Abstract
Cell-based therapies are gaining momentum as promising treatments for rare neurological autoimmune diseases, including neuromyelitis optica spectrum disorders and myelin oligodendrocyte glycoprotein antibody-associated disease. The development of targeted cell therapies is hampered by the lack of adequate animal models that mirror the human disease. Most cell-based treatments, including HSCT, CAR-T cell, tolerogenic dendritic cell and mesenchymal stem cell treatment have entered early stage clinical trials or have been used as rescue treatment in treatment-refractory cases. The development of antigen-specific cell-based immunotherapies for autoimmune diseases is slowed down by the rarity of the diseases, the lack of surrogate outcomes and biomarkers that are able to predict long-term outcomes and/or therapy effectiveness as well as challenges in the manufacturing of cellular products. These challenges are likely to be overcome by future research.
1. Introduction
Various types of cell-based therapies may hold promise for treatment of potentially severe autoimmune neurological diseases, including neuromyelitis optica spectrum disorders (NMOSD) and myelin oligodendrocyte glycoprotein antibody-associated disease (MOGAD). Neuromyelitis optica, or NMO, was first described by Eugène Devic in the late 19th century as a variant of multiple sclerosis (MS), presenting with optic neuritis and myelitis [1]. In 2004, antibodies towards aquaporin-4 (AQP4) were discovered and NMO could clearly be distinguished from MS [2,3]. The term NMOSD was introduced for the first time in 2007 [4]. It was recognised that not only presentations with optic neuritis and/or (longitudinally extensive or short) transverse myelitis could occur. Indeed, a spectrum of clinical presentations including area postrema, diencephalic, brainstem and symptomatic cerebral syndrome has been associated with AQP4-IgG antibodies, leading to renaming of NMO to NMOSD [4,5]. However, in some NMO patients, AQP4-IgG antibodies could not be detected and this category was named ‘seronegative NMO’. Later, it became clear that a proportion of these seronegative NMO patients carried autoantibodies towards another autoantigen, myelin oligodendrocyte glycoprotein (MOG). This disease is now referred to as MOG antibody-associated disease or MOGAD [6]. Clinical presentations include optic neuritis, myelitis, brainstem syndromes, acute disseminated encephalomyelitis (ADEM), but the disease spectrum is expanding with rare clinical presentations such as (but not limited to) unilateral cortical encephalitis [7]. Today, MOGAD is increasingly regarded as a separate disease entity from AQP4-IgG positive (AQP4+) NMOSD [8,9]. Finally, in double seronegative NMOSD, AQP4 or MOG antibodies are not demonstrable and more research is needed to better define this category of patients [10]. While AQP4+ NMOSD has been associated with other non-organ and organ-specific autoantibodies, this is less clearly the case for MOGAD [11]. However, more recently, reports of coexisting NMDAR antibodies [12] and concurrent peripheral and central demyelination with concomitant presence of anti-neurofascin antibodies [13] have challenged this concept. Both NMOSD and MOGAD diseases are accompanied by relapses and episodes of remission that are variable in duration [14].
Relapses are treated with high-dose intravenous methylprednisolone, plasma exchange or intravenous immunoglobulins (IVIg) [15]. To prevent relapses and related disability, these diseases are currently treated off-label with the anti-CD20 (cluster of differentiation) monoclonal antibody rituximab, immunosuppressants such as azathioprine, mycophenolate, methotrexate, tocilizumab—a monoclonal antibody towards interleukin (IL)-6 receptor (Il-6R)—and repeated courses of IVIg, sometimes in combination with low-dose steroids [15]. More recently, the first treatments for NMOSD have been approved by regulatory agencies, based on results of phase III randomised controlled clinical trials: satralizumab (Enspryng®) [16], a monoclonal antibody against the IL-6R, eculizumab (Soliris®) [17], an anti-C5 complement inhibitor and inebilizumab (Uplinza®) [18], a monoclonal antibody leading to lymphocytolysis after binding to CD-19 on B cells and plasmablasts [19]. In severe cases, autologous hematopoietic stem cell transplantation (HSCT) has been used [20]. There is no evidence-based guideline for the treatment of MOGAD, which is based on case series and expert opinion, as in this disease there have been no phase III randomised controlled trials to confirm efficacy of any of the aforementioned treatments [7].
However, chronic immunosuppressive treatments may increase the risk of infections [21] in the long term, highlighting the need for temporary and/or more targeted antigen-specific treatments, leaving protective immunity to fight pathogens and cancer intact. Cell-based therapies aim to do precisely this: either depletion of autoreactive effector cells, or modulation of autoreactive T and B cell responses, resulting in the restoration of tolerance. Regarding the latter, while some cell-based therapies target and try to modulate only the antigen-specific autoreactive T and B cells, immune reconstitution cell-based therapies, such as HSCT, are accompanied by a general and temporary severe immunosuppressive state, aiming to eradicate aberrant immune responses towards self-antigens, while restoring immunity towards non-self-antigens.
After providing an overview of the current knowledge on immunopathogenesis, the fundamental, translational and clinical research approaches in the field of cell-based therapies in NMOSD and MOGAD are reviewed and challenges and areas open to research are discussed.
2. Immunopathogenesis
Both NMOSD and MOGAD are autoimmune central nervous system (CNS) disorders, which share, in part, clinical presentations, but differ in immunopathogenesis and pathological characteristics [22]. In this section, a comprehensive insight into the pathogenesis of both diseases will be given. For a graphical overview, including intervention points for cell therapy, we refer to Figure 1.
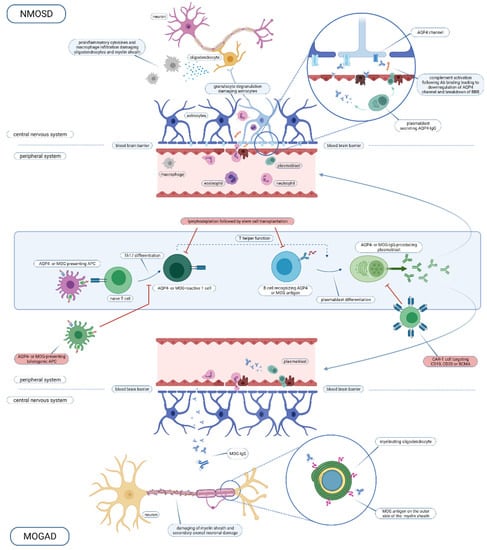
Figure 1.
Overview of the NMOSD and MOGAD immune pathogenesis and intervention points for cell-based therapy. Abbreviations used: NMOSD, neuromyelitis optica spectrum disorder; AQP4, aquaporin-4; Ab, antibody; BBB, blood brain barrier; IgG, immunoglobuline G; MOG, myelin oligodendrocyte glycoprotein; APC, antigen-presenting cell; Th, T helper; CAR-T, chimeric antigen receptor T cell; CD, cluster of differentiation; BCMA, B cell maturation antigen; MOGAD, MOG-antibody disease. Created with BioRender.com.
2.1. AQP4 + NMOSD
In 2003, Peter Agre and Roderick MacKinnon received the Nobel prize for chemistry, honouring their work on the identification and functional characterisation of aquaporines [23]. Aquaporines are selectively permeable to water and thereby control water homeostasis. In 1994, AQP4 was first cloned from rat lung [24]. AQP4 is a cell membrane crossing water channel, which is highly expressed on astrocyte foot processes and ependymal cells in the blood–brain and blood–cerebrospinal fluid barriers in the CNS in the optic nerve, spinal cord, hypothalamus and area postrema [25,26]. However, AQP4 is also expressed in other tissues, such as kidney, stomach, airways, glands and skeletal muscle [27]. The differential expression, both within and outside the CNS, and variation in macroscopic aggregation of tetramers between tissues, are explanations why AQP4 antibodies bind to CNS predilection sites [28,29].
AQP4-IgG antibodies have been demonstrated to be pathogenic [30]. Both AQP4-IgG-producing plasmablasts and AQP4-IgG antibodies are able to cross the blood–brain barrier. Next, antibodies bind to AQP4, leading to complement-dependent cytotoxicity and chemotaxis of immune cells, including T and B lymphocytes, macrophages, neutrophils and eosinophils. Neutrophils degranulate and lead to astrocyte death, in its turn causing oligodendrocyte death. Hence, the severe inflammatory reaction leads to tissue necrosis with demyelination, axonal and neuronal damage [26]. Pathology of acute lesions shows oligodendrocyte and astrocyte loss, confluent demyelination, abundant complement deposition and a predominant CD4+ T lymphocyte inflammatory cell infiltration [22]. Besides antibodies, AQP4-specific T cells play a role in the development of NMOSD lesions. Indeed, AQP4-IgG belongs to a T cell-dependent immunoglobulin subclass (IgG1). A unifying explanation of the pathogenesis is that T cells are involved in the early onset of the disease in the periphery, leading to a breach of tolerance and subsequent antibody production by development of antigen-producing B cells. AQP4-specific T cells are sufficient to induce NMO in a mouse model, independent of antibodies [31]. Moreover, AQP4-specific T cells are amplified in patients with NMOSD versus healthy controls. Both naïve pre-germinal centre B cells (CD19+CD27-IgD+) and post-germinal centre cells (CD19+CD27+) are able to differentiate to secrete AQP4 antibodies [32]. This suggests an early, pre-germinal centre loss of immunological tolerance [32]. However, patients with AQP4+ NMOSD have higher frequency of regulatory B cells (IL-10-producing B cells, CD19+CD39+CD1d+IL-10+) [33,34]. Functional properties of B cells have not been investigated yet. Besides an important role of the adaptive immune system, the innate immune system has also been implicated in lesion initiation. In a mouse model, it has been shown that crosstalk between astrocytes and microglia involving early-activated CNS-intrinsic complement components and microglial C3a receptor signalling is a critical driver in the evolving NMO lesion [35].
Pathologically, demyelination with preferential loss of myelin-associated glycoprotein (MAG) and oligodendrocyte loss, severe astrocytic damage and perivascular-activated complement deposits are the hallmarks of AQP4+ NMOSD [22].
The origin of the AQP4 antibodies remains elusive. Generated most likely in the peripheral immune system, these antibodies can enter the CNS following a break in the blood–brain barrier, again for a yet unresolved reason, and initiate the inflammatory cascade as described above and as depicted in Figure 1. There are several lines of evidence for their generation in the peripheral immune system: (1) AQP4 antibodies have been demonstrated in the serum long before the development of NMOSD [36]), (2) in contrast to MS, oligoclonal bands reflecting intrathecal IgG synthesis are mostly absent in NMOSD [37] and (3) predilection sites are the area postrema and the posterior pituitary, which are not covered by the blood–brain barrier—hence first presenting symptoms consistent with a lesion location in this areas may be regarded as a sign of relocation of AQP4 antibodies from the peripheral system to the brain (as reviewed by [26]). AQP4+ NMOSD has been described as a paraneoplastic disorder [38], which suggests that mechanisms of molecular mimicry with tumour antigens may play a role in the development of the disease. The role of molecular mimicry has also been suggested by the fact that AQP4-specific T cells display cross reactivity towards a bacterial protein (Clostridium perfringens adenosine triphosphate-binding cassette (ABC) transporter permease) [39]. Another Clostridium species (C. bolteae) has recently been implicated in the pathogenesis of NMOSD in Indian patients [40]. Environmental risk factors, especially herpes virus infections, are not clearly associated with NMOSD, unlike MS [41]. HLA associations have been described in Japanese (HLA-DRB1*08:02 and HLA-DRB1*16:02) and Dutch populations (HLA-A*01, -B*08 and -DRB1*03) [42,43,44,45].
While major advances in the knowledge of NMOSD pathophysiology have led to investigating the efficacy of targeted treatments in phase III clinical trials, further elucidation of the immunopathogenesis of AQP4+ NMOSD may lead to novel treatment approaches. Besides IL-6, CCL22 and CCL3, CD16+CD56+ NK cells and CX3CL1 have been identified as potential novel biomarker candidates [46]. Besides the assessment of the frequency of various immune cell types [46], their functional characteristics need further exploration. For instance, the role and function of antigen-presenting cells, such as dendritic cells, have not been clarified so far. Moreover, seronegative NMOSD requires further research.
2.2. MOGAD
Recently, the immunopathology of 11 patients who underwent brain biopsy, and another 24 patients who underwent brain biopsy or for whom autopsy material was available, was described extensively [22,47]. Within the lesions, perivenous demyelination with inflammatory infiltrates consisting of mononuclear cells and/or macrophages, but fewer polymorphonuclear cells, was seen. Axons were relatively preserved and slowly expanding lesions were not present, in contrast to MS pathology [47]. Most lesions showed MOG-predominant myelin loss while other myelin proteins were spared. Myelin phagocytic macrophages were present not only in the lesions but also in the perivascular spaces. The perivascular cellular infiltrates consisted mainly of CD4+ T cells and a lower number of CD8+ T cells and B cells [22]. This is clearly different from MS lesions, where mainly CD8+ T cell infiltrates are found. Only in one MOGAD patient was activated complement seen. In seven patients, IgG staining was seen in the perivenous demyelinated lesions. In the cortico-medullary junction, demyelination and T cell infiltration were also the dominant processes, while axons and oligodendrocytes were relatively preserved [22]. Intracortical demyelination was overrepresented in comparison to classical MS [47]. These pathological observations have to be contrasted with the fact that, to date, no MOG-specific T cells have been found in the peripheral blood of patients with MOGAD [48]. This could be explained by the fact that these MOG-specific T cells are difficult to detect due to their low frequencies. However, this remains to be demonstrated in research. On the other hand, MOG-specific B cells have been detected in samples of patients [25,49]. MOG antibodies seem to have a direct role in the pathogenesis of the disease as suggested by animal models, but cotransfer with myelin-reactive T cells is needed [50,51].
Genetic risk factors for MOGAD remain largely unknown. While an HLA association has been described in NMOSD, no HLA associations have been found in a UK [52] and a Dutch population of MOGAD patients [44]. Only in a Chinese cohort of patients with paediatric-onset MOGAD was an association with HLA-DQB1*05:02-DRB1*16:02 alleles found. MOGAD has been described as occurring after infections [53,54] or more recently as a paraneoplastic syndrome in association with an ovarian teratoma [55]. However, these are only case reports and systematic research on environmental risk factors for MOGAD is lacking. A lack of seasonal variation in MOGAD attacks may argue against a significant role for environmental factors [56].
3. Experimental Animal Models of NMOSD and MOGAD
3.1. Animal Models for NMOSD
As recently reviewed by Duan et al., a multitude of animal models for AQP4+ NMOSD have been developed over the past two decades [57]. These models all partially resemble clinical and pathological features of human NMOSD, which is characterised by spontaneous development of CNS inflammation in a relapsing manner, predominantly targeting spinal cord and optic nerves with relative cerebral sparing. Most of these models are based on human AQP4 IgG administration, with or without pro-inflammatory interventions, or on passive transfer of AQP4-reactive T cells. The earliest models originated from administration of AQP4 IgG in animals with experimental autoimmune encephalomyelitis (EAE), one of the animal models for MS, in which transfer of AQP4 IgG to the CNS was facilitated by breakdown of the blood–brain barrier in the context of EAE. Other NMO-specific animal models were developed later on by administration of AQP4 IgG either directly into the CNS (brain parenchyma or spinal cord) of by systemic administration, both intravenously or intraperitoneally, in combination with manoeuvres to damage the blood–brain barrier, e.g., targeted ultrasound. Finally, passive transfer of AQP4-reactive T cells in rodents induced spinal cord and optic nerve inflammation, albeit without AQP4 loss [58].
Additionally, EAE animal models with NMO resemblance by the specific occurrence of optic neuritis and myelitis have been described without the use of AQP4 IgG or AQP4-reactive T cells for disease induction. For example, opticospinal models of demyelination have been generated in Brown Norway and Lewis rats by administration of recombinant MOG in incomplete Freund’s adjuvant [59,60,61,62]. Apart from the clinical NMO phenotype, some NMO-like pathological features were demonstrated, including astrocyte apoptosis [62]. However, no AQP4 antibodies could be detected, and furthermore, involvement of the periventricular regions was visualised on brain MRI, which are both atypical findings for NMO. Although originally presented as animal models for NMO in terms of their clinical phenotype, these full-length MOG-induced demyelinating models appear to show more resemblance to MOGAD, as discussed further on.
Finally, recently, a Lewis rat model, using mimotopes (peptides, which mimic the conformational AQP4 epitopes), was described as a model to study tolerance induction [63].
Historically, all of these NMOSD animal models have mainly contributed to our understanding of NMO disease pathogenesis. For example, the pathogenicity of AQP4 IgG, as well as the role of complement and antibody-dependent cell cytotoxicity in NMO pathogenesis, has been demonstrated by means of animal models. However, animal models were not involved in recent major therapeutic breakthroughs for NMO, such as eculizumab [17,64]. This is driven by some major limitations of the currently available NMOSD animal models [57], including the lack of animal models with the development of spontaneous AQP4-directed autoimmunity, the intrinsic bias of these animal models towards the cellular or humoral compartment and the fact that most NMOSD animal models are murine, which is problematic due to the fact that mice do not have a functional complement pathway [65]. Finally, no animal model representing seronegative NMO is available up to the present date. Hence, there is a need for more representative NMOSD animal models, in order to facilitate clinical translation.
3.2. Animal Models for MOGAD
To our knowledge, no specific animal model for MOGAD has been developed so far. However, some animal models for MS closely resemble MOGAD pathology, including the full-length MOG-induced opticospinal demyelination in rats described earlier on, as well as the 2D2 EAE model, in which MOG T cell receptor (TCR) transgenic mice spontaneously develop severe optic neuritis [66]. Moreover, when crossed with IgHMOG mice, in which a significant proportion of B cells are MOG-reactive, a transgenic model carrying both MOG-specific B and T cell arises, leading to the spontaneous development of severe myelitis and optic neuritis with relative sparing of the brain [67], similar to MOGAD. A cynomolgus macaque model of EAE, in which administration of recombinant human MOG (rhMOG) elicits brain inflammation mediated by MOG-autoreactive CD4+ lymphocytes and anti-MOG IgG, also mimics the immunopathology of MOGAD [51]. In this model, a recombinant antibody directed against the dendritic cell-asialoglycoprotein receptor (DC-ASGPR) fused to MOG, led to induction of MOG-specific CD4+CD25+FOXP3+CD39+ regulatory lymphocytes and protection from developing EAE [51].
4. Cell-Based Therapies
Various cellular treatment approaches have been investigated in NMOSD and occasionally in MOGAD as well. These have been used either in the controlled setting of clinical trials, or as a rescue therapy for highly aggressive disease in individual patients. The results of these clinical trials and case reports are discussed below. For an overview of registered, completed, ongoing and withdrawn clinical trials in this field, we refer to Table 1.

Table 1.
Overview of completed, ongoing and withdrawn clinical trials with cell therapy in the field of NMOSD and MOGAD, as registered in the ClinicalTrials.gov database. Abbreviations used: MOGAD, MOG antibody-associated disease; AQP4, aquaporine 4; a.o., amongst others; N/A, not applicable; MS, multiple sclerosis; NMOSD, neuromyelitis optica spectrum disorder; CAR, chimeric antigen receptor; CD, cluster of differentiation; EDSS, Expanded Disability Status Scale.
4.1. Tolerance-Inducing Dendritic Cells
One phase Ib, open-label, multiple ascending dose, single-centre clinical trial has investigated the safety and feasibility of intravenously administered autologous tolerogenic peptide-loaded dendritic cells (DC) in four AQP4+ NMOSD patients in Spain [68]. Here, the tolerogenic phenotype of DC was induced by addition of dexamethasone. DC from NMOSD patients were stimulated with seven myelin peptides (MBP13–32, MBP83–99, MBP11–129, MBP146–170, MOG1–20, MOG35–55, PLP139–154) and AQP463–76, which was previously shown to be immunogenic in vitro [39]. Three doses of tolerogenic DC were administered intravenously at week 0, 2 and 4 at progressively increasing doses, including 50 × 106, 100 × 106, 150 × 106 and 300 × 106 DC. Following treatment, the patients entered a safety follow-up phase in which they were followed up to 24 weeks. All NMOSD patients received concomitant treatment with rituximab (3) or mycophenolate (1). One NMOSD patient had elective surgery, not related to the experimental treatment, and this event was classified as a serious adverse event (SAE). All patients remained clinically stable and no relapses occurred. Two patients experienced four adverse events, including back pain, left leg pain, influenza and palpitations. Immunological evaluations demonstrated a trend for decreased T cell proliferation as measured by [3H]-thymidine incorporation in response to AQP4 peptide at week 12 as compared to baseline [68]. A significant increase of IL-10 (interleukin-10) production, measured with ELISA in response to peptide stimulation in PBMC culture supernatant, was demonstrated at week 12 compared to baseline for AQP4 [68]. This was accompanied by an upward trend in the frequency of type 1 regulatory T (Tr1) cells. In conclusion, this tolDC-based therapy was safe in NMOSD patients and immunological analysis demonstrated changes compatible with tolerance induction.
4.2. Hematopoietic Stem Cell Transplantation in NMOSD and MOGAD
In hematopoietic stem cell transplantation for autoimmune diseases, the aim is to destroy the aberrantly functioning immune system with high-dose chemotherapy and rebuild it by hematopoietic stem cell infusion, thereby aiming to induce long-term disease remission. The therapeutic potential lies in aggressive immunosuppression while the HSC are needed for recovery of the immune system. In patients with autoimmune diseases, autologous transplantations have been preferred above allogeneic ones to prevent graft-versus-host reactions and related morbidity and mortality. A recent meta-analysis included three studies (published between 2000 and 2020) on 31 NMOSD patients who underwent AHSCT [69,72,73,74]. The progression-free survival (PFS = survival without progression; progression = worsening of neurologic disability beyond the pre-treatment baseline (increase in EDSS > 1 with a pre-transplant baseline EDSS score of ≤5 or >0.5 with a baseline EDSS score of >5)) was 76% during a follow-up period between 2 and 13 years. Treatment-related mortality (TRM = death within 100 days of AHSCT) was 0% [72]. No results on immunomonitoring of peripheral blood immune cell subsets were available. Guidelines from the European Bone Marrow Transplantation (EBMT) Autoimmune Diseases Working Party (ADWP) recommend the use of AHSCT in NMOSD as a clinical option, with grade II evidence, in therapy-refractory patients [75]. Despite the promising results, a number of patients had persisting AQP4 antibodies and relapsed within 5 years [69,73]. Moreover, the optimal conditioning regimen remains unclear to date.
Only a few cases of NMOSD patients who underwent allogeneic HSCT (alloHSCT) were reported (summarised in [20,76,77,78]). AlloHSCT has the potential to clear all autoreactive lymphocytes by allogeneic donor T lymphocytes (graft versus autoimmunity), thereby leading to a more profound immunotherapeutic effect. However, this needs to be balanced with the more significant risks of morbidity and mortality after alloHSCT. Several immune-mediated peripheral and central nervous system diseases, including a case of MOGAD, have been reported after alloHSCT in haematological patients, urging for prudence when using alloHSCT in patients with autoimmune diseases [79]. Due to limited clinical evidence, alloHSCT in NMO was classified as developmental by the EBMT-ADWP [75] and is currently not recommended as a clinical option.
Only one reported case of a 25-year-old treatment-refractory male with MOGAD was found in the literature, who recovered very well after AHSCT [80]. Follow-up duration was less than one year after AHSCT, while his disease started as early as the age of 10 years [80].
In conclusion, the evidence for HSCT in NMOSD and MOGAD is limited and a proportion of patients will relapse within 5 years. AHSCT can be considered as a clinical option to treat treatment-refractory NMOSD patients according to EBMT [75]. Concerning the use of (allo)HSCT in NMOSD and MOGAD with (allo)HSCT, more research in this field is necessary to determine short- and longterm safety, efficacy and optimal condition regimens.
4.3. CAR-T Cell Therapy
Chimeric antigen receptors (CAR) are receptor proteins carrying both an antigen-binding and a T cell-activating function, allowing T cells to target a specific protein. CARs consist of an extracellular binding domain containing the single-chain variable fragment from an antigen-reactive antibody, allowing antigen-specific binding, and an intracellular domain containing the CD3ζ chain domain, allowing T cell receptor signalling and T cell activation after antigen binding to the extracellular domain [81]. Following in vitro genetic modification of T lymphocytes, mostly using viral transduction but more recently also using the CRISPR/Cas9 technique [82], T cells expressing such CAR proteins can be readministered to a patient, where they induce a protein-specific immune response. CAR-T cell therapy was originally developed in the field of cancer therapy, where it was designed to target specific proteins expressed on tumour cells, inducing a tumour-specific cytotoxic response. Most success has been achieved in the treatment of haematological cancers, including lymphoma and leukaemia [83]. Treatment of solid tumours using CAR-T cell therapy has appeared to be more challenging, mainly due to the lack of identification of tumour-specific antigens and challenges in terms of infiltration and survival of CAR-T cells within the solid tumour microenvironment [84].
B cell targeting using CAR-T cell therapy focuses on B cell markers CD19, CD20 and B cell maturation antigen (BCMA) as antigenic targets, for instance, in the field of B cell leukaemia [85], B cell lymphoma [86,87] and multiple myeloma [88]. Next to the oncological field, B cell overactivation has also been implicated in the field of autoimmunity, where dysregulated B cell activation leads to an antibody-mediated targeting of healthy own body tissue. Hence, B cell targeting using CAR-T cell therapy shows additional promise for the treatment of autoimmune diseases. Breaking the immune tolerance towards autoreactive immune cells induces specific cytotoxic death of these cells, which may downregulate the immune overactivation driving autoimmunity. Indeed, CAR-T cell therapy targeting CD19 has recently been demonstrated to be effective in the prevention and treatment of a murine model of systemic lupus erythematosus [89].
In the field of NMOSD, a first clinical trial evaluating the safety and efficacy of CD19 and CD20 CAR-T cell therapy was withdrawn due to recruitment difficulties (ClinicalTrials.gov NCT03605238). Currently, an open-label phase I clinical trial is ongoing, using BCMA CAR-T cell therapy in patients with refractory AQP4-IgG-seropositive NMOSD (ClinicalTrials.gov NCT04561557). Twelve NMOSD patients will be enrolled, receiving BCMA CAR-T cells following lymphodepletion with cyclophosphamide and fludarabine. Primary outcome measures include the incidence of dose-limiting toxicities and adverse events. The concentration of AQP4-IgG titers in the serum 3 months after infusion and the CAR-T cell proliferation 2 years after infusion will be studied as secondary outcome measures, together with clinical and radiological outcome measures, including annualised relapse rate and active MRI lesions. The first results of this clinical trial are expected by the end of 2023.
In line with the development of CAR-T cells for targeted cytotoxic depletion of autoreactive T cells by breaking of tolerance, another approach is the generation of antigen-specific regulatory T cells (Treg) by transfection with TCRs specific for particular auto-antigens, aiming at antigen-specific tolerance induction. Indeed, in addition to previous clinical trials demonstrating safety and efficacy of polyclonal Treg administration for the treatment of graft-versus-host disease [90,91,92] and type 1 diabetes mellitus [93,94], next-generation antigen-specific CAR-Treg therapies are being developed, albeit still in the preclinical stage (as reviewed by [95]). Although, to our knowledge, no such translational research is currently being performed in the field of NMOSD or MOGAD, this may form an interesting research avenue.
4.4. Mesenchymal Stem Cell Transplantation
Mesenchymal stem cells (MSC) are multipotent stromal progenitor cells, derived from allogeneic umbilical cord tissue, autologous bone marrow or autologous adipose tissue. Although the therapeutic effect of MSC treatment was historically presumed to be driven by the regeneration of damaged tissue, additional beneficial effects have been demonstrated, including an immunomodulatory action by inhibition of the release of pro-inflammatory cytokines from both innate and adaptive immune cells and a neuroprotective action by secretion of neurotrophic and survival-promoting growth factors [96,97,98,99,100]. In the field of NMOSD, clinical trials with both bone marrow-derived MSC (b-MSC) [70] and umbilical cord tissue-derived MSC (hUC-MSC) [71,101] have been conducted.
A pilot study evaluating safety and feasibility of a single intravenous infusion with autologous bMSC in 15 AQP4 IgG+ NMO patients was completed in 2016 [70]. Previous immunomodulatory treatment (cyclophosphamide, azathioprine, with or without corticosteroids) was stopped 30 days before bone marrow harvest and patients were followed for 24 months after the bMSC infusion. No adverse events were observed. At 12 months, the mean annualised relapse rate (ARR) was reduced in comparison to pre-treatment ARR (1.1 versus 0.3, p = 0.002), accompanied by a decrease in T2 or gadolinium-enhancing T1 lesions in the optic nerve and the spinal cord on MR imaging. At 24 months, 13 patients (87%) were still relapse-free and disability had improved in 6 patients (40%), providing encouraging indications of a beneficial clinical effect of bMSC treatment in NMO. Due to lack of tracking experiments or pathological data, the exact mode of action of bMSC transplantation, however, remained unclear. It was postulated that the beneficial effect of bMSC transplantation was driven by both immune response modulation and promotion of tissue recovery and repair, based upon findings of (1) a decrease in T follicular helper cell counts and accompanied attenuation of pro-inflammatory IL-6 and IL-21 cytokine levels and (2) a significant thickening of the retinal nerve fibre layer, an increase in the optic nerve diameter and an enlargement of the upper cervical area [70].
In contrast to bMSC, hUC-MSC are easily collectable and, although not autologous, these MSC have a low risk of induction of allogeneic immune responses and hence transplant rejection [102]. In 2012, a phase I clinical trial evaluated the effect of treatment with hUC-MSC in five AQP4 IgG+ NMO patients [101]. The cells were administered by intravenous and intrathecal route combined, divided over four infusions. Average number of relapses decreased significantly following transplantation, compared to pre-treatment relapse rate (1.4 versus 3.2, p < 0.05). EDSS score improved in four out of five patients 24 months following transplantation. In these four patients, peripheral blood B lymphocyte fraction decreased compared to pre-treatment, although the exact meaning of this finding remains elusive [101]. Recently, the results from the 10-year follow-up of these patients have been published [71]. Importantly, no long-term adverse events were detected, in particular no tumour formation or peripheral organ disorders. In the extended follow-up period, four out of five treated NMO patients demonstrated reduced annual relapse occurrence compared to before treatment. However, only two patients completed the 10-year follow-up period, due to death in two patients and loss-to-follow-up of one patient. The authors stated that these deaths were caused by rapid disease progression and were not a direct consequence of the transplantation [71]. In conclusion, long-term clinical evidence following hUC-MSC transplantation remains scarce and further clinical trials are warranted.
5. Discussion
Autoimmune diseases with well-known target antigens, such as AQP4+ NMOSD and MOGAD, are good candidate diseases to investigate tolerance-inducing cell-based therapies, especially in an antigen-specific way. While next-generation cell-based therapies have entered the arena of cancer treatment [103], they are still in their infancy in the field of autoimmune diseases. Several challenges need to be solved to drive the field of tolerance-inducing cell-based therapies in NMOSD and MOGAD forward.
One major challenge is the lack of adequate experimental animal models that mimic the human disease in such a way that these models can be used for translational research. Hence, it is difficult to obtain preclinical data as incentive towards clinical translation. However, progress is being made in this field.
As both NMOSD and MOGAD are rare diseases, multicentre clinical trials are necessary to achieve sufficient power to detect clinical efficacy. Recent world-wide phase III randomised placebo-controlled clinical trials, investigating the efficacy of satralizumab (Enspryng®) [16], eculizumab (Soliris®) [17] and inebilizumab (Uplinza®) [18] have led to the registration of the first evidence-based treatments for AQP4+ NMOSD. Hence, in the near future, it will become increasingly difficult to design and execute clinical trials investigating tolerance-inducing cell-based therapies in this field. No phase III multicentre randomised clinical trials have been performed to assess treatment effectiveness in MOGAD. Due to the low prevalence of this disease, the wide age range and broad clinical spectrum, it will be a major challenge to set up and conduct a large, multicentre, randomised clinical trial [7].
A reliable biomarker to predict future relapses or disability is not available to date. This makes proof-of-concept clinical trial design, in which biomarkers are important to detect a hint of effectiveness, challenging and prone to selection of very active patients with high relapse activity. Conflicting evidence exists on the clinical usefulness of antibody titers in the follow-up of patients with MOGAD or NMOSD. Antibody titers of AQP4 antibodies [104,105,106] are not predictive of future relapses. MOG antibodies [107,108] are higher during relapse than in remission [109] and conversion to antibody-negative status has been correlated with a higher chance of having a monophasic disease course [110,111]. In children, persisting MOG antibodies have been correlated with a higher risk of relapse [107]. However, this is not absolute: patients can remain clinically stable with positive MOG antibodies for months to years [112,113]. Hence, 6-monthly retesting of MOG antibodies, for up to 2 years or until antibodies become negative, has been recommended by the European Paediatric MOG Consortium [114]. Intrathecal production of MOG antibodies occurs more frequently than that of AQP4 antibodies [115]. The diagnostic and prognostic value of CSF unique MOG antibodies deserves more research [116]. Recently, serum glial fibrillary acidic protein (sGFAP) has gained attention as a potential biomarker to assess disease severity and future disease activity in patients with AQP4+ NMOSD in remission [117]. Biomarkers in MOGAD are the subject of ongoing research [114].
Moreover, it is not clear if cell-based therapy would be sufficient as monotherapy or if combination or adjuvant treatment protocols should be aimed for. Even HSCT is not able to induce long-term remission as many NMOSD patients will continue to relapse (in one study, there was relapse-free survival of 31% and 10% after 3 and 5 years in 16 patients) and they may need other treatments [73]. One strategy of theoretical interest is starting with a strong immunosuppressive treatment, aiming to eradicate disease-causing lymphocytes, followed by a maintenance treatment to maintain immune tolerance over the long term. This concept has been applied in the treatment of MS, albeit with variable results [118,119,120]. As antigen-specific T cells and antibodies are both involved in lesion formation in NMOSD, it is likely that T cell responses should be modulated, but pathogenic antibodies should be cleared as well. In MOGAD, MOG-specific T cells have not been detected in the peripheral blood of patients to date [48]. In this study, antigen-specific T cell responses were measured with carboxyfluorescein diacetate succinimidyl ester proliferation assay and the detection of granulocyte macrophage colony-stimulating factor (GM-CSF), interferon (IFN)-ɤ and IL-4, IL-6 and IL-17A in cell culture supernatants. Nine MOG-peptides were used to stimulate antigen-specific T cells [48]. This result contrasts with the pathological findings, in which CD4+ T cells are abundantly present. Single-cell technology to detect rare immune cell populations or analysing T cells in CSF samples may provide some answers here.
Finally, the manufacturing process of autologous cell-based therapies, such as tolDC and CAR-T cells, is labour intensive and the logistic process from apheresis to administration of the final cell product is complex. On-site manufacturing could be a solution for this; however, harmonization of culture protocols over different study sites may be an issue.
Moreover, it is not clear to date if progenitor cells from patients with NMOSD or MOGAD are of the same quality as healthy controls, e.g., for bMSC (decreased proliferation rate, more prone to senescence) [121], and optimization of culture protocols may be warranted.
6. Conclusions
Cell-based therapies are in the developmental stage for rare neuroinflammatory diseases, such as AQP4+, NMOSD and MOGAD. Numerous challenges in the development and clinical translation lie ahead, which are likely to be overcome by future research.
Author Contributions
Conceptualization, J.D. and B.W.; writing—original draft preparation, J.D. and B.W.; writing—review and editing, T.R., I.W. and N.C. All authors have read and agreed to the published version of the manuscript.
Funding
BW is supported by a personal grant from the Horlait-Dapsens Medical Foundation, awarded in 2020.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
Data availability is not applicable to this article as no new data were created or analysed in this study.
Conflicts of Interest
The authors declare no conflict of interest. The funders had no role in the design of the study; in the writing of the manuscript, or in the decision to publish the results.
References
- Jarius, S.; Wildemann, B. The History of Neuromyelitis Optica. J. Neuroinflamm. 2013, 10, 8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lennon, V.A.; Kryzer, T.J.; Pittock, S.J.; Verkman, A.S.; Hinson, S.R. IgG Marker of Opticspinal Multiple Sclerosis Binds to the Aquaporin-4 Water Channel. J. Exp. Med. 2005, 202, 473–477. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lennon, A.V.; Wingerchuk, D.M.; Kryzer, T.J.; Pittock, S.J.; Lucchinetti, C.F.; Fujihara, K.; Nakashima, I.; Weinshenker, B.G. A Serum Autoantibody Marker of Neuromyelitis Optica: Distinction from Multiple Sclerosis. Lancet 2004, 364, 2106–2112. [Google Scholar] [CrossRef]
- Wingerchuk, D.M.; Lennon, A.V.; Lucchinetti, C.F.; Pittock, S.J.; Weinshenker, B.G. The Spectrum of Neuromyelitis Optica. Lancet Neurol. 2007, 6, 805–815. [Google Scholar] [CrossRef]
- Wingerchuk, D.M.; Banwell, B.; Bennett, J.L.; Cabre, P.; Carroll, W.; Chitnis, T.; De Seze, J.; Fujihara, K.; Greenberg, B.M.; Jacob, A.; et al. International Consensus Diagnostic Criteria for Neuromyelitis Optica Spectrum Disorders. Neurology 2015, 85, 177–189. [Google Scholar] [CrossRef]
- Reindl, M.; Waters, P. Myelin Oligodendrocyte Glycoprotein Antibodies in Neurological Disease. Nat. Rev. Neurol. 2019, 15, 89–102. [Google Scholar] [CrossRef]
- Ambrosius, W.; Michalak, S.; Kozubski, W.; Kalinowska, A. Myelin Oligodendrocyte Glycoprotein Antibody-Associated Disease: Current Insights into the Disease Pathophysiology, Diagnosis and Management. Int. J. Mol. Sci. 2020, 22, 100. [Google Scholar] [CrossRef]
- Fujihara, K. MOG-Antibody-Associated Disease is Different from MS and NMOSD and Should be Classified as a Distinct Disease Entity—Commentary. Mult. Scler. J. 2020, 26, 276–278. [Google Scholar] [CrossRef]
- Fujihara, K.; Cook, L.J. Neuromyelitis Optica Spectrum Disorders and Myelin Oligodendrocyte Glycoprotein Antibody-Associated Disease: Current Topics. Curr. Opin. Neurol. 2020, 33, 300–308. [Google Scholar] [CrossRef]
- Fujihara, K. Neuromyelitis Optica Spectrum Disorders: Still Evolving and Broadening. Curr. Opin. Neurol. 2019, 32, 385–394. [Google Scholar] [CrossRef]
- Kunchok, A.; Flanagan, E.P.; Snyder, M.; Saadeh, R.; Chen, J.J.; Weinshenker, B.G.; McKeon, A.; Pittock, S.J. Coexisting Systemic and Organ-Specific Autoimmunity in MOG-IgG1-Associated Disorders Versus AQP4-IgG + NMOSD. Mult. Scler. J. 2021, 27, 630–635. [Google Scholar] [CrossRef] [PubMed]
- Martinez-Hernandez, E.; Guasp, M.; García-Serra, A.; Maudes, E.; Ariño, H.; Sepulveda, M.; Armangué, T.; Ramos, A.P.; Ben-Hur, T.; Iizuka, T.; et al. Clinical Significance of Anti-NMDAR Concurrent with Glial or Neuronal Surface Antibodies. Neurology 2020, 94, e2302–e2310. [Google Scholar] [CrossRef] [PubMed]
- Rinaldi, S.; Davies, A.; Fehmi, J.; Beadnall, H.N.; Wang, J.; Hardy, T.A.; Barnett, M.H.; Broadley, S.A.; Waters, P.; Reddel, S.W.; et al. Overlapping Central and Peripheral Nervous System Syndromes in MOG Antibody–Associated Disorders. Neurol. Neuroimmunol. Neuroinflamm. 2021, 8, e924. [Google Scholar] [CrossRef]
- Prasad, S.; Chen, J. What You Need to Know About AQP4, MOG, and NMOSD. Semin. Neurol. 2019, 39, 718–731. [Google Scholar] [CrossRef]
- Kitley, J.; Palace, J. Therapeutic Options in Neuromyelitis Optica Spectrum Disorders. Expert Rev. Neurother. 2016, 16, 319–329. [Google Scholar] [CrossRef] [PubMed]
- Traboulsee, A.; Greenberg, B.M.; Bennett, J.L.; Szczechowski, L.; Fox, E.; Shkrobot, S.; Yamamura, T.; Terada, Y.; Kawata, Y.; Wright, P.; et al. Safety and Efficacy of Satralizumab Monotherapy in Neuromyelitis Optica Spectrum Disorder: A Randomised, Double-Blind, Multicentre, Placebo-Controlled Phase 3 Trial. Lancet Neurol. 2020, 19, 402–412. [Google Scholar] [CrossRef]
- Pittock, S.J.; Berthele, A.; Fujihara, K.; Kim, H.J.; Levy, M.; Palace, J.; Nakashima, I.; Terzi, M.; Totolyan, N.; Viswanathan, S.; et al. Eculizumab in Aquaporin-4-Positive Neuromyelitis Optica Spectrum Disorder. N. Engl. J. Med. 2019, 381, 614–625. [Google Scholar] [CrossRef]
- Cree, B.A.C.; Bennett, J.L.; Kim, H.J.; Weinshenker, B.G.; Pittock, S.J.; Wingerchuk, D.M.; Fujihara, K.; Paul, F.; Cutter, G.R.; Marignier, R.; et al. Inebilizumab for the Treatment of Neuromyelitis Optica Spectrum Disorder (N-MOmentum): A Double-Blind, Randomised Placebo-Controlled Phase 2/3 Trial. Lancet 2019, 394, 1352–1363. [Google Scholar] [CrossRef]
- Brod, S.A. Review of Approved NMO Therapies Based on Mechanism of Action, Efficacy and Long-Term Effects. Mult. Scler. Relat. Disord. 2020, 46, 102538. [Google Scholar] [CrossRef]
- Ceglie, G.; Papetti, L.; Valeriani, M.; Merli, P. Hematopoietic Stem Cell Transplantation in Neuromyelitis Optica-Spectrum Disorders (NMO-SD): State-of-the-Art and Future Perspectives. Int. J. Mol. Sci. 2020, 21, 5304. [Google Scholar] [CrossRef]
- Avouac, A.; Maarouf, A.; Stellmann, J.-P.; Rico, A.; Boutiere, C.; Demortiere, S.; Marignier, R.; Pelletier, J.; Audoin, B. Rituximab-Induced Hypogammaglobulinemia and Infections in AQP4 and MOG Antibody–Associated Diseases. Neurol. Neuroimmunol. Neuroinflamm. 2021, 8, e977. [Google Scholar] [CrossRef] [PubMed]
- Takai, Y.; Misu, T.; Kaneko, K.; Chihara, N.; Narikawa, K.; Tsuchida, S.; Nishida, H.; Komori, T.; Seki, M.; Komatsu, T.; et al. Myelin Oligodendrocyte Glycoprotein Antibody-Associated Disease: An Immunopathological Study. Brain 2020, 143, 1431–1446. [Google Scholar] [CrossRef] [PubMed]
- Agre, P. Nobel Lecture. Available online: https://www.nobelprize.org/prizes/chemistry/2003/agre/lecture/ (accessed on 23 July 2021).
- Hasegawa, H.; Ma, T.; Skach, W.; Matthay, A.M.; Verkman, A.S. Molecular Cloning of a Mercurial-Insensitive Water Channel Expressed in Selected Water-Transporting Tissues. J. Biol. Chem. 1994, 269, 5497–5500. [Google Scholar] [CrossRef]
- Mader, S.; Kümpfel, T.; Meinl, E. Novel Insights into Pathophysiology and Therapeutic Possibilities Reveal Further Differences between AQP4-IgG- and MOG-IgG-Associated Diseases. Curr. Opin. Neurol. 2020, 33, 362–371. [Google Scholar] [CrossRef]
- Papadopoulos, M.C.; Verkman, A. Aquaporin 4 and Neuromyelitis Optica. Lancet Neurol. 2012, 11, 535–544. [Google Scholar] [CrossRef] [Green Version]
- Verkman, A. Aquaporins in Clinical Medicine. Annu. Rev. Med. 2012, 63, 303–316. [Google Scholar] [CrossRef] [Green Version]
- Matiello, M.; Schaefer-Klein, J.; Sun, D.; Weinshenker, B.G. Aquaporin 4 Expression and Tissue Susceptibility to Neuromyelitis Optica. JAMA Neurol. 2013, 70, 1118–1125. [Google Scholar] [CrossRef] [Green Version]
- Saini, H.; Fernandez, G.; Kerr, U.; Levy, M. Differential Expression of Aquaporin-4 Isoforms Localizes with Neuromyelitis Optica Disease Activity. J. Neuroimmunol. 2010, 221, 68–72. [Google Scholar] [CrossRef]
- Hillebrand, S.; Schanda, K.; Nigritinou, M.; Tsymala, I.; Böhm, D.; Peschl, P.; Takai, Y.; Fujihara, K.; Nakashima, I.; Misu, T.; et al. Circulating AQP4-Specific Auto-Antibodies Alone can Induce Neuromyelitis Optica Spectrum Disorder in the Rat. Acta Neuropathol. 2019, 137, 467–485. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, M.V.; Huang, H.; Calabresi, P.; Levy, M. Pathogenic Aquaporin-4 Reactive T Cells are Sufficient to induce Mouse Model of Neuromyelitis Optica. Acta Neuropathol. Commun. 2015, 3, 1–8. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, R.; Makuch, M.; Kienzler, A.-K.; Varley, J.; Taylor, J.; Woodhall, M.; Palace, J.; Leite, M.I.; Waters, P.; Irani, S.R. Condition-Dependent Generation of Aquaporin-4 Antibodies from Circulating B Cells in Neuromyelitis Optica. Brain 2018, 141, 1063–1074. [Google Scholar] [CrossRef]
- Cho, E.B.; Cho, H.-J.; Seok, J.M.; Min, J.-H.; Kang, E.-S.; Kim, B.J. The IL-10-Producing Regulatory B Cells (B10 Cells) and Regulatory T Cell Subsets in Neuromyelitis Optica Spectrum Disorder. Neurol. Sci. 2018, 39, 543–549. [Google Scholar] [CrossRef] [PubMed]
- Cotzomi, E.; Stathopoulos, P.; Lee, C.S.; Ritchie, A.M.; Soltys, J.N.; Delmotte, F.R.; Oe, T.; Sng, J.; Jiang, R.; Ma, A.K.; et al. Early B Cell Tolerance Defects in Neuromyelitis Optica Favour Anti-AQP4 Autoantibody Production. Brain 2019, 142, 1598–1615. [Google Scholar] [CrossRef] [PubMed]
- Chen, T.; Lennon, V.A.; Liu, Y.U.; Bosco, D.B.; Li, Y.; Yi, M.-H.; Zhu, J.; Wei, S.; Wu, L.-J. Astrocyte-Microglia Interaction Drives Evolving Neuromyelitis Optica Lesion. J. Clin. Investig. 2020, 130, 4025–4038. [Google Scholar] [CrossRef]
- Nishiyama, S.; Ito, T.; Misu, T.; Takahashi, T.; Kikuchi, A.; Suzuki, N.; Jin, K.; Aoki, M.; Fujihara, K.; Itoyama, Y. A Case of NMO Seropositive for Aquaporin-4 Antibody More than 10 Years before Onset. Neurology 2009, 72, 1960–1961. [Google Scholar] [CrossRef]
- Jarius, S.; Paulb, F.; Franciottac, D.; Ruprechtd, K.; Ringelsteine, M.; Bergamaschic, R.; Pommerf, R.; Kleiterg, I.; Stichh, O.; Reussi, R.; et al. Cerebrospinal Fluid Findings in Aquaporin-4 Antibody Positive Neuromyelitis Optica: Results from 211 Lumbar Punc-tures. J. Neurol. Sci. 2011, 306, 82–90. [Google Scholar]
- Sepúlveda, M.; Sola-Valls, N.; Escudero, D.; Rojc, B.; Barón, M.; Hernández-Echebarría, L.; Gómez, B.; Dalmau, J.; Saiz, A.; Graus, F. Clinical Profile of Patients with Paraneoplastic Neuromyelitis Optica Spectrum Disorder and Aquaporin-4 Anti-Bodies. Mult. Scler. 2018, 24, 1753–1759. [Google Scholar] [CrossRef] [PubMed]
- Varrin-Doyer, M.; Bs, C.M.S.; Schulze-Topphoff, U.; Nelson, P.A.; Stroud, R.M.; Cree, B.A.C.; Zamvil, S.S. Aquaporin 4-Specific T Cells in Neuromyelitis Optica Exhibit a Th17 Bias and Recognize Clostridium ABC Transporter. Ann. Neurol. 2012, 72, 53–64. [Google Scholar] [CrossRef] [Green Version]
- Pandit, L.; Cox, L.M.; Malli, C.; D’Cunha, A.; Rooney, T.; Lokhande, H.; Willocq, V.; Saxena, S.; Chitnis, T. Clostridium Bolteae is Elevated in Neuromyelitis Optica Spectrum Disorder in India and Shares Sequence Similarity with AQP4. Neurol. Neuroimmunol. Neuroinflamm. 2021, 8, e907. [Google Scholar] [CrossRef]
- Graves, J.; Grandhe, S.; Weinfurtner, K.; Krupp, L.; Belman, A.; Chitnis, T.; Ness, J.; Weinstock-Guttman, B.; Gorman, M.; Patterson, M.; et al. Protective Environmental Factors for Neuromyelitis Optica. Neurology 2014, 83, 1923–1929. [Google Scholar] [CrossRef] [Green Version]
- Matsushita, T.; Masaki, K.; Isobe, N.; Sato, S.; Yamamoto, K.; Nakamura, Y.; Watanabe, M.; Suenaga, T.; Kira, J. The Japan Multiple Sclerosis Genetic Consortium Genetic Factors for Susceptibility to and Manifestations of Neuromyelitis Optica. Ann. Clin. Transl. Neurol. 2020, 7, 2082–2093. [Google Scholar] [CrossRef]
- Watanabe, M.; Nakamura, Y.; Sato, S.; Niino, M.; Fukaura, H.; Tanaka, M.; Ochi, H.; Kanda, T.; Takeshita, Y.; Yokota, T.; et al. HLA Genotype-Clinical Phenotype Correlations in Multiple Sclerosis and Neuromyelitis Optica Spectrum Disorders Based on Japan MS/NMOSD Biobank Data. Sci. Rep. 2021, 11, 607. [Google Scholar] [CrossRef] [PubMed]
- Bruijstens, A.L.; Wong, Y.Y.M.; van Pelt, D.E.; van der Linden, P.J.E.; Haasnoot, G.W.; Hintzen, R.Q.; Claas, F.H.J.; Neuteboom, R.F.; Wokke, B.H.A. HLA Association in MOG-IgG- and AQP4-IgG-Related Disorders of the CNS in the Dutch Population. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7. [Google Scholar] [CrossRef] [Green Version]
- Alvarenga, M.P.; do Carmo, L.F.; Vasconcelos, C.C.F.; Alvarenga, M.P.; Alvarenga-Filho, H.; de Melo Bento, C.A.; Paiva, C.L.A.; Leyva-Fernández, L.; Fernández, Ó.; Papais-Alvarenga, R.M. Neuromyelitis Optica is an HLA Associated Disease Different from MULTIPLE SCLEROSIS: A Systematic Review with Meta-Analysis. Sci. Rep. 2021, 11, 152. [Google Scholar] [CrossRef]
- Yandamuri, S.S.; Jiang, R.; Sharma, A.; Cotzomi, E.; Zografou, C.; Ma, A.K.; Alvey, J.S.; Cook, L.J.; Smith, T.J.; Yeaman, M.R.; et al. High-Throughput Investigation of Molecular and Cellular Biomarkers in NMOSD. Neurol. Neuroimmunol. Neuroinflamm. 2020, 7, e852. [Google Scholar] [CrossRef]
- Höftberger, R.; Guo, Y.; Flanagan, E.P.; Lopez-Chiriboga, A.S.; Endmayr, V.; Hochmeister, S.; Joldic, D.; Pittock, S.J.; Tillema, J.M.; Gorman, M.; et al. The Pathology of Central Nervous System Inflammatory Demyelinating Disease Accompanying Myelin Oli-Godendrocyte Glycoprotein Autoantibody. Acta Neuropathol. 2020, 139, 875–892. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hofer, L.S.; Ramberger, M.; Gredler, V.; Pescoller, A.S.; Rostásy, K.; Sospedra, M.; Hegen, H.; Berger, T.; Lutterotti, A.; Reindl, M. Comparative Analysis of T-Cell Responses to Aquaporin-4 and Myelin Oligodendrocyte Glycoprotein in Inflammatory Demyelinating Central Nervous System Diseases. Front. Immunol. 2020, 11, 1188. [Google Scholar] [CrossRef]
- Winklmeier, S.; Schlüter, M.; Spadaro, M.; Thaler, F.S.; Vural, A.; Gerhards, R.; Macrini, C.; Mader, S.; Kurne, A.; Inan, B.; et al. Identification of Circulating MOG-Specific B Cells in Patients with MOG Antibodies. Neurol. Neuroimmunol. Neuroinflamm. 2019, 6, 625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spadaro, M.; Winklmeier, S.; Beltrán, E.; Macrini, C.; Höftberger, R.; Schuh, E.; Thaler, F.S.; Gerdes, L.A.; Laurent, S.; Gerhards, R.; et al. Pathogenicity of Human Antibodies against Myelin Oligodendrocyte Glycoprotein. Ann. Neurol. 2018, 84, 315–328. [Google Scholar] [CrossRef] [Green Version]
- Fovet, C.-M.; Stimmer, L.; Contreras, V.; Horellou, P.; Hubert, A.; Seddiki, N.; Chapon, C.; Tricot, S.; Leroy, C.; Flament, J.; et al. Intradermal Vaccination Prevents Anti-MOG Autoimmune Encephalomyelitis in Macaques. EBioMedicine 2019, 47, 492–505. [Google Scholar] [CrossRef]
- Grant-Peters, M.; Dos Passos, G.R.; Yeung, H.; Jacob, A.; Huda, S.; Leite, M.I.; Dendrou, C.A.; Palace, J. No Strong HLA Association with MOG Antibody Disease in the UK Population. Ann. Clin. Transl. Neurol. 2021. [Google Scholar] [CrossRef]
- Peters, J.; Alhasan, S.; Vogels, C.B.; Grubaugh, N.D.; Farhadian, S.; Longbrake, E.E. MOG-Associated Encephalitis Following SARS-COV-2 Infection. Mult. Scler. Relat. Disord. 2021, 50, 102857. [Google Scholar] [CrossRef]
- Jumah, M.; Rahman, F.; Figgie, M.; Prasad, A.; Zampino, A.; Fadhil, A.; Palmer, K.; Buerki, R.A.; Gunzler, S.; Gundelly, P.; et al. COVID-19, HHV6 and MOG Antibody: A Perfect Storm. J. Neuroimmunol. 2021, 353, 577521. [Google Scholar] [CrossRef]
- Wildemann, B.; Jarius, S.; Franz, J.; Ruprecht, K.; Reindl, M.; Stadelmann, C. MOG-Expressing Teratoma Followed by MOG-IgG-Positive Optic Neuritis. Acta Neuropathol. 2021, 141, 127–131. [Google Scholar] [CrossRef]
- Dos Passos, G.R.; Elsone, L.; Luppe, S.; Kitley, J.; Messina, S.; Cruz, P.M.R.; Harding, K.; Mutch, K.; Leite, M.I.; Robertson, N.; et al. Seasonal Distribution of Attacks in Aquaporin-4 Antibody Disease and Myelin-Oligodendrocyte Antibody Disease. J. Neurol. Sci. 2020, 415, 116881. [Google Scholar] [CrossRef]
- Duan, T.; Verkman, A.S. Experimental Animal Models of Aquaporin-4-IgG-Seropositive Neuromyelitis Optica Spectrum Disorders: Progress and Shortcomings. Brain Pathol. 2020, 30, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Zeka, B.; Hastermann, M.; Hochmeister, S.; Kögl, N.; Kaufmann, N.; Schanda, K.; Mader, S.; Misu, T.; Rommer, P.; Fujihara, K.; et al. Highly Encephalitogenic Aquaporin 4-Specific T Cells and NMO-IgG Jointly Orchestrate Lesion Location and Tissue Damage in the CNS. Acta Neuropathol. 2015, 130, 783–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakuma, H.; Kohyama, K.; Park, I.-K.; Miyakoshi, A.; Tanuma, N.; Matsumoto, Y. Clinicopathological Study of a myelin Oligodendrocyte Glycoprotein-Induced Demyelinating Disease in LEW.1AV1 Rats. Brain 2004, 127, 2201–2213. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stefferl, A.; Brehm, U.; Storch, M.; Lambracht-Washington, D.; Bourquin, C.; Wonigeit, K.; Lassmann, H.; Linington, C.; Stefferl, A.; Brehm, U.; et al. Myelin Oligodendrocyte Glycoprotein Induces Experimental Autoimmune Encephalomyelitis in the “Resistant” Brown Norway Rat: Disease Susceptibility is Determined by MHC and MHC-Linked Effects on the B Cell Response. J. Immunol. 1999, 163, 40–49. [Google Scholar] [PubMed]
- Storch, M.K.; Stefferl, A.; Brehm, U.; Weissert, R.; Wallström, E.; Kerschensteiner, M.; Olsson, T.; Linington, C.; Lassmann, H. Autoimmunity to Myelin Oligodendrocyte Glycoprotein in Rats Mimics the Spectrum of Multiple Sclerosis Pathology. Brain Pathol. 1998, 8, 681–694. [Google Scholar] [CrossRef] [PubMed]
- Collongues, N.; Chanson, J.; Blanc, F.; Steibel, J.; Lam, C.; Shabbir, A.; Trifilieff, E.; Honnorat, J.; Pham-Dinh, D.; Ghandour, M.; et al. The Brown Norway Opticospinal Model of Demyelination: Does it Mimic Multiple Sclerosis or Neuromyelitis Optica? Int. J. Dev. Neurosci. 2012, 30, 487–497. [Google Scholar] [CrossRef]
- Tsymala, I.; Nigritinou, M.; Zeka, B.; Schulz, R.; Niederschick, F.; Matković, M.; Bauer, I.J.; Szalay, M.; Schanda, K.; Lerch, M.; et al. Induction of Aquaporin 4-Reactive Antibodies in Lewis Rats Immunized with Aquaporin 4 Mimotopes. Acta Neuropathol. Commun. 2020, 8, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Pittock, S.J.; Lennon, V.A.; McKeon, A.; Mandrekar, J.; Weinshenker, B.G.; Lucchinetti, C.F.; Toole, O.O.; Wingerchuk, D.M. Eculizumab in AQP4-IgG-Positive Relapsing Neuromyelitis Optica Spectrum Disorders: An Open-Label Pilot Study. Lancet Neurol. 2013, 12, 554–562. [Google Scholar] [CrossRef]
- Ratelade, J.; Verkman, A. Inhibitor(s) of the Classical Complement Pathway in Mouse Serum Limit the Utility of Mice as Experimental Models of Neuromyelitis Optica. Mol. Immunol. 2014, 62, 104–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bettelli, E.; Pagany, M.; Weiner, H.L.; Linington, C.; Sobel, R.A.; Kuchroo, V.K. Myelin Oligodendrocyte Glycoprotein–Specific T Cell Receptor Transgenic Mice Develop Spontaneous Autoimmune Optic Neuritis. J. Exp. Med. 2003, 197, 1073–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bettelli, E.; Baeten, M.; Jäger, A.; Sobel, R.A.; Kuchroo, V.K. Myelin Oligodendrocyte Glycoprotein–Specific T and B Cells Cooperate to Induce a Devic-Like Disease in Mice. J. Clin. Investig. 2006, 116, 2393–2402. [Google Scholar] [CrossRef]
- Zubizarreta, I.; Flórez-Grau, G.; Vila, G.; Cabezón, R.; España, C.; Andorra, M.; Saiz, A.; Llufriu, S.; Sepulveda, M.; Sola-Valls, N.; et al. Immune Tolerance in Multiple Sclerosis and Neuromyelitis Optica with Peptide-Loaded Tolerogenic Dendritic Cells in a Phase 1b Trial. Proc. Natl. Acad. Sci. USA 2019, 116, 8463–8470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burt, R.K.; Balabanov, R.; Han, X.; Burns, C.; Gastala, J.; Jovanovic, B.; Helenowski, I.; Jitprapaikulsan, J.; Fryer, J.P.; Pittock, S.J. Autologous Nonmyeloablative Hematopoietic Stem Cell Transplantation for Neuromyelitis Optica. Neurology 2019, 93, e1732–e1741. [Google Scholar] [CrossRef]
- Fu, Y.; Yan, Y.; Qi, Y.; Yang, L.; Li, T.; Zhang, N.; Yu, C.; Su, L.; Zhang, R.; Shen, Y.; et al. Impact of Autologous Mesenchymal Stem Cell Infusion on Neuromyelitis Optica Spectrum Disorder: A Pilot, 2-Year Observational Study. CNS Neurosci. Ther. 2016, 22, 677–685. [Google Scholar] [CrossRef] [PubMed]
- Lu, Z.; Zhu, L.; Liu, Z.; Wu, J.; Xu, Y.; Zhang, C.J. IV/IT hUC-MSCs Infusion in RRMS and NMO: A 10-Year Follow-Up Study. Front. Neurol. 2020, 11, 967. [Google Scholar] [CrossRef]
- Zhang, P.; Liu, B. Effect of Autologous Hematopoietic Stem Cell Transplantation on Multiple Sclerosis and Neuromyelitis Optica Spectrum Disorder: A PRISMA-Compliant Meta-Analysis. Bone Marrow Transplant. 2020, 55, 1928–1934. [Google Scholar] [CrossRef] [PubMed]
- Greco, R.; Bondanza, A.; Oliveira, M.C.; Badoglio, M.; Burman, J.; Piehl, F.; Hagglund, H.; Krasulová, E.; Simões, B.P.; Carlson, K.; et al. Autologous Hematopoietic Stem Cell Transplantation in Neuromyelitis Optica: A Registry Study of the EBMT Autoimmune Diseases Working Party. Mult. Scler. J. 2015, 21, 189–197. [Google Scholar] [CrossRef]
- Hoay, K.Y.; Ratnagopal, P. Autologous Hematopoietic Stem Cell Transplantation for the Treatment of Neuromyelitis Optica in Singapore. Acta Neurol. Taiwanica 2018, 27, 26–32. [Google Scholar]
- Sharrack, B.; For the European Society for Blood and Marrow Transplantation (EBMT) Autoimmune Diseases Working Party (ADWP) and the Joint Accreditation Committee of the International Society for Cellular Therapy (ISCT) and EBMT (JACIE); Saccardi, R.; Alexander, T.; Badoglio, M.; Burman, J.; Farge, D.; Greco, R.; Jessop, H.; Kazmi, M.; et al. Autologous Haematopoietic Stem Cell Transplantation and Other Cellular Therapy in Multiple Sclerosis and Immune-Mediated Neurological Diseases: Updated Guidelines and Recommendations from the EBMT Autoimmune Diseases Working Party (ADWP) and the Joint Accreditation Committee of EBMT and ISCT (JACIE). Bone Marrow Transplant. 2020, 55, 283–306. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hau, L.; Kállay, K.; Kertész, G.; Goda, V.; Kassa, C.; Horváth, O.; Liptai, Z.; Constantin, T.; Kriván, G. Allogeneic Haematopoietic Stem Cell Transplantation in a Refractory Case of Neuromyelitis Optica Spectrum Disorder. Mult. Scler. Relat. Disord. 2020, 42, 102110. [Google Scholar] [CrossRef] [PubMed]
- Ceglie, G.; Papetti, L.; Talamanca, L.F.; Lucarelli, B.; Algeri, M.; Gaspari, S.; Pira, G.L.; Colafati, G.; Montanari, M.; Valeriani, M.; et al. T-Cell Depleted HLA-Haploidentical HSCT in a Child with Neuromyelitis Optica. Ann. Clin. Transl. Neurol. 2019, 6, 2110–2113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greco, R.; Bondanza, A.; Vago, L.; Moiola, L.; Rossi, P.; Furlan, R.; Martino, G.; Radaelli, M.; Martinelli, V.; Carbone, M.R.; et al. Allogeneic Hematopoietic Stem Cell Transplantation for Neuromyelitis Optica. Ann. Neurol. 2014, 75, 447–453. [Google Scholar] [CrossRef]
- Hümmert, M.W.; Stadler, M.; Hambach, L.; Gingele, S.; Bredt, M.; Wattjes, M.P.; Göhring, G.; Venturini, L.; Möhn, N.; Stangel, M.; et al. Severe Allo-Immune Antibody-Associated Peripheral and Central Nervous System Diseases after Allogeneic Hematopoietic Stem Cell Transplantation. Sci. Rep. 2021, 11, 1–8. [Google Scholar] [CrossRef]
- Rush, C.; Atkins, H. Aggressive and refractory MOG IgG Associated Encephalomyelitis Treated with Autologous Hematopoeitic Stem Cell Transplantation: First Case Worldwide. Mult. Scler. J. 2019, 25, 367–368. [Google Scholar]
- Dotti, G.; Gottschalk, S.; Savoldo, B.; Brenner, M.K. Design and Development of Therapies Using Chimeric Antigen Receptor-expressing T Cells. Immunol. Rev. 2014, 257, 107–126. [Google Scholar] [CrossRef]
- Jensen, T.I.; Axelgaard, E.; Bak, R.O. Therapeutic Gene Editing in Haematological Disorders with CRISPR/Cas9. Br. J. Haematol. 2019, 185, 821–835. [Google Scholar] [CrossRef] [PubMed]
- Atrash, S.; Bano, K.; Harrison, B.; Abdallah, A.-O. CAR-T Treatment for Hematological Malignancies. J. Investig. Med. 2020, 68, 956–964. [Google Scholar] [CrossRef] [PubMed]
- Martinez, M.; Moon, E.K. CAR T Cells for Solid Tumors: New Strategies for Finding, Infiltrating, and Surviving in the Tumor Microenvironment. Front. Immunol. 2019, 10, 128. [Google Scholar] [CrossRef] [Green Version]
- Vairy, S.; Garcia, J.L.; Teira, P.; Bittencourt, H. CTL019 (Tisagenlecleucel): CAR-T Therapy for Relapsed and Refractory B-Cell Acute Lymphoblastic Leukemia. Drug Des. Dev. Ther. 2018, 12, 3885–3898. [Google Scholar] [CrossRef] [Green Version]
- Abramson, J.S. Anti-CD19 CAR T-Cell Therapy for B-Cell Non-Hodgkin Lymphoma. Transfus. Med. Rev. 2020, 34, 29–33. [Google Scholar] [CrossRef]
- Kersten, M.J.; Spanjaart, A.M.; Thieblemont, C. CD19-Directed CAR T-Cell Therapy in B-Cell NHL. Curr. Opin. Oncol. 2020, 32, 408–417. [Google Scholar] [CrossRef]
- Roex, G.; Timmers, M.; Wouters, K.; Campillo-Davo, D.; Flumens, D.; Schroyens, W.; Chu, Y.; Berneman, Z.N.; Lion, E.; Luo, F.; et al. Safety and Clinical Efficacy of BCMA CAR-T-Cell Therapy in Multiple Myeloma. J. Hematol. Oncol. 2020, 13, 1–14. [Google Scholar] [CrossRef]
- Jin, X.; Xu, Q.; Pu, C.; Zhu, K.; Lu, C.; Jiang, Y.; Xiao, L.; Han, Y.; Lu, L. Therapeutic Efficacy of Anti-CD19 CAR-T Cells in a Mouse Model of Systemic Lupus Erythematosus. Cell. Mol. Immunol. 2020, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Brunstein, C.G.; Miller, J.S.; Cao, Q.; McKenna, D.H.; Hippen, K.L.; Curtsinger, J.; DeFor, T.; Levine, B.L.; June, C.; Rubinstein, P.; et al. Infusion of Ex Vivo Expanded T Regulatory Cells in Adults Transplanted with Umbilical Cord Blood: Safety Profile and Detection Kinetics. Blood 2011, 117, 1061–1070. [Google Scholar] [CrossRef]
- Martelli, M.F.; Di Ianni, M.; Ruggeri, L.; Falzetti, F.; Carotti, A.; Terenzi, A.; Pierini, A.; Massei, M.S.; Amico, L.; Urbani, E.; et al. HLA-Haploidentical Transplantation with Regulatory and Conventional T-Cell Adoptive Immunotherapy Prevents Acute Leukemia Relapse. Blood 2014, 124, 638–644. [Google Scholar] [CrossRef]
- Trzonkowski, P.; Dukat-Mazurek, A.; Bieniaszewska, M.; Marek-Trzonkowska, N.; Dobyszuk, A.; Juścińska, J.; Dutka, M.; Myśliwska, J.; Hellmann, A. Treatment of Graft-Versus-Host Disease with Naturally Occurring T Regulatory Cells. BioDrugs 2013, 27, 605–614. [Google Scholar] [CrossRef] [Green Version]
- Marek-Trzonkowska, N.; Myśliwiec, M.; Dobyszuk, A.; Grabowska, M.; Techmańska, I.; Juścińska, J.; Wujtewicz, M.A.; Witkowski, P.; Młynarski, W.; Balcerska, A.; et al. Administration of CD4+CD25highCD127- Regulatory T cells Preserves β-Cell Function in Type 1 Diabetes in Children. Diabetes Care 2012, 35, 1817–1820. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bluestone, J.A.; Buckner, J.H.; Fitch, M.D.; Gitelman, S.E.; Gupta, S.; Hellerstein, M.K.; Herold, K.C.; Lares, A.; Lee, M.R.; Li, K.; et al. Type 1 Diabetes Immunotherapy Using Polyclonal Regulatory T Cells. Sci. Transl. Med. 2015, 7, 315ra189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohseni, Y.R.; Tung, S.L.; Dudreuilh, C.; Lechler, R.I.; Fruhwirth, G.O.; Lombardi, G. The Future of Regulatory T Cell Therapy: Promises and Challenges of Implementing CAR Technology. Front. Immunol. 2020, 11, 1608. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Zhang, Z.; Lu, Z.; Borlongan, C.; Pan, J.; Chen, J.; Qian, L.; Liu, Z.; Zhu, L.; Zhang, J.; et al. Human Umbilical Cord Stem Cells Ameliorate Experimental Autoimmune Encephalomyelitis by Regulating Immunoinflammation and Remyelination. Stem Cells Dev. 2013, 22, 1053–1062. [Google Scholar] [CrossRef]
- Kokaia, Z.; Martino, G.; Schwartz, M.; Lindvall, O. Cross-Talk Between Neural Stem Cells and Immune Cells: The Key to Better Brain Repair? Nat. Neurosci. 2012, 15, 1078–1087. [Google Scholar] [CrossRef]
- Le Blanc, K.; Mougiakakos, D. Multipotent Mesenchymal Stromal Cells and the Innate Immune system. Nat. Rev. Immunol. 2012, 12, 383–396. [Google Scholar] [CrossRef]
- Pittenger, M.F.; Mackay, A.M.; Beck, S.C.; Jaiswal, R.K.; Douglas, R.; Mosca, J.D.; Moorman, M.A.; Simonetti, D.W.; Craig, S.; Marshak, D.R. Multilineage Potential of Adult Human Mesenchymal Stem Cells. Science 1999, 284, 143–147. [Google Scholar] [CrossRef] [Green Version]
- Prockop, D.J. Marrow Stromal Cells as Stem Cells for Nonhematopoietic Tissues. Science 1997, 276, 71–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lu, Z.; Ye, D.; Qian, L.; Zhu, L.; Wang, C.; Guan, D.; Zhang, X.; Xu, Y. Human Umbilical Cord Mesenchymal Stem Cell Therapy on Neuromyelitis Optica. Curr. Neurovasc. Res. 2012, 9, 250–255. [Google Scholar] [CrossRef]
- Wang, H.; Qiu, X.; NI, P.; Qiu, X.; Lin, X.; Wu, W.; Xie, L.; Lin, L.; Min, J.; Lai, X.; et al. Immunological Characteristics of Human Umbilical Cord Mesenchymal Stem Cells and the Therapeutic Effects of Their Transplantion on Hyperglycemia in Diabetic Rats. Int. J. Mol. Med. 2014, 33, 263–270. [Google Scholar] [CrossRef]
- Hayes, C. Cellular Immunotherapies for Cancer. Ir. J. Med Sci. 2021, 190, 41–57. [Google Scholar] [CrossRef]
- Akaishi, T.; Takahashi, T.; Nakashima, I.; Abe, M.; Ishii, T.; Aoki, M.; Fujihara, K. Repeated Follow-Up of AQP4-IgG Titer by Cell-Based Assay in Neuromyelitis Optica Spectrum Disorders (NMOSD). J. Neurol. Sci. 2020, 410, 116671. [Google Scholar] [CrossRef]
- Kessler, R.A.; Mealy, M.A.; Jimenez-Arango, J.A.; Quan, C.; Paul, F.; López, R.; Hopkins, S.; Levy, M. Anti-Aquaporin-4 Titer is Not Predictive of Disease Course in Neuromyelitis Optica Spectrum Disorder: A Multicenter Cohort Study. Mult. Scler. Relat. Disord. 2017, 17, 198–201. [Google Scholar] [CrossRef]
- Takahashi, T.; Fujihara, K.; Nakashima, I.; Misu, T.; Miyazawa, I.; Nakamura, M.; Watanabe, S.; Shiga, Y.; Kanaoka, C.; Fujimori, J.; et al. Anti-Aquaporin-4 Antibody is Involved in the Pathogenesis of NMO: A Study on Antibody Titre. Brain 2007, 130, 1235–1243. [Google Scholar] [CrossRef] [Green Version]
- Hennes, E.-M.; Baumann, M.; Schanda, K.; Anlar, B.; Bajer-Kornek, B.; Blaschek, A.; Brantner-Inthaler, S.; Diepold, K.; Eisenkölbl, A.; Gotwald, T.; et al. Prognostic Relevance of MOG Antibodies in Children with an Acquired Demyelinating Syndrome. Neurology 2017, 89, 900–908. [Google Scholar] [CrossRef] [Green Version]
- Cobo-Calvo, A.; The OFSEP Group; Sepúlveda, M.; D’Indy, H.; Armangue, T.; Ruiz, A.; Maillart, E.; Papeix, C.; Audoin, B.; Zephir, H.; et al. Usefulness of MOG-Antibody Titres at First Episode to Predict the Future Clinical Course in Adults. J. Neurol. 2019, 266, 806–815. [Google Scholar] [CrossRef] [PubMed]
- Cobo-Calvo, A.; Ruiz, A.; Maillart, E.; Audoin, B.; Zephir, H.; Bourre, B.; Ciron, J.; Collongues, N.; Brassat, D.; Cottonetal, F. Clinical Spectrum and Prognostic Value Of CNS MOG autoimmunity in Adults: The MOGADOR Study. Neurology 2018, 90, e1858–e1869. [Google Scholar] [CrossRef] [PubMed]
- Mariotto, S.; Ferrari, S.; Monaco, S.; Benedetti, M.D.; Schanda, K.; Alberti, D.; Farinazzo, A.; Capra, R.; Mancinelli, C.; De Rossi, N.; et al. Clinical Spectrum and IgG Subclass Analysis of Anti-Myelin Oligodendrocyte Glycoprotein Antibody-Associated Syndromes: A Multicenter Study. J. Neurol. 2017, 264, 2420–2430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jurynczyk, M.; Messina, S.; Woodhall, M.R.; Raza, N.; Everett, R.; Roca-Fernandez, A.; Tackley, G.; Hamid, S.; Sheard, A.; Reynolds, G.; et al. Clinical Presentation and Prognosis in MOG-Antibody Disease: A UK Study. Brain 2017, 140, 3128–3138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Armangue, T.; Olivé-Cirera, G.; Martínez-Hernandez, E.; Sepulveda, M.; Ruiz-García, R.; Muñoz-Batista, M.; Ariño, H.; González-Álvarez, V.; Felipe-Rucian, A.; Martínez-González, M.J.; et al. Associations of Paediatric Demyelinating and Encephalitic Syndromes with Myelin Oligodendrocyte Glycoprotein Antibodies: A Multicentre Observational Study. Lancet Neurol. 2020, 19, 234–246. [Google Scholar] [CrossRef]
- Waters, P.; Fadda, G.; Woodhall, M.; O’Mahony, J.; Brown, R.A.; Castro, D.A.; Longoni, G.; Irani, S.R.; Sun, B.; Yeh, E.A.; et al. Serial Anti–Myelin Oligodendrocyte Glycoprotein Antibody Analyses and Outcomes in Children With Demyelinating Syndromes. JAMA Neurol. 2020, 77, 82. [Google Scholar] [CrossRef] [Green Version]
- Armangue, T.; Capobianco, M.; de Chalus, A.; Laetitia, G.; Deiva, K.; Bruijstens, A.L.; Wendel, E.-M.; Lechner, C.; Bartels, F.; Finke, C.; et al. E.U. Paediatric MOG Consortium Consensus: Part 3—Biomarkers of Paediatric Myelin Oligodendrocyte Glycoprotein Antibody-Associated Disorders. Eur. J. Paediatr. Neurol. 2020, 29, 22–31. [Google Scholar] [CrossRef]
- Akaishi, T.; Takahashi, T.; Misu, T.; Kaneko, K.; Takai, Y.; Nishiyama, S.; Ogawa, R.; Fujimori, J.; Ishii, T.; Aoki, M.; et al. Difference in the Source of Anti-AQP4-IgG and Anti-MOG-IgG Antibodies in CSF in Patients With Neuromyelitis Optica Spectrum Disorder. Neurology 2021. [Google Scholar] [CrossRef]
- Carta, S.; Höftberger, R.; Bolzan, A.; Bozzetti, S.; Bonetti, B.; Scarpelli, M.; Ottaviani, S.; Ghimenton, C.; Alberti, D.; Schanda, K.; et al. Antibodies to MOG in CSF Only: Pathological Findings Support the Diagnostic Value. Acta Neuropathol. 2021, 141, 801–804. [Google Scholar] [CrossRef]
- Schindler, P.; Grittner, U.; Oechtering, J.; Leppert, D.; Siebert, N.; Duchow, A.S.; Oertel, F.C.; Asseyer, S.; Kuchling, J.; Zimmermann, H.G.; et al. Serum GFAP and NfL as Disease Severity and Prognostic Biomarkers in Patients with Aquaporin-4 Antibody-Positive Neuromyelitis Optica Spectrum Disorder. J. Neuroinflamm. 2021, 18, 1–14. [Google Scholar] [CrossRef]
- Lünemann, J.D.; Ruck, T.; Muraro, P.A.; Bar-Or, A.; Wiendl, H. Immune Reconstitution Therapies: Concepts for Durable Remission in Multiple Sclerosis. Nat. Rev. Neurol. 2020, 16, 56–62. [Google Scholar] [CrossRef]
- Honce, J.M.; Nair, K.V.; Sillau, S.; Valdez, B.; Miravalle, A.; Alvarez, E.; Schreiner, T.; Corboy, J.R.; Vollmer, T.L. Rituximab vs Placebo Induction Prior to Glatiramer Acetate Monotherapy in Multiple Sclerosis. Neurology 2019, 92, e723–e732. [Google Scholar] [CrossRef]
- Harrison, D.M.; E. Gladstone, D.; Hammond, E.; Cheng, J.; Jones, R.J.; A. Brodsky, R.; Kerr, D.; McArthur, J.C.; Kaplin, A. Treatment of Relapsing–Remitting Multiple Sclerosis with High-Dose Cyclophosphamide Induction Followed by Glatiramer Acetate Maintenance. Mult. Scler. J. 2011, 18, 202–209. [Google Scholar] [CrossRef] [Green Version]
- Yang, C.; Yang, Y.; Ma, L.; Zhang, G.; Shi, F.; Yan, Y.; Chang, G. Study of the Cytological Features of Bone Marrow Mesenchymal Stem Cells from Patients with Neuromyelitis Optica. Int. J. Mol. Med. 2019, 43, 1395–1405. [Google Scholar] [CrossRef] [Green Version]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).