection by AUY954 is closely associated with attenuation of microglial activation, proliferation, and morphological transformation into ameboid cells in the brains of mice challenged with tMCAO [77]. Furthermore, S1P1 can regulate the activation of mitogen-activated protein kinases (MAPKs) and microglial NF-κB signaling pathways in post-ischemic brains [78]. Both MAPKs and NF-κB signaling pathways are associated with the regulation of neuroinflammation in cerebral ischemia [79,80,81]. In MS pathogenesis, modulation of S1P1, presumably inhibition, can lead to the attenuation of microglial activation [82,83,84]. FTY720 modulates microglial activation in the injured spinal cords of EAE mice, in which FTY720 administration dramatically attenuates mRNA upregulation of proinflammatory mediators, including C-X-C motif chemokine ligand 9 (CXCL9), CXCL11, CXCL13, CCL1, CCL2, CCL4, CCL5, CCL7, CCL17, Axl, FosB, Fos, and TNF-α, while it increases the upregulation of colony-stimulating factor 2 (CSF2), Chi3l3, IL-10, IGF-1, Rentla, and CD206 [84]. In the corpus callosum of cuprizone-administered mouse, in another model of MS with a feature of demyelination [85,86], FTY720 administration can dramatically decrease the number of Iba1-immunopositive cells and upregulate mRNA expression levels of proinflammatory cytokines and chemokines [83]. These effects of FTY720 might be mediated through downregulation of S1P1 in the brain because FTY720 administration completely abolishes cuprizone-induced protein expression levels of S1P1 in the brain [83]. Such possible involvement of S1P1 has been reaffirmed with CYM5442, another S1P1 selective modulator and a functional antagonist of S1P1 [82], indicating that S1P receptor modulation, possibly S1P1 modulation, could dramatically attenuate neuroinflammatory responses of microglia in MS.
S1P2 also plays a critical role in the activation of microglia and their inflammatory responses in post-ischemic brains [87]. JTE013, an antagonist for S1P2 [88,89], can dramatically attenuate microglial activation at day 1 and day 3 after ischemic challenge (tMCAO challenge) [87]. It can also attenuate microglial proliferation and mRNA upregulation of proinflammatory mediators in post-ischemic brains [87]. Importantly, JTE013 administration into tMCAO-challenged mice can lead to the attenuation of NF-κB signaling in activated microglia, suggesting that suppression of S1P2 activity can attenuate transcriptional activation of proinflammatory mediators in post-ischemic brains [87]. The inhibition of inflammatory microglial activation upon pharmacological inhibition of S1P2 in a post-ischemic brain could be the associated neuroprotective mechanism achieved by either JTE013 administration or genetic deletion of S1P2 against tMCAO challenge [87,90]. JTE013 can also decrease mRNA expression levels of proinflammatory cytokines in BV2 murine microglia upon LPS stimulation [87], suggesting that S1P2 is a critical regulator of microglial activation. However, how S1P2 regulates microglial activation in other CNS pathologies remains unclear.
Roles of S1P3 in the activation of microglia and their proinflammatory responses have also been reported in cerebral ischemia. Blockade of S1P3 with CAY10444, a specific S1P3 antagonist [91,92,93], can decrease the number of Iba1-positive cells, microglial proliferation, morphological transformation into neurotoxic ameboid shape, and inflammatory M1 polarization of microglia [94]. In addition, S1P3 can influence the activation of microglial NF-κB and the production of inflammatory cytokines in injured brains after an ischemic challenge [94]. In primary microglia, suppressing S1P3 activity with either CAY10444 or S1P3-specific shRNA lentivirus particles can dramatically attenuate mRNA upregulation of proinflammatory cytokines, suggesting that S1P3 is involved in the proinflammatory activation of microglia [94]. Unsurprisingly, S1P3 has been validated as a therapeutic target for drug development to treat cerebral ischemia because CAY10444 administration can attenuate brain damages such as brain infarction, neurological functional deficit, and neural cell death after tMCAO challenge [94]. However, the role of S1P3 in microglial activation-associated other CNS disorders is yet to be uncovered.
S1P4 is poorly expressed in the brain. Therefore, brain cells do not express S1P4 under normal conditions [95,96]. However, under disease conditions, expression levels of S1P4 in the brain are increased because infiltrated cells, particularly T cells, express S1P4 [95,96]. S1P4 is also expressed on neutrophils known to play important roles in neuroinflammatory responses under disease conditions [97,98]. S1P4 can regulate the migration and infiltration of neutrophils and their inflammatory responses through 5-lipoxygenase activity [97]. In injured brains after an ischemic challenge, mRNA expression levels of S1P4 are increased and peaked at 4 days after tMCAO challenge [99]. Similarly, in injured spinal cords of EAE mice, mRNA expression levels of S1P4 are upregulated [100]. These independent in vivo studies indicate that S1P4 plays a critical role in the pathogenesis of stroke and MS. However, whether S1P4 can directly regulate the activation of microglia and their neuroinflammatory responses requires further investigation.
S1P5 is moderately expressed on microglia [101]. Being a target of FTY720, S1P5 must have certain roles in microglial biology, including microglial activation and proinflammatory responses. In fact, S1P5 is associated with microglial activation because an S1P5 agonist can significantly increase the number of Iba1-positive cells in organotypic cerebellar slice cultures [102]. Upon stimulation with Th1 cytokines, ozanimod can significantly reduce mRNA expression levels of proinflammatory cytokines including IL-6, RANTES, and TNF-α in striatal slice cultures from EAE mice as well as in Th1-stimulated BV2 microglial cells [64]. In addition, ozanimod can decrease the number of Iba1/IL-1β-double immunopositive cells in corticostriatal slices from EAE mice [64], suggesting that it can inhibit inflammatory microglial activation. Considering that ozanimod can decrease mRNA expression levels of IL-1β and TNF-α while increasing FIZZ1 in Th1 cytokines-activated BV2 cells [64], ozanimod not only can inhibit M1 microglial phenotype but also can promote M2 microglial phenotype in EAE.
3. S1P Receptors in Activation of Astrocytes and Their Neuroinflammatory Responses
Receptor-mediated S1P signaling has been well characterized in astrogliosis (astrocyte activation) under different disease conditions. In the blood, platelets are considered the major source of S1P production [103,104]. In the CNS, astrocytes play such a role [105,106]. In fact, cerebellar astrocytes can release S1P [105,106] to influence the proliferation of astrocytes by activating ERK1/2 signaling [106]. In spinal cord injuries, reactive astrocytes mainly found in injured areas can produce S1P, indicating that astrocytes are major sources of S1P in an injured spinal cord [107]. In addition to the role of cells producing S1P, astrocytes themselves can be activated by S1P. In addition, S1P can induce migration of astrocytes and promote the secretion of CXCL1, a chemokine, from reactive astrocytes [108], suggesting that S1P can regulate the biology of astrocytes and their inflammatory responses. Therefore, S1P-influenced inflammatory astrogliosis can trigger the pathogenesis of diverse neuroinflammatory diseases. Indeed, it has been reported that S1P microinjection can result in a significant increase in astrogliosis in the corpus callosum of either normal or stroke-challenged mice [27]. The observed astrogliosis has been thought to occur possibly via S1P1 activation because the administration of FTY720 with the highest binding affinity to S1P1 among S1P receptors [109,110] can decrease the number of reactive astrocytes in the corpus callosum of normal or stroke-challenged mice [27]. FTY720 administration can also reduce mRNA expression levels of proinflammatory mediators (IL-6, IFN-γ, IL-1β, TNF-α, IL-12A, IL-23A, CXCL10, CCL2, CCL20, and NOS2) in astrocytes isolated from EAE mice [84]. Conversely, mRNA expression levels of anti-inflammatory mediators such as CXCL12 and IL-33 are upregulated after FTY720 administration [84]. In addition, microinjection of exogenous S1P into the striatum of a mouse brain can increase astrogliosis [111], suggesting that S1P can induce neuroinflammation by promoting astrogliosis. Most S1P receptors (S1P1, S1P2, S1P3, and S1P5) are expressed on astrocytes [95,105,112]. They can influence diverse biological events of astrocytes, such as proliferation, migration, communication between astrocytes and the blood–brain barrier gap junction, and production of growth factors [95]. Among S1P receptors expressed on astrocytes, S1P1 is the highest expressed receptor type, followed by S1P3 [112,113]. Although the basal gene expression level of S1P5 is undetectable, its expression has been found to be increased in cultured astrocytes upon growth factor supplementation for 4–6 days [112,113]. Growth factor supplement (containing epidermal growth factor, basic fibroblast growth factor, insulin, biotin, human transferrin, and hydrocortisone) can dramatically increase mRNA expression levels of S1P5 in cultured astrocytes [113]. Among S1P receptors, S1P1 and S1P3 can promote inflammatory activation of astrocytes, which has been well characterized in different disease conditions [13,114,115].
The neuroinflammatory roles of S1P1 in astrocytes have been thoroughly studied in MS [84,116]. Protein expression levels of S1P1 and S1P3 are significantly increased in active MS lesions [117]. Upregulated S1P1 and S1P3 are mainly detected in reactive astrocytes [117], indicating that activation of these receptors on astrocytes may trigger inflammatory responses, thereby playing a role in the pathogenesis of MS. In human autopsy brain samples from MS patients, S1P1 has been found to be mainly upregulated in astrocytes and blood vessels, but not in myelin sheath or microglia/macrophages [118], suggesting that S1P1 on astrocytes is a critical pathogenic player in MS. In fact, neuroprotective effects of FTY720 in MS are mainly associated with modulation of astrocytic S1P1 activity [43,82,119,120]. In EAE, selective deletion of S1P1 from astrocytes, but not from neurons, can improve disease severity, demonstrating that astrocytic S1P1 is responsible for the pathogenesis of MS [43]. S1P1 deletion from astrocytes can also reduce protein expression levels of proinflammatory cytokines [43]. FTY720 can reduce migration and proliferation of S1P-stimulated cultured astrocytes mainly by downregulating S1P1 [121]. These effects of FTY720 can ameliorate spinal cord injury-induced functional deficits in rats [121]. In addition, FTY720 can influence astrocytes proliferation and reduce the secretion of inflammatory chemokines, astrogliosis, and neuroinflammation (reviewed well in [122]), all of which are associated with MS pathogenesis [123,124,125]. Furthermore, FTY720 treatment can decrease mRNA expression levels of proinflammatory mediators such as CCL2, NOS2, CSF2, IL-6, and TNF-α, while increasing mRNA expression levels of anti-inflammatory cytokines such as IL-10 in LPS-activated astrocytes culture [84], indicating its potent anti-inflammatory efficacy in activated astrocytes. In EAE mice, FTY720 can attenuate the upregulation of IL-1 receptor (IL-1R), S1P1, S1P3, iNOS, and nitrotyrosine in astrocytes [126]. Neuroprotective effects of FTY720 in EAE are also mediated through the blockade of NO production and nuclear translocation of NF-κB in astrocytes [126]. These effects of FTY720 on inflammatory astrogliosis and MS pathogenesis might be due to its ability to suppress S1P1 activity [127]. Similarly, CYM-5442, a modulator of S1P1, can attenuate the severity of EAE and reduce amounts of proinflammatory mediators such as monocyte chemoattractant protein 1 (MCP-1), IL-17, IL-10, and IL-1β in blood plasma [82]. These effects of CYM-5442 are comparable to those of FTY720 [82]. S1P1 is upregulated on neurons and astrocytes during EAE. Such upregulation is significantly attenuated by CYM-5442 treatment, indicating that downregulating S1P1 activity in astrocytes can reduce neuroinflammation and the severity of MS pathogenesis, at least in part [82]. Matrine, a natural alkaloid component extracted from Sophorae flavescens, can also attenuate astrogliosis in the brains of EAE mice [128]. This effect is mediated by lowering S1P amounts and suppressing SphK1/2 activity [128]. Matrine can also attenuate S1P1 upregulation in astrocytes in the brains of EAE mice [128], suggesting that the neuroprotective effects of Matrine in EAE mice are mediated through downregulation of S1P1 in astrocytes, which is reaffirmed in cultured astrocytes stimulated with IFN-γ [128]. S1P1 activation can also trigger ERK1/2 phosphorylation in mixed glial culture and astrocytes [129]. Since ERK1/2 signaling could contribute to MAPK-mediated inflammatory responses [130,131,132], S1P1 activation can promote such inflammatory responses. All these findings clearly indicate that astrocytic S1P1 is the major S1P receptor subtype responsible for neuroinflammation and disease pathogenesis of MS by modulating astrogliosis.
Roles of receptor-mediated S1P signaling, possibly through S1P1, in astrocytes have been indicated in other CNS diseases as well. Siponimod can block inflammatory activation of astrocytes as it can reduce nuclear translocation of NF-κB in IL-1β- or IL-17-stimulated astrocytes [133]. On the other hand, siponimod significantly increases expression levels of nuclear factor erythroid 2-related factor 2 (Nrf2), a potent antioxidant regulator [134]. These independent studies suggest its dual effects on astrocytes. Siponimod can also attenuate cell death of spinal neurons exposed to conditioned medium of S1P- or IL-1-treated astrocytes and induce S1P1 internalization [135]. Although whether siponimod can block S1P1 recycling is not determined in that study [135], siponimod might reduce S1P-induced neurodegeneration via a functional antagonism of S1P1 in astrocytes. FTY720 can enhance the recovery after contusion-induced spinal cord injury in mice by attenuating the accumulation of reactive astrocytes in injured areas [136], which could be mediated by suppressing S1P1 activity because FTY720 has been proven to have a functional antagonistic effect on S1P1 [43,44,45]. Similarly, in APP/PS1 mice, FTY720 administration can attenuate astrocyte activation as evidenced by attenuated glial fibrillary acidic protein (GFAP) expression in the hippocampus and neocortex [73,74]. In addition, it can increase astrocytic phagocytosis of Aβ, suggesting that it not only attenuates neuroinflammatory activation of astrocytes but also contributes to the clearance of Aβ plaque in AD pathogenesis [74]. FTY720 can also attenuate astrocyte activation in experimental models of PD [74,75]. It can decrease the number of GFAP-immunopositive cells in the substantia nigra and striatum at 21 days after exposing mice to 6-hydroxydopamine (6-OHDA) [75]. These studies indicate that FTY720 can exert its neuroprotective effects by attenuating astrocyte activation in different CNS pathogenesis. Such anti-neuroinflammatory activities of FTY720 in astrocytes might be mediated by downregulation of S1P1 activity as FTY720 has a functional antagonistic effect on S1P1 [43,44,45].
S1P can trigger the upregulation of mRNA and protein expression levels of COX-2 in astrocytes through the Gα12/13-Rho pathway [137]. This S1P-triggered COX-2 upregulation is mediated through S1P3 activation because such upregulation is attenuated upon S1P3 knockdown or its pharmacological inhibition [137]. Since COX-2 is considered a proinflammatory mediator in diverse disease conditions [138], suppressing S1P3 activity could lead to attenuation of COX-2-mediated inflammatory responses. S1P3-mediated inflammatory responses in astrocytes have been reported by other independent studies as well. When murine or rat astrocytes are stimulated with TNF-α or LPS, mRNA expression levels of S1P3 are dramatically increased [139]. In addition, upon LPS stimulation, protein expression levels of S1P3 are increased in rat primary astrocytes [108]. Similarly, in cultured human astrocytes, mRNA expression levels of S1P3 are significantly increased upon TNF-α stimulation [117]. These independent studies indicate that astrocytic S1P3 can regulate inflammatory responses. Indeed, TNF-α-stimulated astrocytes can secrete MCP-1, an inflammatory mediator [117] involved in the pathogenesis of diverse neuroinflammatory diseases [140]. Moreover, astrocytic S1P3 has been reported to participate in the pathogenesis of several neuroinflammatory diseases. In MS lesions, S1P3 has been found to be dramatically upregulated in reactive astrocytes [108]. A recent study has revealed that S1P3 can regulate gene expression of inflammatory mediators in astrocytes through RhoA signaling pathways [137]. In a post-ischemic brain, pharmacological inhibition of S1P3 activity through administration of CAY10444 can attenuate astrogliosis, as evidenced by reduced GFAP immunoreactivity and astrocyte proliferation [94]. Particularly, at 3 days after the ischemic challenge, the number of reactive astrocytes is increased in injured brains, which is then reduced upon CAY10444 administration [94]. These results from a study using an in vivo cerebral ischemia model suggest that S1P3 can promote reactive astrogliosis in the post-ischemic brain. Similarly, in a mouse model of Sandhoff disease mainly characterized by reactive astrogliosis [141], genetic deletion of S1P3 can improve disease symptoms and attenuate astrogliosis [142]. In brain cancer, S1P3 is also upregulated in highly permeable metastases, mainly in astrocytes, where S1P3 influences the infiltration of peripheral immune cells and the production of inflammatory cytokines [143]. When astrocytic S1P3 activity is attenuated through its genetic knockdown, most inflammatory mediators (CCL2, CXCL1, CXCL10, intercellular adhesion molecule-1 (ICAM-1), IL-6, and IL-8) are downregulated at mRNA expression levels [143], reaffirming the neuroinflammatory roles of astrocytic S1P3. These studies clearly reflect the roles of S1P3 in reactive astrocytes and their neuroinflammatory responses during diverse pathological conditions. Mechanistically, neuroinflammatory roles of astrocytic S1P3 can be mediated through increased intracellular calcium signaling [91]. S1P-induced increase in intracellular calcium concentration is reduced upon pharmacological inhibition of S1P3 in rat primary cortical astrocytes, suggesting that S1P-influenced intracellular calcium stress is mediated by S1P3. As intracellular calcium overload is associated with diverse cellular responses, including neuroinflammation [144,145], suppressing S1P3 activity can reduce astrocyte-dependent neuroinflammatory events. Indeed, S1P-stimulated CXCL1 chemokine production from cultured astrocytes is attenuated upon pharmacological inhibition of S1P3 [91], further supporting the neuroinflammatory roles of S1P3 in astrocytes.
In a normal brain, S1P4 expression is negligible, whereas it is upregulated in an injured brain after an ischemic challenge [99]. However, how neuroglial S1P4 influences disease pathogenesis has not been reported yet. S1P5 is also highly expressed on astrocytes and oligodendrocytes of active MS lesions as much as S1P1 [118], indicating that S1P5 might also modulate neuroinflammatory responses of astrocytes. A previous study has revealed that siponimod can inhibit NF-κB activation in IL-1β-stimulated astrocytes [133], indicating possible roles of S1P5 in the activation of astrocytes and subsequent inflammatory responses. However, whether astrocytic S1P5 can influence neuroinflammatory events remains unclear. Instead, in oligodendrocytes, a main locus of S1P5 in the brain, S1P5 is known to have roles in regulating biological functions, including maturation, differentiation from neural stem cells to oligodendrocytes, and survival [146,147]. Further detailed studies about the roles of S1P4 and S1P5 in activation of astrocytes and their neuroinflammatory responses are needed.
Table 1 presents the biological roles of S1P receptors in the activation of neuroglia and their neuroinflammatory responses.

Table 1.
S1P receptors in activation of neuroglia and their neuroinflammatory responses. N/A: not available.
Although we discussed the role of receptor-mediated S1P signaling in the activation of microglia and/or astrocytes and their neuroinflammatory responses, there could be S1P-mediated glial crosstalk, resulting in neuroinflammatory responses. The proinflammatory mediators secreted by S1P-activated astrocytes or microglia might be able to further activate each other and prolong glial activation cascades. In fact, S1P signaling in both microglia and astrocytes has been reported to promote neuronal apoptosis [148], indicating a possible influence of S1P on neuron–glial crosstalk. However, whether S1P signaling is involved in the crosstalk between microglia and astrocytes remains unknown, which would be of interest for future studies.
4. LPA Receptors in Activation of Microglia and Their Neuroinflammatory Responses
LPA plays significant roles in microglial biology, such as motility, cytoskeletal architecture, membrane ruffling, glycolysis, brain-derived neurotrophic factor (BDNF) production, and ATP release [149,150]. Recent reports have suggested that LPA is one of the critical mediators of microglial activation [28,151,152], further indicating that LPA and its receptor-mediated signaling might have critical roles in the pathogenies of diverse CNS disorders. LPA exposure can increase the metabolic activities of cultured microglia [29]. Especially, when microglia are exposed to LPA, their morphology is changed to a round shape with shorter and more thickened processes than resting microglia [149]. These morphological changes of microglia are characteristic features of activated ameboid microglia playing important roles in the pathogenesis of diverse CNS disorders [71,72]. In addition to functions demonstrated in vitro, LPA can trigger microglial activation in vivo. In neuropathic pain, intrathecal injection of LPA can induce microglial MAPK phosphorylation and upregulation of genes involved in microglial activation [29,149,153]. Minocycline, a specific inhibitor of microglial activation, can decrease LPA-induced mechanical allodynia and thermal hyperalgesia [154]. These effects of minocycline are also observed in sciatic inflammatory neuropathy [155]. These two independent studies clearly suggest that LPA-mediated neuropathic pain is caused by microglial activation, at least in part. In addition, intrathecal LPA injection not only can increase the number of activated microglia in the dorsal horn of the spinal cord but also can skew ramified microglia towards more toxic ameboid microglia [154]. Such microglial activation and morphological transformation by intrathecal LPA injection are associated with p38 MAPK phosphorylation [154]. In fact, it has been well demonstrated that LPA-mediated microglial activation can lead to the development of neuropathic pain at an early stage and that LPA1 plays a critical role in this process [156,157].
Among LPA receptors, LPA1 is expressed at the highest level in the brain and spinal cord [28,158]. While LPA1 is well expressed on primary microglia, it is undetectable in immortalized cell lines, for example, murine BV2 microglial cells [29,149,152,158,159]. It is clear that LPA1 is associated with the activation of microglia and their inflammatory responses in diverse neuroinflammatory diseases. In post-ischemic brains, LPA1 is associated with microglial activation, proliferation, and inflammatory responses [160,161,162]. Suppressing LPA1 activity with LPA1-specific shRNA lentivirus or AM152 (a specific antagonist for LPA1, also known as BMS-986020 [163,164,165]) can decrease the number of Iba1-positive microglia in the brains of mice challenged with tMCAO [160,162]. It can also reduce the soma sizes of microglia and attenuate microglial proliferation and morphological transformation from ramified microglia into ameboid cells in injured brains after tMCAO challenge [160,162]. These data indicate that LPA1 is associated with microglial activation in post-ischemic brains. Importantly, suppressing LPA1 activity can dramatically attenuate microglial NF-κB expression in post-ischemic brains and decrease mRNA expression levels of proinflammatory cytokines in mouse primary microglia exposed to LPS [162]. These findings clearly reflect that LPA1 can influence the inflammatory activation of microglia. A recent report has also demonstrated that LPA1 is upregulated in the brains of mice challenged with intracerebral hemorrhage (ICH) and that such upregulation is mainly observed in microglia [166]. Suppressing LPA1 activity with AM966 (another specific antagonist for LPA1 [167]) not only can reverse microglial activation but can also improve long-term neurobehavioral functions following ICH [166], indicating that microglial LPA1 is critically involved in the pathogenesis of ICH as in that of cerebral ischemia [160,161,162]. In addition, in a mouse model of spinal cord injury caused by an intraspinal injection of LPA, microglial activation and demyelination are significantly increased [28]. Either LPA1 knockout or pharmacological inhibition of LPA1 with AM095, an LPA1-selective antagonist [168], can attenuate LPA-induced demyelination in the spinal cord and promote cell survival when oligodendrocytes are exposed to microglial conditioned medium treated with LPA, indicating that LPA1 is a critical mediator of LPA-induced microglial activation [28]. Microglial activation is a hallmark pathogenic event in neuropathic pain, in which LPA/LPA1 signaling is considered as a major factor in the pathogenesis of neuropathic pain [156,169,170]. LPA1 is also associated with microglial activation in an LPS-induced sepsis animal model [158]. In septic brains, LPA1 is associated with activation of microglia and their TNF-α production, which is recapitulated in LPS-stimulated rat primary microglia [158]. Suppressing LPA1 activity with LPA1-specific shRNA lentivirus can decrease the number of activated microglia and attenuate their proliferation in septic brains [158]. Similarly, in rat primary microglia, LPA1 knockdown with its specific siRNA can attenuate mRNA upregulation and protein release of TNF-α after LPS stimulation [158]. In SH-SY5Y cells co-cultured with BV2 microglia, saikosaponin-d, a natural triterpenoid saponin that acts as an inhibitor of LPA1 [171], can suppress neuronal LPA1 and MAP2 expression and reduce neuronal apoptosis as evidenced by increased protein expression levels of Bcl-2 with decreased Bax expression [171], revealing that microglial LPA1 is responsible for neuronal apoptosis. Saikosaponin-d not only can reduce microglial activation in the hippocampus of LPS-induced septic mouse, but also can attenuate proinflammatory cytokines production from LPS-stimulated primary microglia [172]. These aforementioned studies clearly suggest that LPA1 is a critical regulator of microglial activation, thus contributing to the pathogenesis of diverse CNS diseases.
LPA2 is expressed on microglia of the spinal cord, cultured primary microglia, BV2 murine microglia, and C13NJ microglia [149,152,173]. Microglial LPA2 is responsible for oligodendrocyte cell death, possibly by enhancing the production of purines and subsequent activation of P2X7 signaling [173]. Conditioned medium obtained from LPA-stimulated microglia can decrease the number of myelin basic protein (MBP)-positive cells and induce cell death of oligodendrocytes, suggesting that microglial LPA2 is responsible for this death of oligodendrocytes [173]. In fact, such regulatory roles of LPA2 are linked to the pathogenesis of spinal cord injury [173].
LPA3 is abundantly expressed on microglia [29]. Its expression is further increased in activated microglia [101]. Interestingly, microglial LPA3 is associated with LPA production [154,156]. The LPA/LPA3 signaling axis can influence the pathogenesis of neuropathic pain [154,156]. In addition, LPA3 can trigger the production of IL-1β from microglia, resulting in inflammatory cascades [154,170]. LPA3 is also associated with increased ERK1/2 phosphorylation in LPA-stimulated microglia [149]. Considering that ERK1/2 activation can trigger the production of inflammatory mediators [130,131,132], LPA3-regulated ERK1/2 activation might be an underlying event that promotes neuroinflammatory responses in microglia. In addition, LPA-induced membrane ruffling of activated microglia can be mediated through LPA3 [150] because it is attenuated by DGPP [150], a potent LPA3 antagonist [174]. Such findings [150] suggest that LPA3 is a critical modulator of microglial activation, indirectly supporting the previously reported roles of LPA3 in microglial neuroinflammatory responses [154,170]. In addition, Ki16425, a dual inhibitor of LPA1 and LPA3 [175], can attenuate LPA-induced ROS production in EOC mouse microglial cells, indicating that LPA3 can influence microglial oxidative stress [176]. Collectively, these studies indicate that LPA3 is associated with inflammatory microglial activation and oxidative stress under pathological conditions.
LPA4 is moderately expressed in a normal mouse brain, primary microglia, and BV2 microglia [158], suggesting its certain role in microglial biology. However, its role in microglial biology and related disease pathogenies has not been reported yet.
LPA5 is well expressed on microglia [177]. The association of LPA5 with microglial activation has been validated in both in vitro microglial culture and in vivo animal models. In LPA-stimulated primary microglia or BV2 microglial cell line, TCLPA5, a selective antagonist for LPA5 [178], can attenuate the upregulation of M1-related markers (such as iNOS, COX-2, CD40, and CD86) and proinflammatory cytokines (such as IL-1β and IL-6) [152]. In BV2 microglial cells, LPA5 knockdown with its specific siRNA can attenuate the production of proinflammatory cytokines (TNF-α, IL-1β, and IL-6) [179]. In addition, other LPA5 antagonists, such as AS2717638 and compound 3, can attenuate LPA-induced production of proinflammatory cytokines (TNF-α, IL-1β, and IL-6) and chemokines (CXCL10, CXCL2, and CCL5), all of which are markers of M1 microglia [180], indicating that the LPA/LPA5 signaling axis is important for the proinflammatory M1 activation of microglia. The LPA/LPA5 signaling axis is also associated with the activation of microglial MAPK signaling pathways, which subsequently promotes the activation of NF-κB and STAT1/3 signaling pathways in LPA-stimulated microglial cultures [181]. Importantly, LPA5 activation can increase the sizes of microglia and promote morphological transformation into ameboid-shaped microglia [181], further suggesting that LPA5 can promote inflammatory microglial activation in cultured microglia. This notion is also supported by studies employing an in vivo stroke model. In fact, LPA5 has been demonstrated to be a novel pathogenic factor for both acute and chronic ischemic injuries in mice subjected to tMCAO challenge because TCLPA5 not only can attenuate acute brain damage but also exert long-term neuroprotection in these mice [179,182]. Such a pathogenic role of LPA5 is closely associated with microglial activation in injured brains [179]. LPA5 can influence microglial activation and proliferation in post-ischemic brains [179]. The pharmacological inhibition of LPA5 with TCLPA5 can decrease the number of activated microglia in peri-ischemic and ischemic core regions and attenuate microglial proliferation in the penumbra region after tMCAO challenge [179]. TCLPA5 administration can also attenuate the morphological transformation of activated microglia from ramified into ameboid shape in post-ischemic brains [179]. In addition, it can attenuate the upregulation of M1-relevant proinflammatory markers (TNF-α, IL-1β, and IL-6) and microglial NF-κB activation in injured brains after tMCAO challenge [179]. The latter indicates that the observed LPA5-driven upregulation of M1-like proinflammatory cytokines may occur in microglia. This notion has been reaffirmed in cultured microglia in vitro [179,180]. Interestingly, LPA5 is upregulated in post-ischemic brains. Such upregulation is observed mostly in Iba1-positive cells (activated microglia) [179], indicating that LPA5 is the critical regulator of microglial activation in post-ischemic brains.
LPA6 is also highly expressed on mouse primary microglia and BV2 murine microglia [158]. However, its role in neuroinflammatory disease conditions remains unclear.
5. LPA Receptors in Activation of Astrocytes and Their Neuroinflammatory Responses
Receptor-mediated LPA signaling can also influence the activation of astrocytes in different CNS disease conditions. Autotaxin, a key enzyme of LPA synthesis [183], can promote adhesion of astrocytes and glioma invasion during glioblastoma multiforme pathogenesis [184], indicating that LPA can regulate astrocyte biology. LPA can induce the formation of stress fibers and focal adhesions [185] and increase intracellular calcium concentration in astrocytes, [186], all of which can stimulate astrocytic proliferation and migration [187,188,189,190] that are featured biological events of reactive astrocytes [190,191,192].
In a normal brain, LPA1 is highly expressed on astrocytes [113,166,193,194], indicating that LPA1 can influence astrocyte activation. A few recent studies have demonstrated such a role. In cultured astrocytes that express LPA1 abundantly, LPA can induce cell proliferation via LPA1 [195]. LPA can also promote the migration of LPS- or IL-1β-stimulated rat primary astrocytes mediated through LPA1 [196]. In C6 glioma cells, an immortalized astrocyte cell line, inhibition of LPA1 activity by Ki16425 (an LPA1/3 antagonist) or AM966 (an LPA1 antagonist) can attenuate LPA-triggered ERK1/2 activation, which is reaffirmed by LPA1 knockdown with its specific siRNA [197]. Inactivation of ERK1/2 by suppressing LPA1 activity could lead to attenuated proinflammatory responses in diverse CNS diseases such as cerebral ischemia [161,162]. In fact, inhibiting LPA1 signaling through its specific antagonist, AM095, can attenuate the activation and proliferation of astrocytes in post-ischemic brains, which might ultimately contribute to neuroprotection [162]. Besides cerebral ischemia, LPA1 may have certain roles in the activation of astrocytes and their neuroinflammatory responses in other disease types. As discussed earlier, either AM966 administration or genetic deletion of LPA1 can attenuate brain damages and neuroinflammatory responses in ICH-challenged mice [166]. In the injured brains of these mice, LPA1 is upregulated on reactive astrocytes, activated microglia, and neurons [166]. However, whether LPA1 could regulate astrocyte activation and whether the observed neuroinflammation is dependent on any of those cell types in ICH-challenged mice remains unknown. In a neuropathic pain model by a partial sciatic nerve ligation, LPA1 has been indicated as a possible mediator for the activation of astrocytes and their production of chemokine (CXCL1) in injured spinal dorsal horns, leading to the aggravation of pathogenesis because LPA1/3 antagonist (Ki16425) can attenuate CXCL1 upregulation in activated astrocytes [198]. Although LPA1 can influence a variety of functions of astrocytes [195,196], many of its roles in astrocyte-dependent neuroinflammation remain unclear.
LPA2 is also highly expressed on astrocytes [193], indicating that LPA2 might also play a critical role in neuroinflammatory responses. Similarly, other LPA receptors, including LPA3, LPA4, and LPA6, are also expressed on astrocytes [195,199], suggesting that these receptors might have certain roles in the neuroinflammatory responses of astrocytes. However, the roles of these LPA receptor subtypes in astrocyte activation and their neuroinflammatory responses have not been reported yet.
Table 2 presents the biological roles of LPA receptors in the activation of neuroglia and their neuroinflammatory responses.
Table 2.
LPA receptors in neuroglial activation and their neuroinflammatory responses. N/A: not available.
6. Conclusions
Most LPA and S1P receptors can modulate neuroinflammatory responses by influencing the activation of microglia and astrocytes. S1P and LPA receptors are expressed on both cell types, and most of these expressed receptors are upregulated during CNS pathogenesis. Table 1 and Table 2 present the biological roles of S1P and LPA receptors in glial activation and their neuroinflammatory responses. Figure 1 schematically illustrates the neuroinflammatory responses of S1P and LPA receptors. Up to date, it has been reported that S1P1, S1P2, S1P3, S1P5, LPA1, and LPA5 can promote the activation of microglia and/or astrocytes and their neuroinflammatory responses. However, the roles of other receptors such as S1P4, LPA2, LPA4, and LPA6 remain unclear, possibly due to the lack of pharmacological modulators for these receptor subtypes. Therefore, future studies should focus on the development of receptor-specific modulators and the use of genetic tools for verifying the roles of specific receptor subtypes in the activation of microglia and/or astrocytes. Since the roles of LPA and S1P receptors in glial activation have been mainly studied in cerebral ischemia and MS, future studies should also focus on whether these receptors can influence the activation of microglia and/or astrocytes in other CNS diseases such as PD, AD, and Huntington’s disease. Moreover, developing new modulators of S1P and LPA receptor subtypes could be an appealing therapeutic option to manage CNS diseases featured by the activation of microglia and astrocytes and their inflammatory responses.
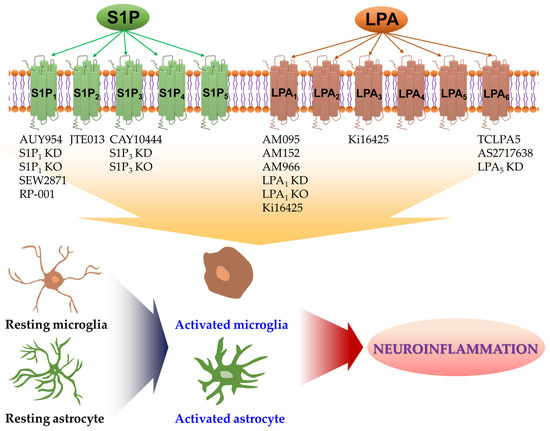
Figure 1.
Either pharmacological or genetic approaches have been employed to identify roles of S1P and LPA receptors in glial activation under neuroinflammatory conditions. Receptor-mediated S1P and LPA signaling can promote the activation of microglia and astrocytes in various CNS pathologies. Activated microglia or activated astrocytes can secrete diverse proinflammatory mediators, including cytokines and chemokines, all of which can trigger neuroinflammatory events and contribute to CNS pathogenesis. Pharmacological or genetic tools listed below each receptor subtype can suppress the activity of the respective receptor. They have been reported to attenuate neuroinflammatory responses of activated microglia or activated astrocytes. Up to date, S1P1, S1P2, S1P3, S1P5, LPA1, and LPA5 have been identified as the receptors involved in such events. See text for details and references. KD: knockdown; KO: knockout.
Author Contributions
Writing—original draft preparation, B.P.G. and J.-W.C.; writing—review and editing, B.P.G. and J.-W.C.; visualization, B.P.G. and J.-W.C.; supervision, J.-W.C.; funding acquisition, J.-W.C. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by grants (NRF-2021R1A2C1005520 and NRF-2020R1A6A1A03043708 to J.-W.C.) of the National Research Foundation (NRF) of Korea.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Jakel, S.; Dimou, L. Glial Cells and Their Function in the Adult Brain: A Journey through the History of Their Ablation. Front. Cell. Neurosci. 2017, 11, 24. [Google Scholar] [CrossRef]
- Fields, R.D.; Stevens-Graham, B. New insights into neuron-glia communication. Science 2002, 298, 556–562. [Google Scholar] [CrossRef]
- Meyer, K.; Kaspar, B.K. Glia-neuron interactions in neurological diseases: Testing non-cell autonomy in a dish. Brain Res. 2017, 1656, 27–39. [Google Scholar] [CrossRef] [PubMed]
- Tognatta, R.; Miller, R.H. Contribution of the oligodendrocyte lineage to CNS repair and neurodegenerative pathologies. Neuropharmacology 2016, 110, 539–547. [Google Scholar] [CrossRef] [PubMed]
- Kabba, J.A.; Xu, Y.; Christian, H.; Ruan, W.; Chenai, K.; Xiang, Y.; Zhang, L.; Saavedra, J.M.; Pang, T. Microglia: Housekeeper of the Central Nervous System. Cell. Mol. Neurobiol. 2018, 38, 53–71. [Google Scholar] [CrossRef]
- Gopinath, A.; Collins, A.; Khoshbouei, H.; Streit, W.J. Microglia and Other Myeloid Cells in Central Nervous System Health and Disease. J. Pharmacol. Exp. Ther. 2020, 375, 154–160. [Google Scholar] [CrossRef]
- Nedergaard, M.; Ransom, B.; Goldman, S.A. New roles for astrocytes: Redefining the functional architecture of the brain. Trends Neurosci. 2003, 26, 523–530. [Google Scholar] [CrossRef] [PubMed]
- Colonna, M.; Butovsky, O. Microglia Function in the Central Nervous System During Health and Neurodegeneration. Annu. Rev. Immunol. 2017, 35, 441–468. [Google Scholar] [CrossRef]
- Liddelow, S.; Barres, B. SnapShot: Astrocytes in Health and Disease. Cell 2015, 162, 1170. [Google Scholar] [CrossRef] [PubMed]
- Griffiths, M.R.; Gasque, P.; Neal, J.W. The regulation of the CNS innate immune response is vital for the restoration of tissue homeostasis (repair) after acute brain injury: A brief review. Int. J. Inflam. 2010, 2010, 151097. [Google Scholar] [CrossRef][Green Version]
- Saxena, S.; Kruys, V.; Vamecq, J.; Maze, M. The Role of Microglia in Perioperative Neuroinflammation and Neurocognitive Disorders. Front. Aging Neurosci. 2021, 13, 671499. [Google Scholar] [CrossRef]
- Anderson, M.A.; Burda, J.E.; Ren, Y.; Ao, Y.; O’Shea, T.M.; Kawaguchi, R.; Coppola, G.; Khakh, B.S.; Deming, T.J.; Sofroniew, M.V. Astrocyte scar formation aids central nervous system axon regeneration. Nature 2016, 532, 195–200. [Google Scholar] [CrossRef]
- Kwon, H.S.; Koh, S.H. Neuroinflammation in neurodegenerative disorders: The roles of microglia and astrocytes. Transl. Neurodegener. 2020, 9, 42. [Google Scholar] [CrossRef] [PubMed]
- Chun, J.; Rosen, H. Lysophospholipid receptors as potential drug targets in tissue transplantation and autoimmune diseases. Curr. Pharm. Des. 2006, 12, 161–171. [Google Scholar] [CrossRef]
- Rosen, H.; Goetzl, E.J. Sphingosine 1-phosphate and its receptors: An autocrine and paracrine network. Nat. Rev. Immunol. 2005, 5, 560–570. [Google Scholar] [CrossRef]
- Ross, B.M.; Kish, S.J. Characterization of lysophospholipid metabolizing enzymes in human brain. J. Neurochem. 1994, 63, 1839–1848. [Google Scholar] [CrossRef]
- Choi, J.W.; Chun, J. Lysophospholipids and their receptors in the central nervous system. Biochim. Biophys. Acta 2013, 1831, 20–32. [Google Scholar] [CrossRef]
- Choi, J.W.; Herr, D.R.; Noguchi, K.; Yung, Y.C.; Lee, C.W.; Mutoh, T.; Lin, M.E.; Teo, S.T.; Park, K.E.; Mosley, A.N.; et al. LPA receptors: Subtypes and biological actions. Annu. Rev. Pharmacol. Toxicol. 2010, 50, 157–186. [Google Scholar] [CrossRef] [PubMed]
- Rao, P.V. Bioactive lysophospholipids: Role in regulation of aqueous humor outflow and intraocular pressure in the context of pathobiology and therapy of glaucoma. J. Ocul. Pharmacol. Ther. 2014, 30, 181–190. [Google Scholar] [CrossRef] [PubMed]
- Rivera, R.; Chun, J. Biological effects of lysophospholipids. Rev. Physiol. Biochem. Pharmacol. 2008, 160, 25–46. [Google Scholar] [CrossRef]
- Shao, Y.; Nanayakkara, G.; Cheng, J.; Cueto, R.; Yang, W.Y.; Park, J.Y.; Wang, H.; Yang, X. Lysophospholipids and Their Receptors Serve as Conditional DAMPs and DAMP Receptors in Tissue Oxidative and Inflammatory Injury. Antioxid. Redox. Signal. 2018, 28, 973–986. [Google Scholar] [CrossRef]
- Binder, B.Y.; Williams, P.A.; Silva, E.A.; Leach, J.K. Lysophosphatidic Acid and Sphingosine-1-Phosphate: A Concise Review of Biological Function and Applications for Tissue Engineering. Tissue Eng. Part B Rev. 2015, 21, 531–542. [Google Scholar] [CrossRef]
- Tan, S.T.; Ramesh, T.; Toh, X.R.; Nguyen, L.N. Emerging roles of lysophospholipids in health and disease. Prog. Lipid. Res. 2020, 80, 101068. [Google Scholar] [CrossRef]
- Hao, Y.; Guo, M.; Feng, Y.; Dong, Q.; Cui, M. Lysophospholipids and Their G-Coupled Protein Signaling in Alzheimer’s Disease: From Physiological Performance to Pathological Impairment. Front. Mol. Neurosci. 2020, 13, 58. [Google Scholar] [CrossRef]
- Crack, P.J.; Zhang, M.; Morganti-Kossmann, M.C.; Morris, A.J.; Wojciak, J.M.; Fleming, J.K.; Karve, I.; Wright, D.; Sashindranath, M.; Goldshmit, Y.; et al. Anti-lysophosphatidic acid antibodies improve traumatic brain injury outcomes. J. Neuroinflamm. 2014, 11, 37. [Google Scholar] [CrossRef]
- Goldshmit, Y.; Matteo, R.; Sztal, T.; Ellett, F.; Frisca, F.; Moreno, K.; Crombie, D.; Lieschke, G.J.; Currie, P.D.; Sabbadini, R.A.; et al. Blockage of lysophosphatidic acid signaling improves spinal cord injury outcomes. Am. J. Pathol. 2012, 181, 978–992. [Google Scholar] [CrossRef] [PubMed]
- Moon, E.; Han, J.E.; Jeon, S.; Ryu, J.H.; Choi, J.W.; Chun, J. Exogenous S1P Exposure Potentiates Ischemic Stroke Damage That Is Reduced Possibly by Inhibiting S1P Receptor Signaling. Mediat. Inflamm. 2015, 2015, 492659. [Google Scholar] [CrossRef] [PubMed]
- Santos-Nogueira, E.; Lopez-Serrano, C.; Hernandez, J.; Lago, N.; Astudillo, A.M.; Balsinde, J.; Estivill-Torrus, G.; de Fonseca, F.R.; Chun, J.; Lopez-Vales, R. Activation of Lysophosphatidic Acid Receptor Type 1 Contributes to Pathophysiology of Spinal Cord Injury. J. Neurosci. 2015, 35, 10224–10235. [Google Scholar] [CrossRef]
- Moller, T.; Contos, J.J.; Musante, D.B.; Chun, J.; Ransom, B.R. Expression and function of lysophosphatidic acid receptors in cultured rodent microglial cells. J. Biol. Chem. 2001, 276, 25946–25952. [Google Scholar] [CrossRef] [PubMed]
- Suckau, O.; Gross, I.; Schrotter, S.; Yang, F.; Luo, J.; Wree, A.; Chun, J.; Baska, D.; Baumgart, J.; Kano, K.; et al. LPA1, LPA2, LPA4, and LPA6 receptor expression during mouse brain development. Dev. Dyn. 2019, 248, 375–395. [Google Scholar] [CrossRef]
- Kajitani, N.; Okada-Tsuchioka, M.; Kano, K.; Omori, W.; Boku, S.; Aoki, J.; Takebayashi, M. Differential anatomical and cellular expression of lysophosphatidic acid receptor 1 in adult mouse brain. Biochem. Biophys. Res. Commun. 2020, 531, 89–95. [Google Scholar] [CrossRef]
- Healy, L.M.; Antel, J.P. Sphingosine-1-Phosphate Receptors in the Central Nervous and Immune Systems. Curr. Drug Targets 2016, 17, 1841–1850. [Google Scholar] [CrossRef]
- Gaire, B.P.; Choi, J.W. Sphingosine 1-Phosphate Receptors in Cerebral Ischemia. Neuromolecular Med. 2021, 23, 211–223. [Google Scholar] [CrossRef] [PubMed]
- Karunakaran, I.; Alam, S.; Jayagopi, S.; Frohberger, S.J.; Hansen, J.N.; Kuehlwein, J.; Holbling, B.V.; Schumak, B.; Hubner, M.P.; Graler, M.H.; et al. Neural sphingosine 1-phosphate accumulation activates microglia and links impaired autophagy and inflammation. Glia 2019, 67, 1859–1872. [Google Scholar] [CrossRef] [PubMed]
- Zahiri, D.; Burow, P.; Grossmann, C.; Muller, C.E.; Klapperstuck, M.; Markwardt, F. Sphingosine-1-phosphate induces migration of microglial cells via activation of volume-sensitive anion channels, ATP secretion and activation of purinergic receptors. Biochim. Biophys. Acta Mol. Cell. Res. 2021, 1868, 118915. [Google Scholar] [CrossRef] [PubMed]
- Juntunen, M.; Hagman, S.; Moisan, A.; Narkilahti, S.; Miettinen, S. In Vitro Oxygen-Glucose Deprivation-Induced Stroke Models with Human Neuroblastoma Cell- and Induced Pluripotent Stem Cell-Derived Neurons. Stem. Cells Int. 2020, 2020, 8841026. [Google Scholar] [CrossRef]
- Ryou, M.G.; Mallet, R.T. An In Vitro Oxygen-Glucose Deprivation Model for Studying Ischemia-Reperfusion Injury of Neuronal Cells. Methods Mol. Biol. 2018, 1717, 229–235. [Google Scholar] [CrossRef] [PubMed]
- Lv, M.; Zhang, D.; Dai, D.; Zhang, W.; Zhang, L. Sphingosine kinase 1/sphingosine-1-phosphate regulates the expression of interleukin-17A in activated microglia in cerebral ischemia/reperfusion. Inflamm. Res. 2016, 65, 551–562. [Google Scholar] [CrossRef] [PubMed]
- Blondeau, N.; Lai, Y.; Tyndall, S.; Popolo, M.; Topalkara, K.; Pru, J.K.; Zhang, L.; Kim, H.; Liao, J.K.; Ding, K.; et al. Distribution of sphingosine kinase activity and mRNA in rodent brain. J. Neurochem. 2007, 103, 509–517. [Google Scholar] [CrossRef]
- Nayak, D.; Huo, Y.; Kwang, W.X.; Pushparaj, P.N.; Kumar, S.D.; Ling, E.A.; Dheen, S.T. Sphingosine kinase 1 regulates the expression of proinflammatory cytokines and nitric oxide in activated microglia. Neuroscience 2010, 166, 132–144. [Google Scholar] [CrossRef] [PubMed]
- Brinkmann, V.; Davis, M.D.; Heise, C.E.; Albert, R.; Cottens, S.; Hof, R.; Bruns, C.; Prieschl, E.; Baumruker, T.; Hiestand, P.; et al. The immune modulator FTY720 targets sphingosine 1-phosphate receptors. J. Biol. Chem. 2002, 277, 21453–21457. [Google Scholar] [CrossRef]
- Pitman, M.R.; Woodcock, J.M.; Lopez, A.F.; Pitson, S.M. Molecular targets of FTY720 (fingolimod). Curr. Mol. Med. 2012, 12, 1207–1219. [Google Scholar] [CrossRef]
- Choi, J.W.; Gardell, S.E.; Herr, D.R.; Rivera, R.; Lee, C.W.; Noguchi, K.; Teo, S.T.; Yung, Y.C.; Lu, M.; Kennedy, G.; et al. FTY720 (fingolimod) efficacy in an animal model of multiple sclerosis requires astrocyte sphingosine 1-phosphate receptor 1 (S1P1) modulation. Proc. Natl. Acad. Sci. USA 2011, 108, 751–756. [Google Scholar] [CrossRef] [PubMed]
- Graler, M.H.; Goetzl, E.J. The immunosuppressant FTY720 down-regulates sphingosine 1-phosphate G-protein-coupled receptors. FASEB J. 2004, 18, 551–553. [Google Scholar] [CrossRef]
- Oo, M.L.; Thangada, S.; Wu, M.T.; Liu, C.H.; Macdonald, T.L.; Lynch, K.R.; Lin, C.Y.; Hla, T. Immunosuppressive and anti-angiogenic sphingosine 1-phosphate receptor-1 agonists induce ubiquitinylation and proteasomal degradation of the receptor. J. Biol. Chem. 2007, 282, 9082–9089. [Google Scholar] [CrossRef] [PubMed]
- Nussbaum, C.; Bannenberg, S.; Keul, P.; Graler, M.H.; Goncalves-de-Albuquerque, C.F.; Korhonen, H.; von Wnuck Lipinski, K.; Heusch, G.; de Castro Faria Neto, H.C.; Rohwedder, I.; et al. Sphingosine-1-phosphate receptor 3 promotes leukocyte rolling by mobilizing endothelial P-selectin. Nat. Commun. 2015, 6, 6416. [Google Scholar] [CrossRef] [PubMed]
- Bascunana, P.; Mohle, L.; Brackhan, M.; Pahnke, J. Fingolimod as a Treatment in Neurologic Disorders Beyond Multiple Sclerosis. Drugs R&D 2020, 20, 197–207. [Google Scholar] [CrossRef]
- Noda, H.; Takeuchi, H.; Mizuno, T.; Suzumura, A. Fingolimod phosphate promotes the neuroprotective effects of microglia. J. Neuroimmunol. 2013, 256, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Jackson, S.J.; Giovannoni, G.; Baker, D. Fingolimod modulates microglial activation to augment markers of remyelination. J. Neuroinflamm. 2011, 8, 76. [Google Scholar] [CrossRef] [PubMed]
- Cipriani, R.; Chara, J.C.; Rodriguez-Antiguedad, A.; Matute, C. FTY720 attenuates excitotoxicity and neuroinflammation. J. Neuroinflamm. 2015, 12, 86. [Google Scholar] [CrossRef] [PubMed]
- Guo, Y.; Gan, X.; Zhou, H.; Zhou, H.; Pu, S.; Long, X.; Ren, C.; Feng, T.; Tang, H. Fingolimod suppressed the chronic unpredictable mild stress-induced depressive-like behaviors via affecting microglial and NLRP3 inflammasome activation. Life Sci. 2020, 263, 118582. [Google Scholar] [CrossRef] [PubMed]
- Gustin, A.; Kirchmeyer, M.; Koncina, E.; Felten, P.; Losciuto, S.; Heurtaux, T.; Tardivel, A.; Heuschling, P.; Dostert, C. NLRP3 Inflammasome Is Expressed and Functional in Mouse Brain Microglia but Not in Astrocytes. PLoS ONE 2015, 10, e0130624. [Google Scholar] [CrossRef] [PubMed]
- Sanna, M.G.; Liao, J.; Jo, E.; Alfonso, C.; Ahn, M.Y.; Peterson, M.S.; Webb, B.; Lefebvre, S.; Chun, J.; Gray, N.; et al. Sphingosine 1-phosphate (S1P) receptor subtypes S1P1 and S1P3, respectively, regulate lymphocyte recirculation and heart rate. J. Biol. Chem. 2004, 279, 13839–13848. [Google Scholar] [CrossRef]
- Pepin, E.; Jalinier, T.; Lemieux, G.L.; Massicotte, G.; Cyr, M. Sphingosine-1-Phosphate Receptors Modulators Decrease Signs of Neuroinflammation and Prevent Parkinson’s Disease Symptoms in the 1-Methyl-4-Phenyl-1,2,3,6-Tetrahydropyridine Mouse Model. Front. Pharmacol. 2020, 11, 77. [Google Scholar] [CrossRef]
- Ji, J.; Wang, J.; Yang, J.; Wang, X.P.; Huang, J.J.; Xue, T.F.; Sun, X.L. The Intra-nuclear SphK2-S1P Axis Facilitates M1-to-M2 Shift of Microglia via Suppressing HDAC1-Mediated KLF4 Deacetylation. Front. Immunol. 2019, 10, 1241. [Google Scholar] [CrossRef]
- Das, A.; Arifuzzaman, S.; Kim, S.H.; Lee, Y.S.; Jung, K.H.; Chai, Y.G. FTY720 (fingolimod) regulates key target genes essential for inflammation in microglial cells as defined by high-resolution mRNA sequencing. Neuropharmacology 2017, 119, 1–14. [Google Scholar] [CrossRef]
- Aytan, N.; Choi, J.K.; Carreras, I.; Brinkmann, V.; Kowall, N.W.; Jenkins, B.G.; Dedeoglu, A. Fingolimod modulates multiple neuroinflammatory markers in a mouse model of Alzheimer’s disease. Sci. Rep. 2016, 6, 24939. [Google Scholar] [CrossRef]
- Serdar, M.; Herz, J.; Kempe, K.; Lumpe, K.; Reinboth, B.S.; Sizonenko, S.V.; Hou, X.; Herrmann, R.; Hadamitzky, M.; Heumann, R.; et al. Fingolimod protects against neonatal white matter damage and long-term cognitive deficits caused by hyperoxia. Brain Behav. Immun. 2016, 52, 106–119. [Google Scholar] [CrossRef]
- Bechet, S.; O’Sullivan, S.A.; Yssel, J.; Fagan, S.G.; Dev, K.K. Fingolimod Rescues Demyelination in a Mouse Model of Krabbe’s Disease. J. Neurosci. 2020, 40, 3104–3118. [Google Scholar] [CrossRef]
- Wang, Z.; Kawabori, M.; Houkin, K. FTY720 (Fingolimod) Ameliorates Brain Injury through Multiple Mechanisms and is a Strong Candidate for Stroke Treatment. Curr. Med. Chem. 2020, 27, 2979–2993. [Google Scholar] [CrossRef]
- Constantinescu, C.S.; Farooqi, N.; O’Brien, K.; Gran, B. Experimental autoimmune encephalomyelitis (EAE) as a model for multiple sclerosis (MS). Br. J. Pharmacol. 2011, 164, 1079–1106. [Google Scholar] [CrossRef]
- Burrows, D.J.; McGown, A.; Jain, S.A.; De Felice, M.; Ramesh, T.M.; Sharrack, B.; Majid, A. Animal models of multiple sclerosis: From rodents to zebrafish. Mult. Scler. 2019, 25, 306–324. [Google Scholar] [CrossRef] [PubMed]
- Scott, F.L.; Clemons, B.; Brooks, J.; Brahmachary, E.; Powell, R.; Dedman, H.; Desale, H.G.; Timony, G.A.; Martinborough, E.; Rosen, H.; et al. Ozanimod (RPC1063) is a potent sphingosine-1-phosphate receptor-1 (S1P1 ) and receptor-5 (S1P5 ) agonist with autoimmune disease-modifying activity. Br. J. Pharmacol. 2016, 173, 1778–1792. [Google Scholar] [CrossRef]
- Musella, A.; Gentile, A.; Guadalupi, L.; Rizzo, F.R.; De Vito, F.; Fresegna, D.; Bruno, A.; Dolcetti, E.; Vanni, V.; Vitiello, L.; et al. Central Modulation of Selective Sphingosine-1-Phosphate Receptor 1 Ameliorates Experimental Multiple Sclerosis. Cells 2020, 9, 1290. [Google Scholar] [CrossRef] [PubMed]
- Behrangi, N.; Fischbach, F.; Kipp, M. Mechanism of Siponimod: Anti-Inflammatory and Neuroprotective Mode of Action. Cells 2019, 8, 24. [Google Scholar] [CrossRef]
- Ward, L.A.; Lee, D.S.; Sharma, A.; Wang, A.; Naouar, I.; Ma, X.I.; Pikor, N.; Nuesslein-Hildesheim, B.; Ramaglia, V.; Gommerman, J.L. Siponimod therapy implicates Th17 cells in a preclinical model of subpial cortical injury. JCI Insight. 2020, 5. [Google Scholar] [CrossRef] [PubMed]
- Cahalan, S.M.; Gonzalez-Cabrera, P.J.; Sarkisyan, G.; Nguyen, N.; Schaeffer, M.T.; Huang, L.; Yeager, A.; Clemons, B.; Scott, F.; Rosen, H. Actions of a picomolar short-acting S1P(1) agonist in S1P(1)-eGFP knock-in mice. Nat. Chem. Biol. 2011, 7, 254–256. [Google Scholar] [CrossRef]
- Li, R.; Venkat, P.; Chopp, M.; Zhang, Q.; Yan, T.; Chen, J. RP001 hydrochloride improves neurological outcome after subarachnoid hemorrhage. J. Neurol. Sci. 2019, 399, 6–14. [Google Scholar] [CrossRef]
- Asle-Rousta, M.; Kolahdooz, Z.; Dargahi, L.; Ahmadiani, A.; Nasoohi, S. Prominence of central sphingosine-1-phosphate receptor-1 in attenuating abeta-induced injury by fingolimod. J. Mol. Neurosci. 2014, 54, 698–703. [Google Scholar] [CrossRef]
- Carreras, I.; Aytan, N.; Choi, J.K.; Tognoni, C.M.; Kowall, N.W.; Jenkins, B.G.; Dedeoglu, A. Dual dose-dependent effects of fingolimod in a mouse model of Alzheimer’s disease. Sci. Rep. 2019, 9, 10972. [Google Scholar] [CrossRef]
- Sawano, T.; Watanabe, F.; Ishiguchi, M.; Doe, N.; Furuyama, T.; Inagaki, S. Effect of Sema4D on microglial function in middle cerebral artery occlusion mice. Glia 2015, 63, 2249–2259. [Google Scholar] [CrossRef]
- Tam, W.Y.; Ma, C.H. Bipolar/rod-shaped microglia are proliferating microglia with distinct M1/M2 phenotypes. Sci. Rep. 2014, 4, 7279. [Google Scholar] [CrossRef]
- Kartalou, G.I.; Salgueiro-Pereira, A.R.; Endres, T.; Lesnikova, A.; Casarotto, P.; Pousinha, P.; Delanoe, K.; Edelmann, E.; Castren, E.; Gottmann, K.; et al. Anti-Inflammatory Treatment with FTY720 Starting after Onset of Symptoms Reverses Synaptic Deficits in an AD Mouse Model. Int. J. Mol. Sci. 2020, 21, 8957. [Google Scholar] [CrossRef]
- McManus, R.M.; Finucane, O.M.; Wilk, M.M.; Mills, K.H.G.; Lynch, M.A. FTY720 Attenuates Infection-Induced Enhancement of Abeta Accumulation in APP/PS1 Mice by Modulating Astrocytic Activation. J. Neuroimmune Pharmacol. 2017, 12, 670–681. [Google Scholar] [CrossRef]
- Ren, M.; Han, M.; Wei, X.; Guo, Y.; Shi, H.; Zhang, X.; Perez, R.G.; Lou, H. FTY720 Attenuates 6-OHDA-Associated Dopaminergic Degeneration in Cellular and Mouse Parkinsonian Models. Neurochem. Res. 2017, 42, 686–696. [Google Scholar] [CrossRef] [PubMed]
- Yao, S.; Li, L.; Sun, X.; Hua, J.; Zhang, K.; Hao, L.; Liu, L.; Shi, D.; Zhou, H. FTY720 Inhibits MPP(+)-Induced Microglial Activation by Affecting NLRP3 Inflammasome Activation. J. Neuroimmune Pharm. 2019, 14, 478–492. [Google Scholar] [CrossRef]
- Gaire, B.P.; Lee, C.H.; Sapkota, A.; Lee, S.Y.; Chun, J.; Cho, H.J.; Nam, T.G.; Choi, J.W. Identification of Sphingosine 1-Phosphate Receptor Subtype 1 (S1P1) as a Pathogenic Factor in Transient Focal Cerebral Ischemia. Mol. Neurobiol. 2018, 55, 2320–2332. [Google Scholar] [CrossRef]
- Gaire, B.P.; Bae, Y.J.; Choi, J.W. S1P1 Regulates M1/M2 Polarization toward Brain Injury after Transient Focal Cerebral Ischemia. Biomol. Ther. 2019, 522–529. [Google Scholar] [CrossRef] [PubMed]
- Jayaraj, R.L.; Azimullah, S.; Beiram, R.; Jalal, F.Y.; Rosenberg, G.A. Neuroinflammation: Friend and foe for ischemic stroke. J. Neuroinflamm. 2019, 16, 142. [Google Scholar] [CrossRef]
- Kunz, A.; Abe, T.; Hochrainer, K.; Shimamura, M.; Anrather, J.; Racchumi, G.; Zhou, P.; Iadecola, C. Nuclear factor-kappaB activation and postischemic inflammation are suppressed in CD36-null mice after middle cerebral artery occlusion. J. Neurosci. 2008, 28, 1649–1658. [Google Scholar] [CrossRef]
- Xie, W.; Zhu, T.; Dong, X.; Nan, F.; Meng, X.; Zhou, P.; Sun, G.; Sun, X. HMGB1-triggered inflammation inhibition of notoginseng leaf triterpenes against cerebral ischemia and reperfusion injury via MAPK and NF-kappaB signaling pathways. Biomolecules 2019, 9, 512. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Cabrera, P.J.; Cahalan, S.M.; Nguyen, N.; Sarkisyan, G.; Leaf, N.B.; Cameron, M.D.; Kago, T.; Rosen, H. S1P(1) receptor modulation with cyclical recovery from lymphopenia ameliorates mouse model of multiple sclerosis. Mol. Pharmacol. 2012, 81, 166–174. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Bielawski, J.; Yang, H.; Kong, Y.; Zhou, B.; Li, J. Functional antagonism of sphingosine-1-phosphate receptor 1 prevents cuprizone-induced demyelination. Glia 2018, 66, 654–669. [Google Scholar] [CrossRef]
- Rothhammer, V.; Kenison, J.E.; Tjon, E.; Takenaka, M.C.; de Lima, K.A.; Borucki, D.M.; Chao, C.C.; Wilz, A.; Blain, M.; Healy, L.; et al. Sphingosine 1-phosphate receptor modulation suppresses pathogenic astrocyte activation and chronic progressive CNS inflammation. Proc. Natl. Acad. Sci. USA 2017, 114, 2012–2017. [Google Scholar] [CrossRef] [PubMed]
- Oveland, E.; Ahmad, I.; Lereim, R.R.; Kroksveen, A.C.; Barsnes, H.; Guldbrandsen, A.; Myhr, K.M.; Bo, L.; Berven, F.S.; Wergeland, S. Cuprizone and EAE mouse frontal cortex proteomics revealed proteins altered in multiple sclerosis. Sci. Rep. 2021, 11, 7174. [Google Scholar] [CrossRef]
- Hillis, J.M.; Davies, J.; Mundim, M.V.; Al-Dalahmah, O.; Szele, F.G. Cuprizone demyelination induces a unique inflammatory response in the subventricular zone. J. Neuroinflamm. 2016, 13, 190. [Google Scholar] [CrossRef] [PubMed]
- Sapkota, A.; Gaire, B.P.; Kang, M.G.; Choi, J.W. S1P2 contributes to microglial activation and M1 polarization following cerebral ischemia through ERK1/2 and JNK. Sci. Rep. 2019, 9, 12106. [Google Scholar] [CrossRef]
- Sanchez, T.; Skoura, A.; Wu, M.T.; Casserly, B.; Harrington, E.O.; Hla, T. Induction of vascular permeability by the sphingosine-1-phosphate receptor-2 (S1P2R) and its downstream effectors ROCK and PTEN. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 1312–1318. [Google Scholar] [CrossRef]
- Arikawa, K.; Takuwa, N.; Yamaguchi, H.; Sugimoto, N.; Kitayama, J.; Nagawa, H.; Takehara, K.; Takuwa, Y. Ligand-dependent inhibition of B16 melanoma cell migration and invasion via endogenous S1P2 G protein-coupled receptor. Requirement of inhibition of cellular RAC activity. J. Biol. Chem. 2003, 278, 32841–32851. [Google Scholar] [CrossRef]
- Kim, G.S.; Yang, L.; Zhang, G.; Zhao, H.; Selim, M.; McCullough, L.D.; Kluk, M.J.; Sanchez, T. Critical role of sphingosine-1-phosphate receptor-2 in the disruption of cerebrovascular integrity in experimental stroke. Nat. Commun. 2015, 6, 7893. [Google Scholar] [CrossRef]
- Shirakawa, H.; Katsumoto, R.; Iida, S.; Miyake, T.; Higuchi, T.; Nagashima, T.; Nagayasu, K.; Nakagawa, T.; Kaneko, S. Sphingosine-1-phosphate induces Ca(2+) signaling and CXCL1 release via TRPC6 channel in astrocytes. Glia 2017, 65, 1005–1016. [Google Scholar] [CrossRef]
- Li, C.; Li, J.N.; Kays, J.; Guerrero, M.; Nicol, G.D. Sphingosine 1-phosphate enhances the excitability of rat sensory neurons through activation of sphingosine 1-phosphate receptors 1 and/or 3. J. Neuroinflamm. 2015, 12, 70. [Google Scholar] [CrossRef]
- Tang, H.B.; Jiang, X.J.; Wang, C.; Liu, S.C. S1P/S1PR3 signaling mediated proliferation of pericytes via Ras/pERK pathway and CAY10444 had beneficial effects on spinal cord injury. Biochem. Biophys. Res. Commun. 2018, 498, 830–836. [Google Scholar] [CrossRef]
- Gaire, B.P.; Song, M.R.; Choi, J.W. Sphingosine 1-phosphate receptor subtype 3 (S1P3) contributes to brain injury after transient focal cerebral ischemia via modulating microglial activation and their M1 polarization. J. Neuroinflamm. 2018, 15, 284. [Google Scholar] [CrossRef]
- Groves, A.; Kihara, Y.; Chun, J. Fingolimod: Direct CNS effects of sphingosine 1-phosphate (S1P) receptor modulation and implications in multiple sclerosis therapy. J. Neurol. Sci. 2013, 328, 9–18. [Google Scholar] [CrossRef] [PubMed]
- Graeler, M.; Goetzl, E.J. Activation-regulated expression and chemotactic function of sphingosine 1-phosphate receptors in mouse splenic T cells. FASEB J. 2002, 16, 1874–1878. [Google Scholar] [CrossRef] [PubMed]
- Fettel, J.; Kuhn, B.; Guillen, N.A.; Surun, D.; Peters, M.; Bauer, R.; Angioni, C.; Geisslinger, G.; Schnutgen, F.; Meyer Zu Heringdorf, D.; et al. Sphingosine-1-phosphate (S1P) induces potent anti-inflammatory effects in vitro and in vivo by S1P receptor 4-mediated suppression of 5-lipoxygenase activity. FASEB J. 2019, 33, 1711–1726. [Google Scholar] [CrossRef] [PubMed]
- Allende, M.L.; Bektas, M.; Lee, B.G.; Bonifacino, E.; Kang, J.; Tuymetova, G.; Chen, W.; Saba, J.D.; Proia, R.L. Sphingosine-1-phosphate lyase deficiency produces a pro-inflammatory response while impairing neutrophil trafficking. J. Biol. Chem. 2011, 286, 7348–7358. [Google Scholar] [CrossRef]
- Salas-Perdomo, A.; Miro-Mur, F.; Gallizioli, M.; Brait, V.H.; Justicia, C.; Meissner, A.; Urra, X.; Chamorro, A.; Planas, A.M. Role of the S1P pathway and inhibition by fingolimod in preventing hemorrhagic transformation after stroke. Sci. Rep. 2019, 9, 8309. [Google Scholar] [CrossRef]
- Foster, C.A.; Mechtcheriakova, D.; Storch, M.K.; Balatoni, B.; Howard, L.M.; Bornancin, F.; Wlachos, A.; Sobanov, J.; Kinnunen, A.; Baumruker, T. FTY720 rescue therapy in the dark agouti rat model of experimental autoimmune encephalomyelitis: Expression of central nervous system genes and reversal of blood-brain-barrier damage. Brain Pathol. 2009, 19, 254–266. [Google Scholar] [CrossRef]
- Tham, C.S.; Lin, F.F.; Rao, T.S.; Yu, N.; Webb, M. Microglial activation state and lysophospholipid acid receptor expression. Int. J. Dev. Neurosci. 2003, 21, 431–443. [Google Scholar] [CrossRef]
- Miron, V.E.; Ludwin, S.K.; Darlington, P.J.; Jarjour, A.A.; Soliven, B.; Kennedy, T.E.; Antel, J.P. Fingolimod (FTY720) enhances remyelination following demyelination of organotypic cerebellar slices. Am. J. Pathol. 2010, 176, 2682–2694. [Google Scholar] [CrossRef] [PubMed]
- Jonnalagadda, D.; Sunkara, M.; Morris, A.J.; Whiteheart, S.W. Granule-mediated release of sphingosine-1-phosphate by activated platelets. Biochim. Biophys. Acta 2014, 1841, 1581–1589. [Google Scholar] [CrossRef] [PubMed]
- Weth, D.; Benetti, C.; Rauch, C.; Gstraunthaler, G.; Schmidt, H.; Geisslinger, G.; Sabbadini, R.; Proia, R.L.; Kress, M. Activated platelets release sphingosine 1-phosphate and induce hypersensitivity to noxious heat stimuli in vivo. Front. Neurosci. 2015, 9, 140. [Google Scholar] [CrossRef] [PubMed]
- Anelli, V.; Bassi, R.; Tettamanti, G.; Viani, P.; Riboni, L. Extracellular release of newly synthesized sphingosine-1-phosphate by cerebellar granule cells and astrocytes. J. Neurochem. 2005, 92, 1204–1215. [Google Scholar] [CrossRef]
- Bassi, R.; Anelli, V.; Giussani, P.; Tettamanti, G.; Viani, P.; Riboni, L. Sphingosine-1-phosphate is released by cerebellar astrocytes in response to bFGF and induces astrocyte proliferation through Gi-protein-coupled receptors. Glia 2006, 53, 621–630. [Google Scholar] [CrossRef]
- Kimura, A.; Ohmori, T.; Ohkawa, R.; Madoiwa, S.; Mimuro, J.; Murakami, T.; Kobayashi, E.; Hoshino, Y.; Yatomi, Y.; Sakata, Y. Essential roles of sphingosine 1-phosphate/S1P1 receptor axis in the migration of neural stem cells toward a site of spinal cord injury. Stem. Cells 2007, 25, 115–124. [Google Scholar] [CrossRef]
- Fischer, I.; Alliod, C.; Martinier, N.; Newcombe, J.; Brana, C.; Pouly, S. Sphingosine kinase 1 and sphingosine 1-phosphate receptor 3 are functionally upregulated on astrocytes under pro-inflammatory conditions. PLoS ONE 2011, 6, e23905. [Google Scholar] [CrossRef]
- Sanada, Y.; Mizushima, T.; Kai, Y.; Nishimura, J.; Hagiya, H.; Kurata, H.; Mizuno, H.; Uejima, E.; Ito, T. Therapeutic effects of novel sphingosine-1-phosphate receptor agonist W-061 in murine DSS colitis. PLoS ONE 2011, 6, e23933. [Google Scholar] [CrossRef] [PubMed]
- Volpi, C.; Orabona, C.; Macchiarulo, A.; Bianchi, R.; Puccetti, P.; Grohmann, U. Preclinical discovery and development of fingolimod for the treatment of multiple sclerosis. Expert. Opin. Drug. Discov. 2019, 14, 1199–1212. [Google Scholar] [CrossRef]
- Sorensen, S.D.; Nicole, O.; Peavy, R.D.; Montoya, L.M.; Lee, C.J.; Murphy, T.J.; Traynelis, S.F.; Hepler, J.R. Common signaling pathways link activation of murine PAR-1, LPA, and S1P receptors to proliferation of astrocytes. Mol. Pharmacol. 2003, 64, 1199–1209. [Google Scholar] [CrossRef]
- Rao, T.S.; Lariosa-Willingham, K.D.; Lin, F.F.; Palfreyman, E.L.; Yu, N.; Chun, J.; Webb, M. Pharmacological characterization of lysophospholipid receptor signal transduction pathways in rat cerebrocortical astrocytes. Brain Res. 2003, 990, 182–194. [Google Scholar] [CrossRef]
- Rao, T.S.; Lariosa-Willingham, K.D.; Lin, F.F.; Yu, N.; Tham, C.S.; Chun, J.; Webb, M. Growth factor pre-treatment differentially regulates phosphoinositide turnover downstream of lysophospholipid receptor and metabotropic glutamate receptors in cultured rat cerebrocortical astrocytes. Int. J. Dev. Neurosci. 2004, 22, 131–135. [Google Scholar] [CrossRef]
- Giovannoni, F.; Quintana, F.J. The Role of Astrocytes in CNS Inflammation. Trends Immunol. 2020, 41, 805–819. [Google Scholar] [CrossRef] [PubMed]
- Linnerbauer, M.; Wheeler, M.A.; Quintana, F.J. Astrocyte Crosstalk in CNS Inflammation. Neuron 2020, 108, 608–622. [Google Scholar] [CrossRef]
- Farez, M.F.; Correale, J. Sphingosine 1-phosphate signaling in astrocytes: Implications for progressive multiple sclerosis. J. Neurol. Sci. 2016, 361, 60–65. [Google Scholar] [CrossRef]
- Van Doorn, R.; Van Horssen, J.; Verzijl, D.; Witte, M.; Ronken, E.; Van Het Hof, B.; Lakeman, K.; Dijkstra, C.D.; Van Der Valk, P.; Reijerkerk, A.; et al. Sphingosine 1-phosphate receptor 1 and 3 are upregulated in multiple sclerosis lesions. Glia 2010, 58, 1465–1476. [Google Scholar] [CrossRef]
- Brana, C.; Frossard, M.J.; Pescini Gobert, R.; Martinier, N.; Boschert, U.; Seabrook, T.J. Immunohistochemical detection of sphingosine-1-phosphate receptor 1 and 5 in human multiple sclerosis lesions. Neuropathol. Appl. Neurobiol. 2014, 40, 564–578. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.A.; Chun, J. Mechanisms of fingolimod’s efficacy and adverse effects in multiple sclerosis. Ann. Neurol. 2011, 69, 759–777. [Google Scholar] [CrossRef]
- Hunter, S.F.; Bowen, J.D.; Reder, A.T. The Direct Effects of Fingolimod in the Central Nervous System: Implications for Relapsing Multiple Sclerosis. CNS Drugs 2016, 30, 135–147. [Google Scholar] [CrossRef]
- Wang, J.; Wang, J.; Lu, P.; Cai, Y.; Wang, Y.; Hong, L.; Ren, H.; Heng, B.C.; Liu, H.; Zhou, J.; et al. Local delivery of FTY720 in PCL membrane improves SCI functional recovery by reducing reactive astrogliosis. Biomaterials 2015, 62, 76–87. [Google Scholar] [CrossRef] [PubMed]
- Cruz, V.T.; Fonseca, J. Central effects of fingolimod. Rev. Neurol. 2014, 59, 121–128. [Google Scholar]
- Cheng, W.; Chen, G. Chemokines and chemokine receptors in multiple sclerosis. Mediat. Inflamm. 2014, 2014, 659206. [Google Scholar] [CrossRef]
- Ponath, G.; Park, C.; Pitt, D. The Role of Astrocytes in Multiple Sclerosis. Front. Immunol. 2018, 9, 217. [Google Scholar] [CrossRef]
- Bjelobaba, I.; Savic, D.; Lavrnja, I. Multiple Sclerosis and Neuroinflammation: The Overview of Current and Prospective Therapies. Curr. Pharm. Des. 2017, 23, 693–730. [Google Scholar] [CrossRef] [PubMed]
- Colombo, E.; Di Dario, M.; Capitolo, E.; Chaabane, L.; Newcombe, J.; Martino, G.; Farina, C. Fingolimod may support neuroprotection via blockade of astrocyte nitric oxide. Ann. Neurol. 2014, 76, 325–337. [Google Scholar] [CrossRef]
- Brinkmann, V. FTY720 (fingolimod) in Multiple Sclerosis: Therapeutic effects in the immune and the central nervous system. Br. J. Pharmacol. 2009, 158, 1173–1182. [Google Scholar] [CrossRef] [PubMed]
- Ma, W.; Zhang, M.; Liu, S.; Wang, M.; Shi, Y.; Yang, T.; Li, X.; Zhu, L. Matrine alleviates astrogliosis through sphingosine 1-phosphate signaling in experimental autoimmune encephalomyelitis. Neurosci. Lett. 2020, 715, 134599. [Google Scholar] [CrossRef]
- Osinde, M.; Mullershausen, F.; Dev, K.K. Phosphorylated FTY720 stimulates ERK phosphorylation in astrocytes via S1P receptors. Neuropharmacology 2007, 52, 1210–1218. [Google Scholar] [CrossRef] [PubMed]
- Lu, N.; Malemud, C.J. Extracellular Signal-Regulated Kinase: A Regulator of Cell Growth, Inflammation, Chondrocyte and Bone Cell Receptor-Mediated Gene Expression. Int. J. Mol. Sci. 2019, 20, 3792. [Google Scholar] [CrossRef]
- Sun, J.; Nan, G. The extracellular signal-regulated kinase 1/2 pathway in neurological diseases: A potential therapeutic target (Review). Int. J. Mol. Med. 2017, 39, 1338–1346. [Google Scholar] [CrossRef] [PubMed]
- Okazaki, R.; Doi, T.; Hayakawa, K.; Morioka, K.; Imamura, O.; Takishima, K.; Hamanoue, M.; Sawada, Y.; Nagao, M.; Tanaka, S.; et al. The crucial role of Erk2 in demyelinating inflammation in the central nervous system. J. Neuroinflamm. 2016, 13, 235. [Google Scholar] [CrossRef]
- Colombo, E.; Bassani, C.; De Angelis, A.; Ruffini, F.; Ottoboni, L.; Comi, G.; Martino, G.; Farina, C. Siponimod (BAF312) Activates Nrf2 While Hampering NFkappaB in Human Astrocytes, and Protects From Astrocyte-Induced Neurodegeneration. Front. Immunol. 2020, 11, 635. [Google Scholar] [CrossRef] [PubMed]
- Vomund, S.; Schafer, A.; Parnham, M.J.; Brune, B.; von Knethen, A. Nrf2, the Master Regulator of Anti-Oxidative Responses. Int. J. Mol. Sci. 2017, 18, 2772. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, C.; Schubart, A.; Mir, A.K.; Dev, K.K. The dual S1PR1/S1PR5 drug BAF312 (Siponimod) attenuates demyelination in organotypic slice cultures. J. Neuroinflamm. 2016, 13, 31. [Google Scholar] [CrossRef] [PubMed]
- Norimatsu, Y.; Ohmori, T.; Kimura, A.; Madoiwa, S.; Mimuro, J.; Seichi, A.; Yatomi, Y.; Hoshino, Y.; Sakata, Y. FTY720 improves functional recovery after spinal cord injury by primarily nonimmunomodulatory mechanisms. Am. J. Pathol. 2012, 180, 1625–1635. [Google Scholar] [CrossRef]
- Dusaban, S.S.; Chun, J.; Rosen, H.; Purcell, N.H.; Brown, J.H. Sphingosine 1-phosphate receptor 3 and RhoA signaling mediate inflammatory gene expression in astrocytes. J. Neuroinflamm. 2017, 14, 111. [Google Scholar] [CrossRef]
- Ferrer, M.D.; Busquets-Cortes, C.; Capo, X.; Tejada, S.; Tur, J.A.; Pons, A.; Sureda, A. Cyclooxygenase-2 Inhibitors as a Therapeutic Target in Inflammatory Diseases. Curr. Med. Chem. 2019, 26, 3225–3241. [Google Scholar] [CrossRef]
- Janssen, S.; Schlegel, C.; Gudi, V.; Prajeeth, C.K.; Skripuletz, T.; Trebst, C.; Stangel, M. Effect of FTY720-phosphate on the expression of inflammation-associated molecules in astrocytes in vitro. Mol. Med. Rep. 2015, 12, 6171–6177. [Google Scholar] [CrossRef]
- Conductier, G.; Blondeau, N.; Guyon, A.; Nahon, J.L.; Rovere, C. The role of monocyte chemoattractant protein MCP1/CCL2 in neuroinflammatory diseases. J. Neuroimmunol. 2010, 224, 93–100. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, Y.; Sano, T.; Irisa, M.; Kodama, T.; Saito, T.; Furusawa, E.; Kaizu, K.; Yanagi, Y.; Tsukimura, T.; Togawa, T.; et al. FcRgamma-dependent immune activation initiates astrogliosis during the asymptomatic phase of Sandhoff disease model mice. Sci. Rep. 2017, 7, 40518. [Google Scholar] [CrossRef]
- Wu, Y.P.; Mizugishi, K.; Bektas, M.; Sandhoff, R.; Proia, R.L. Sphingosine kinase 1/S1P receptor signaling axis controls glial proliferation in mice with Sandhoff disease. Hum. Mol. Genet. 2008, 17, 2257–2264. [Google Scholar] [CrossRef] [PubMed]
- Gril, B.; Paranjape, A.N.; Woditschka, S.; Hua, E.; Dolan, E.L.; Hanson, J.; Wu, X.; Kloc, W.; Izycka-Swieszewska, E.; Duchnowska, R.; et al. Reactive astrocytic S1P3 signaling modulates the blood-tumor barrier in brain metastases. Nat. Commun. 2018, 9, 2705. [Google Scholar] [CrossRef]
- Mustaly-Kalimi, S.; Littlefield, A.M.; Stutzmann, G.E. Calcium Signaling Deficits in Glia and Autophagic Pathways Contributing to Neurodegenerative Disease. Antioxid Redox Signal. 2018, 29, 1158–1175. [Google Scholar] [CrossRef]
- Yu, Z.; Dou, F.; Wang, Y.; Hou, L.; Chen, H. Ca(2+)-dependent endoplasmic reticulum stress correlation with astrogliosis involves upregulation of KCa3.1 and inhibition of AKT/mTOR signaling. J. Neuroinflamm. 2018, 15, 316. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Li, X.; Ciric, B.; Ma, C.G.; Gran, B.; Rostami, A.; Zhang, G.X. Effect of Fingolimod on Neural Stem Cells: A Novel Mechanism and Broadened Application for Neural Repair. Mol. Ther. 2017, 25, 401–415. [Google Scholar] [CrossRef] [PubMed]
- Mutoh, T.; Rivera, R.; Chun, J. Insights into the pharmacological relevance of lysophospholipid receptors. Br. J. Pharmacol. 2012, 165, 829–844. [Google Scholar] [CrossRef]
- Karunakaran, I.; van Echten-Deckert, G. Sphingosine 1-phosphate—A double edged sword in the brain. Biochim. Biophys. Acta Biomembr. 2017, 1859, 1573–1582. [Google Scholar] [CrossRef]
- Bernhart, E.; Kollroser, M.; Rechberger, G.; Reicher, H.; Heinemann, A.; Schratl, P.; Hallstrom, S.; Wintersperger, A.; Nusshold, C.; DeVaney, T.; et al. Lysophosphatidic acid receptor activation affects the C13NJ microglia cell line proteome leading to alterations in glycolysis, motility, and cytoskeletal architecture. Proteomics 2010, 10, 141–158. [Google Scholar] [CrossRef]
- Fujita, R.; Ma, Y.; Ueda, H. Lysophosphatidic acid-induced membrane ruffling and brain-derived neurotrophic factor gene expression are mediated by ATP release in primary microglia. J. Neurochem. 2008, 107, 152–160. [Google Scholar] [CrossRef]
- Plastira, I.; Bernhart, E.; Joshi, L.; Koyani, C.N.; Strohmaier, H.; Reicher, H.; Malle, E.; Sattler, W. MAPK signaling determines lysophosphatidic acid (LPA)-induced inflammation in microglia. J. Neuroinflamm. 2020, 17, 127. [Google Scholar] [CrossRef]
- Plastira, I.; Bernhart, E.; Goeritzer, M.; Reicher, H.; Kumble, V.B.; Kogelnik, N.; Wintersperger, A.; Hammer, A.; Schlager, S.; Jandl, K.; et al. 1-Oleyl-lysophosphatidic acid (LPA) promotes polarization of BV-2 and primary murine microglia towards an M1-like phenotype. J. Neuroinflamm. 2016, 13, 205. [Google Scholar] [CrossRef] [PubMed]
- Uchida, H.; Matsumoto, M.; Ueda, H. Profiling of BoNT/C3-reversible gene expression induced by lysophosphatidic acid: ephrinB1 gene up-regulation underlying neuropathic hyperalgesia and allodynia. Neurochem. Int. 2009, 54, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Nagai, J.; Ueda, H. Microglial activation mediates de novo lysophosphatidic acid production in a model of neuropathic pain. J. Neurochem. 2010, 115, 643–653. [Google Scholar] [CrossRef] [PubMed]
- Ledeboer, A.; Sloane, E.M.; Milligan, E.D.; Frank, M.G.; Mahony, J.H.; Maier, S.F.; Watkins, L.R. Minocycline attenuates mechanical allodynia and proinflammatory cytokine expression in rat models of pain facilitation. Pain 2005, 115, 71–83. [Google Scholar] [CrossRef] [PubMed]
- Ueda, H. Lysophosphatidic acid as the initiator of neuropathic pain. Biol. Pharm. Bull. 2011, 34, 1154–1158. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ueda, H.; Ueda, M. Lysophosphatidic acid as an initiator of neuropathic pain: Biosynthesis and demyelination. Clin. Lipidol. 2011, 6, 147–158. [Google Scholar] [CrossRef]
- Kwon, J.H.; Gaire, B.P.; Park, S.J.; Shin, D.Y.; Choi, J.W. Identifying lysophosphatidic acid receptor subtype 1 (LPA1) as a novel factor to modulate microglial activation and their TNF-alpha production by activating ERK1/2. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2018, 1863, 1237–1245. [Google Scholar] [CrossRef]
- Amaral, R.F.; Geraldo, L.H.M.; Einicker-Lamas, M.; TCLS, E.S.; Mendes, F.; Lima, F.R.S. Microglial lysophosphatidic acid promotes glioblastoma proliferation and migration via LPA1 receptor. J. Neurochem. 2021, 156, 499–512. [Google Scholar] [CrossRef]
- Gaire, B.P.; Sapkota, A.; Choi, J.W. BMS-986020, a Specific LPA1 Antagonist, Provides Neuroprotection against Ischemic Stroke in Mice. Antioxidants 2020, 9, 1097. [Google Scholar] [CrossRef] [PubMed]
- Lee, C.H.; Sapkota, A.; Gaire, B.P.; Choi, J.W. NLRP3 Inflammasome Activation Is Involved in LPA1-Mediated Brain Injury after Transient Focal Cerebral Ischemia. Int. J. Mol. Sci. 2020, 21, 8595. [Google Scholar] [CrossRef] [PubMed]
- Gaire, B.P.; Sapkota, A.; Song, M.R.; Choi, J.W. Lysophosphatidic acid receptor 1 (LPA1) plays critical roles in microglial activation and brain damage after transient focal cerebral ischemia. J. Neuroinflamm. 2019, 16, 170. [Google Scholar] [CrossRef]
- Kihara, Y.; Mizuno, H.; Chun, J. Lysophospholipid receptors in drug discovery. Exp. Cell. Res. 2015, 333, 171–177. [Google Scholar] [CrossRef]
- Smith, M.J.; Lawler, M.J.; Kopp, N.; Mcleod, D.D.; Davulcu, A.H.; Lin, D.; Katipally, K.; Sfouggatakis, C. Development of a Concise Multikilogram Synthesis of LPA-1 Antagonist BMS-986020 via a Tandem Borylation–Suzuki Procedure. Org. Process. Res. Dev. 2017, 21, 1859–1863. [Google Scholar] [CrossRef]
- Pena, A.; kim, J.; Donnelly, D.; Murphy, B.; Shuster, D.; Watson, L.; Bonacorsi, S.; Hayes, W.; Chow, P.; Du, S. Autoradiographic evaluation of [18F]BMT-083133, a lysophosphatidic acid receptor 1 (LPA1) radioligand. J. Nucl. Med. 2014, 55, 1207. [Google Scholar]
- Gao, L.; Shi, H.; Sherchan, P.; Tang, H.; Peng, L.; Xie, S.; Liu, R.; Hu, X.; Tang, J.; Xia, Y.; et al. Inhibition of lysophosphatidic acid receptor 1 attenuates neuroinflammation via PGE2/EP2/NOX2 signalling and improves the outcome of intracerebral haemorrhage in mice. Brain Behav. Immun. 2021, 91, 615–626. [Google Scholar] [CrossRef] [PubMed]
- Swaney, J.S.; Chapman, C.; Correa, L.D.; Stebbins, K.J.; Bundey, R.A.; Prodanovich, P.C.; Fagan, P.; Baccei, C.S.; Santini, A.M.; Hutchinson, J.H.; et al. A novel, orally active LPA(1) receptor antagonist inhibits lung fibrosis in the mouse bleomycin model. Br. J. Pharmacol. 2010, 160, 1699–1713. [Google Scholar] [CrossRef] [PubMed]
- Swaney, J.S.; Chapman, C.; Correa, L.D.; Stebbins, K.J.; Broadhead, A.R.; Bain, G.; Santini, A.M.; Darlington, J.; King, C.D.; Baccei, C.S.; et al. Pharmacokinetic and pharmacodynamic characterization of an oral lysophosphatidic acid type 1 receptor-selective antagonist. J. Pharmacol. Exp. Ther. 2011, 336, 693–700. [Google Scholar] [CrossRef]
- Rivera, R.R.; Lin, M.E.; Bornhop, E.C.; Chun, J. Conditional Lpar1 gene targeting identifies cell types mediating neuropathic pain. FASEB J. 2020, 34, 8833–8842. [Google Scholar] [CrossRef]
- Ueda, H. LPA receptor signaling as a therapeutic target for radical treatment of neuropathic pain and fibromyalgia. Pain Manag. 2020, 10, 43–53. [Google Scholar] [CrossRef]
- Xu, L.; Su, J.; Guo, L.; Wang, S.; Deng, X.; Ma, S. Modulation of LPA1 receptor-mediated neuronal apoptosis by Saikosaponin-d: A target involved in depression. Neuropharmacology 2019, 155, 150–161. [Google Scholar] [CrossRef] [PubMed]
- Su, J.; Pan, Y.W.; Wang, S.Q.; Li, X.Z.; Huang, F.; Ma, S.P. Saikosaponin-d attenuated lipopolysaccharide-induced depressive-like behaviors via inhibiting microglia activation and neuroinflammation. Int. Immunopharmacol. 2020, 80, 106181. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Serrano, C.; Santos-Nogueira, E.; Francos-Quijorna, I.; Coll-Miro, M.; Chun, J.; Lopez-Vales, R. Lysophosphatidic acid receptor type 2 activation contributes to secondary damage after spinal cord injury in mice. Brain Behav. Immun. 2019, 76, 258–267. [Google Scholar] [CrossRef]
- Fischer, D.J.; Nusser, N.; Virag, T.; Yokoyama, K.; Wang, D.; Baker, D.L.; Bautista, D.; Parrill, A.L.; Tigyi, G. Short-chain phosphatidates are subtype-selective antagonists of lysophosphatidic acid receptors. Mol. Pharmacol. 2001, 60, 776–784. [Google Scholar]
- Ohta, H.; Sato, K.; Murata, N.; Damirin, A.; Malchinkhuu, E.; Kon, J.; Kimura, T.; Tobo, M.; Yamazaki, Y.; Watanabe, T.; et al. Ki16425, a subtype-selective antagonist for EDG-family lysophosphatidic acid receptors. Mol. Pharmacol. 2003, 64, 994–1005. [Google Scholar] [CrossRef] [PubMed]
- Awada, R.; Rondeau, P.; Gres, S.; Saulnier-Blache, J.S.; Lefebvre d’Hellencourt, C.; Bourdon, E. Autotaxin protects microglial cells against oxidative stress. Free Radic. Biol. Med. 2012, 52, 516–526. [Google Scholar] [CrossRef] [PubMed]
- Kozian, D.H.; von Haeften, E.; Joho, S.; Czechtizky, W.; Anumala, U.R.; Roux, P.; Dudda, A.; Evers, A.; Nazare, M. Modulation of Hexadecyl-LPA-Mediated Activation of Mast Cells and Microglia by a Chemical Probe for LPA5. Chembiochem 2016, 17, 861–865. [Google Scholar] [CrossRef]
- Kozian, D.H.; Evers, A.; Florian, P.; Wonerow, P.; Joho, S.; Nazare, M. Selective non-lipid modulator of LPA5 activity in human platelets. Bioorg. Med. Chem. Lett. 2012, 22, 5239–5243. [Google Scholar] [CrossRef]
- Sapkota, A.; Lee, C.H.; Park, S.J.; Choi, J.W. Lysophosphatidic Acid Receptor 5 Plays a Pathogenic Role in Brain Damage after Focal Cerebral Ischemia by Modulating Neuroinflammatory Responses. Cells 2020, 9, 1446. [Google Scholar] [CrossRef]
- Plastira, I.; Joshi, L.; Bernhart, E.; Schoene, J.; Specker, E.; Nazare, M.; Sattler, W. Small-Molecule Lysophosphatidic Acid Receptor 5 (LPAR5) Antagonists: Versatile Pharmacological Tools to Regulate Inflammatory Signaling in BV-2 Microglia Cells. Front. Cell Neurosci. 2019, 13, 531. [Google Scholar] [CrossRef]
- Plastira, I.; Bernhart, E.; Goeritzer, M.; DeVaney, T.; Reicher, H.; Hammer, A.; Lohberger, B.; Wintersperger, A.; Zucol, B.; Graier, W.F.; et al. Lysophosphatidic acid via LPA-receptor 5/protein kinase D-dependent pathways induces a motile and pro-inflammatory microglial phenotype. J. Neuroinflamm. 2017, 14, 253. [Google Scholar] [CrossRef]
- Sapkota, A.; Park, S.J.; Choi, J.W. Inhibition of LPA5 Activity Provides Long-Term Neuroprotection in Mice with Brain Ischemic Stroke. Biomol. Ther. 2020, 28, 512–518. [Google Scholar] [CrossRef] [PubMed]
- Nakanaga, K.; Hama, K.; Aoki, J. Autotaxin—an LPA producing enzyme with diverse functions. J. Biochem. 2010, 148, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Hoelzinger, D.B.; Nakada, M.; Demuth, T.; Rosensteel, T.; Reavie, L.B.; Berens, M.E. Autotaxin: A secreted autocrine/paracrine factor that promotes glioma invasion. J. Neurooncol. 2008, 86, 297–309. [Google Scholar] [CrossRef]
- Ramakers, G.J.; Moolenaar, W.H. Regulation of astrocyte morphology by RhoA and lysophosphatidic acid. Exp. Cell Res. 1998, 245, 252–262. [Google Scholar] [CrossRef] [PubMed]
- Defize, L.H.; Boonstra, J.; Meisenhelder, J.; Kruijer, W.; Tertoolen, L.G.; Tilly, B.C.; Hunter, T.; van Bergen en Henegouwen, P.M.; Moolenaar, W.H.; de Laat, S.W. Signal transduction by epidermal growth factor occurs through the subclass of high affinity receptors. J. Cell Biol. 1989, 109, 2495–2507. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.V.; Raymond, M.; Pace, F.; Certo, A.; Zuidema, J.M.; McKay, C.A.; Gilbert, R.J.; Lu, X.L.; Wan, L.Q. Astrocytes increase ATP exocytosis mediated calcium signaling in response to microgroove structures. Sci. Rep. 2015, 5, 7847. [Google Scholar] [CrossRef]
- Takenaga, K.; Kozlova, E.N. Role of intracellular S100A4 for migration of rat astrocytes. Glia 2006, 53, 313–321. [Google Scholar] [CrossRef]
- Lagos-Cabre, R.; Burgos-Bravo, F.; Avalos, A.M.; Leyton, L. Connexins in Astrocyte Migration. Front. Pharmacol. 2019, 10, 1546. [Google Scholar] [CrossRef]
- Avalos, A.M.; Arthur, W.T.; Schneider, P.; Quest, A.F.; Burridge, K.; Leyton, L. Aggregation of integrins and RhoA activation are required for Thy-1-induced morphological changes in astrocytes. J. Biol. Chem. 2004, 279, 39139–39145. [Google Scholar] [CrossRef]
- Sofroniew, M.V.; Vinters, H.V. Astrocytes: Biology and pathology. Acta Neuropathol. 2010, 119, 7–35. [Google Scholar] [CrossRef] [PubMed]
- Sofroniew, M.V. Molecular dissection of reactive astrogliosis and glial scar formation. Trends Neurosci. 2009, 32, 638–647. [Google Scholar] [CrossRef]
- Goldshmit, Y.; Munro, K.; Leong, S.Y.; Pebay, A.; Turnley, A.M. LPA receptor expression in the central nervous system in health and following injury. Cell Tissue Res. 2010, 341, 23–32. [Google Scholar] [CrossRef] [PubMed]
- Cervera, P.; Tirard, M.; Barron, S.; Allard, J.; Trottier, S.; Lacombe, J.; Daumas-Duport, C.; Sokoloff, P. Immunohistological localization of the myelinating cell-specific receptor LP(A1). Glia 2002, 38, 126–136. [Google Scholar] [CrossRef]
- Shano, S.; Moriyama, R.; Chun, J.; Fukushima, N. Lysophosphatidic acid stimulates astrocyte proliferation through LPA1. Neurochem. Int. 2008, 52, 216–220. [Google Scholar] [CrossRef] [PubMed]
- Sato, K.; Horiuchi, Y.; Jin, Y.; Malchinkhuu, E.; Komachi, M.; Kondo, T.; Okajima, F. Unmasking of LPA1 receptor-mediated migration response to lysophosphatidic acid by interleukin-1beta-induced attenuation of Rho signaling pathways in rat astrocytes. J. Neurochem. 2011, 117, 164–174. [Google Scholar] [CrossRef] [PubMed]
- Olianas, M.C.; Dedoni, S.; Onali, P. LPA1 Mediates Antidepressant-Induced ERK1/2 Signaling and Protection from Oxidative Stress in Glial Cells. J. Pharmacol. Exp. Ther. 2016, 359, 340–353. [Google Scholar] [CrossRef]
- Ueda, H.; Neyama, H.; Nagai, J.; Matsushita, Y.; Tsukahara, T.; Tsukahara, R. Involvement of lysophosphatidic acid-induced astrocyte activation underlying the maintenance of partial sciatic nerve injury-induced neuropathic pain. Pain 2018, 159, 2170–2178. [Google Scholar] [CrossRef]
- Spohr, T.C.; Choi, J.W.; Gardell, S.E.; Herr, D.R.; Rehen, S.K.; Gomes, F.C.; Chun, J. Lysophosphatidic acid receptor-dependent secondary effects via astrocytes promote neuronal differentiation. J. Biol. Chem. 2008, 283, 7470–7479. [Google Scholar] [CrossRef]
- Sapkota, A.; Park, S.J.; Choi, J.W. Receptor for Advanced Glycation End Products Is Involved in LPA5-Mediated Brain Damage after a Transient Ischemic Stroke. Life 2021, 11, 80. [Google Scholar] [CrossRef]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).