
Activation of Peripheral Blood Mononuclear Cells and Leptin Secretion: New Potential Role of Interleukin-2 and High Mobility Group Box (HMGB)1


 ,
, 

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
2.1. PBMCs Pro-Inflammatory Cytokines Secretion in Response to IL-2 and HMGB1
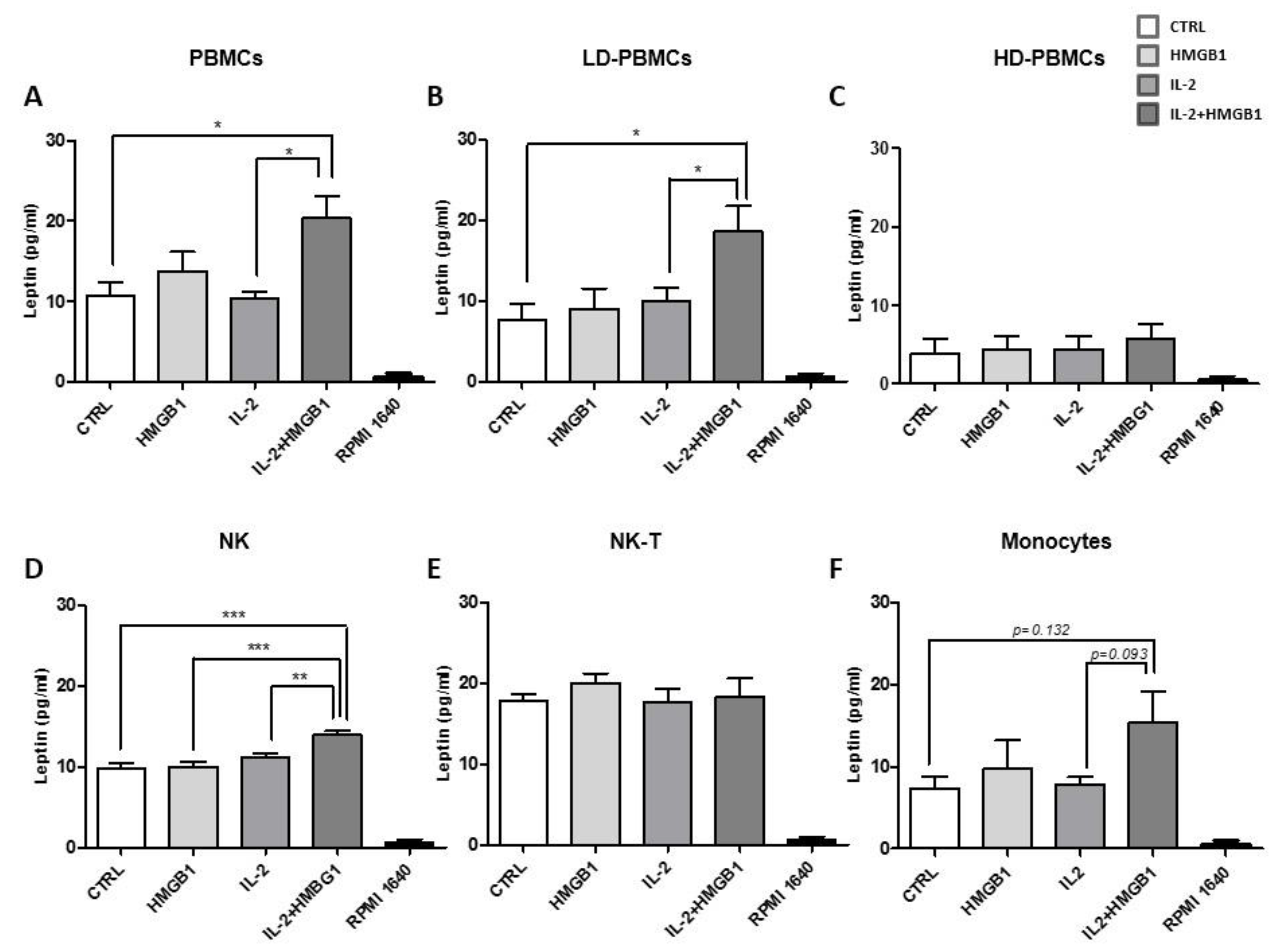
2.2. Leptin Secretion from PBMC Subpopulations
2.3. HMGB1 and IL-2 Co-Treatment Increased Leptin Secretion
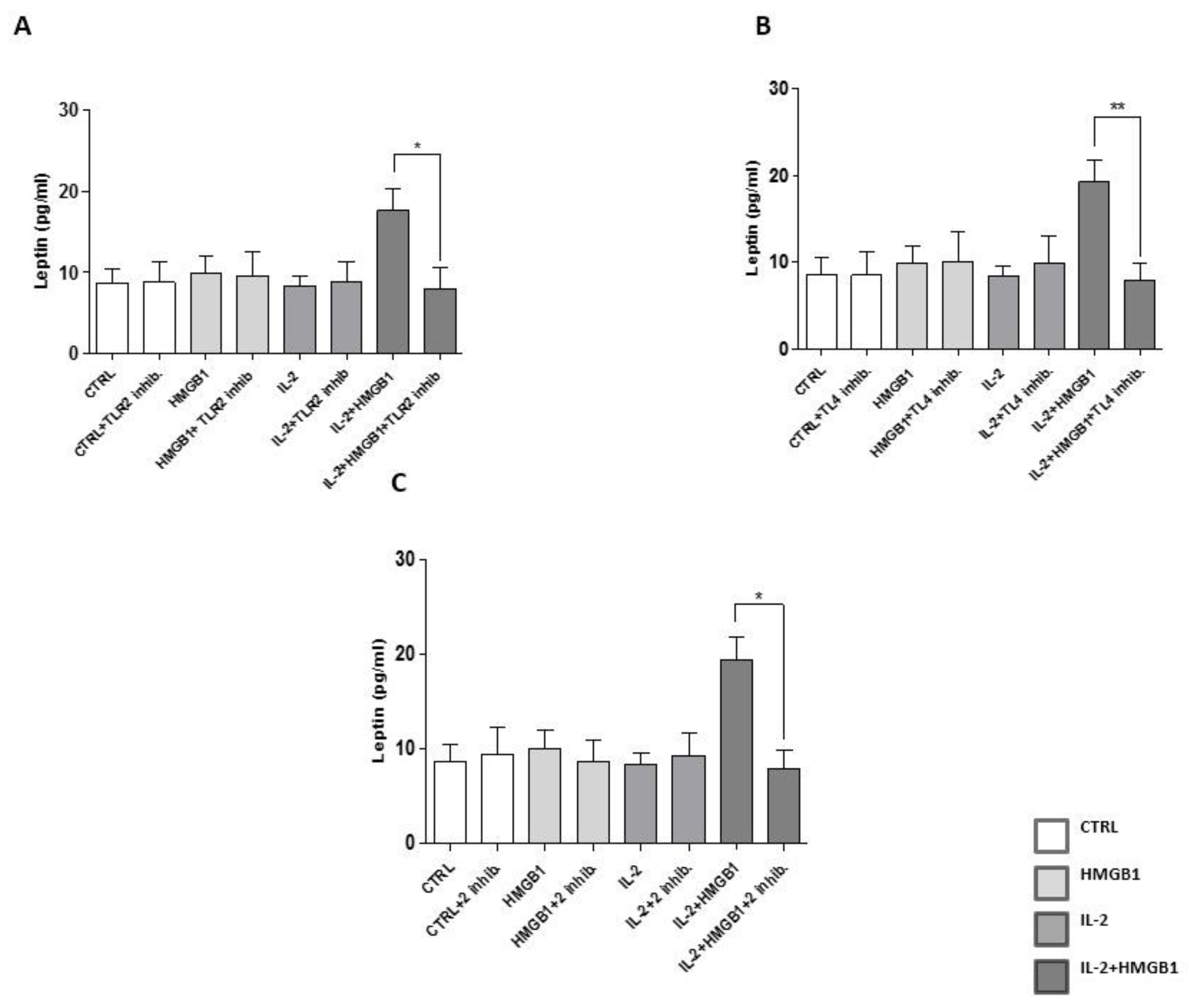
2.4. Leptin Secretion from PBMCs Was Mediated by TLR2 and TLR4
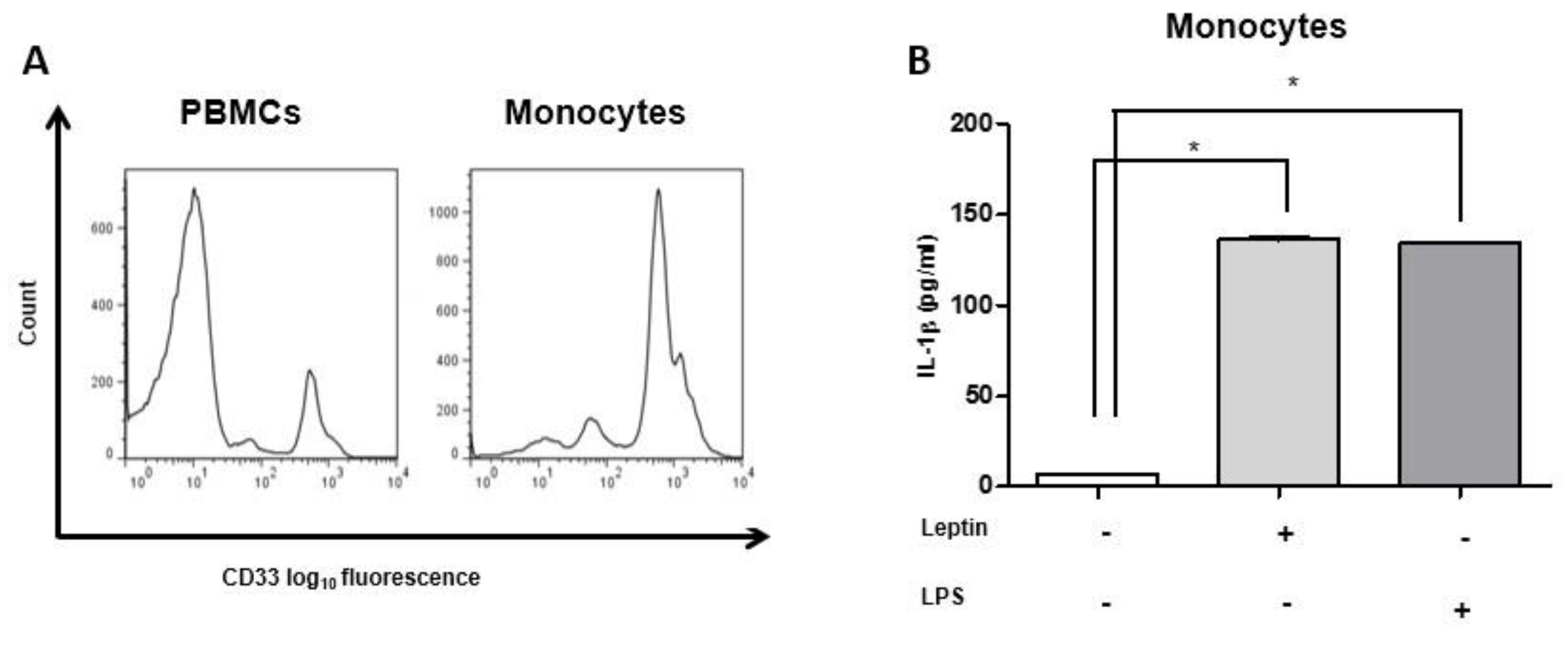
2.5. IL-1β Secretion Was Induced by Leptin and IL-2/HMGB1 Co-Treatment
2.6. Glucose Modulates Leptin Secretion
2.7. Insulin Treatment and Leptin Secretion
2.8. IL-2 and HMGB1 and Leptin Secretion in T2DM Patients’ PBMCs
3. Discussion
4. Materials and Methods
4.1. Isolation of PBMCs and Their Subpopulations
4.2. NK Cells Isolation
4.3. NK-T Cells Isolation
4.4. CD14 Monocytes Isolation
4.5. Cell Culture
4.6. Western Blotting Analysis
4.7. Enzyme-Linked Immunosorbent Assay
4.8. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Donath, M.Y.; Dalmas, E.; Sauter, N.S.; Boni-Schnetzler, M. Inflammation in obesity and diabetes: Islet dysfunction and therapeutic opportunity. Cell Metab. 2013, 17, 860–872. [Google Scholar] [CrossRef] [Green Version]
- Pollack, R.M.; Donath, M.Y.; LeRoith, D.; Leibowitz, G. Anti-inflammatory Agents in the Treatment of Diabetes and Its Vascular Complications. Diabetes Care 2016, 39, S244–S252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tesauro, M.; Schinzari, F.; Rovella, V.; Melina, D.; Mores, N.; Barini, A.; Mettimano, M.; Lauro, D.; Iantorno, M.; Quon, M.J.; et al. Tumor necrosis factor-alpha antagonism improves vasodilation during hyperinsulinemia in metabolic syndrome. Diabetes Care 2008, 31, 1439–1441. [Google Scholar] [CrossRef] [Green Version]
- Dasu, M.R.; Devaraj, S.; Park, S.; Jialal, I. Increased toll-like receptor (TLR) activation and TLR ligands in recently diagnosed type 2 diabetic subjects. Diabetes Care 2010, 33, 861–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Donath, M.Y.; Dinarello, C.A.; Mandrup-Poulsen, T. Targeting innate immune mediators in type 1 and type 2 diabetes. Nat. Rev. Immunol. 2019, 19, 734–746. [Google Scholar] [CrossRef]
- Boni-Schnetzler, M.; Thorne, J.; Parnaud, G.; Marselli, L.; Ehses, J.A.; Kerr-Conte, J.; Pattou, F.; Halban, P.A.; Weir, G.C.; Donath, M.Y. Increased interleukin (IL)-1beta messenger ribonucleic acid expression in beta-cells of individuals with type 2 diabetes and regulation of IL-1beta in human islets by glucose and autostimulation. J. Clin. Endocrinol. Metab. 2008, 93, 4065–4074. [Google Scholar] [CrossRef] [Green Version]
- Shi, H.; Kokoeva, M.V.; Inouye, K.; Tzameli, I.; Yin, H.; Flier, J.S. TLR4 links innate immunity and fatty acid-induced insulin resistance. J. Clin. Investig. 2006, 116, 3015–3025. [Google Scholar] [CrossRef] [PubMed]
- Wong, F.S.; Wen, L. Toll-like receptors and diabetes. Ann. N. Y. Acad. Sci. 2008, 1150, 123–132. [Google Scholar] [CrossRef]
- Bagarolli, R.A.; Saad, M.J.; Saad, S.T. Toll-like receptor 4 and inducible nitric oxide synthase gene polymorphisms are associated with Type 2 diabetes. J. Diab. Complicat. 2010, 24, 192–198. [Google Scholar] [CrossRef]
- Kim, H.S.; Han, M.S.; Chung, K.W.; Kim, S.; Kim, E.; Kim, M.J.; Jang, E.; Lee, H.A.; Youn, J.; Akira, S.; et al. Toll-like receptor 2 senses beta-cell death and contributes to the initiation of autoimmune diabetes. Immunity 2007, 27, 321–333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Devaraj, S.; Dasu, M.R.; Park, S.H.; Jialal, I. Increased levels of ligands of Toll-like receptors 2 and 4 in type 1 diabetes. Diabetologia 2009, 52, 1665–1668. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, M.E.; Agresti, A. HMG proteins: Dynamic players in gene regulation and differentiation. Curr. Opin. Genet. Dev. 2005, 15, 496–506. [Google Scholar] [CrossRef] [PubMed]
- Lotze, M.T.; Tracey, K.J. High-mobility group box 1 protein (HMGB1): Nuclear weapon in the immune arsenal. Nat. Rev. Immunol. 2005, 5, 331–342. [Google Scholar] [CrossRef]
- Guzman-Ruiz, R.; Ortega, F.; Rodriguez, A.; Vazquez-Martinez, R.; Diaz-Ruiz, A.; Garcia-Navarro, S.; Giralt, M.; Garcia-Rios, A.; Cobo-Padilla, D.; Tinahones, F.J.; et al. Alarmin high-mobility group B1 (HMGB1) is regulated in human adipocytes in insulin resistance and influences insulin secretion in beta-cells. Int. J. Obes. 2014, 38, 1545–1554. [Google Scholar] [CrossRef]
- Volz, H.C.; Seidel, C.; Laohachewin, D.; Kaya, Z.; Muller, O.J.; Pleger, S.T.; Lasitschka, F.; Bianchi, M.E.; Remppis, A.; Bierhaus, A.; et al. HMGB1: The missing link between diabetes mellitus and heart failure. Basic Res. Cardiol. 2010, 105, 805–820. [Google Scholar] [CrossRef] [PubMed]
- Sims, G.P.; Rowe, D.C.; Rietdijk, S.T.; Herbst, R.; Coyle, A.J. HMGB1 and RAGE in inflammation and cancer. Annu. Rev. Immunol. 2010, 28, 367–388. [Google Scholar] [CrossRef] [PubMed]
- Capuani, B.; Della-Morte, D.; Donadel, G.; Caratelli, S.; Bova, L.; Pastore, D.; De Canio, M.; D’Aguanno, S.; Coppola, A.; Pacifici, F.; et al. Liver protein profiles in insulin receptor-knockout mice reveal novel molecules involved in the diabetes pathophysiology. Am. J. Physiol. Endocrinol. Metab. 2015, 308, E744–E755. [Google Scholar] [CrossRef] [PubMed]
- Yu, T.K.; Caudell, E.G.; Smid, C.; Grimm, E.A. IL-2 activation of NK cells: Involvement of MKK1/2/ERK but not p38 kinase pathway. J. Immunol. 2000, 164, 6244–6251. [Google Scholar] [CrossRef] [Green Version]
- Liu, H.; Yao, Y.M.; Ding, L.H.; Zhang, H.; Yuan, B.; Song, Q.; Ye, Q.N.; Huang, C.F.; Sheng, Z.Y. High mobility group box-1 protein acts as a coactivator of nuclear factor of activated T cells-2 in promoting interleukin-2 transcription. Int. J. Biochem. Cell Biol. 2009, 41, 641–648. [Google Scholar] [CrossRef]
- Oncul, O.; Top, C.; Ozkan, S.; Cavuslu, S.; Danaci, M. Serum interleukin 2 levels in patients with rheumatoid arthritis and correlation with insulin sensitivity. J. Int Med. Res. 2002, 30, 386–390. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Zhang, L.; Zhang, S.; Yu, Q.; Xiong, F.; Huang, K.; Wang, C.Y.; Yang, P. HMGB1, an innate alarmin, plays a critical role in chronic inflammation of adipose tissue in obesity. Mol. Cell. Endocrinol. 2017, 454, 103–111. [Google Scholar] [CrossRef] [PubMed]
- Morton, G.J.; Cummings, D.E.; Baskin, D.G.; Barsh, G.S.; Schwartz, M.W. Central nervous system control of food intake and body weight. Nature 2006, 443, 289–295. [Google Scholar] [CrossRef] [PubMed]
- Lord, G.M.; Matarese, G.; Howard, J.K.; Baker, R.J.; Bloom, S.R.; Lechler, R.I. Leptin modulates the T-cell immune response and reverses starvation-induced immunosuppression. Nature 1998, 394, 897–901. [Google Scholar] [CrossRef]
- Samara, A.; Marie, B.; Pfister, M.; Visvikis-Siest, S. Leptin expression in Peripheral Blood Mononuclear Cells (PBMCs) is related with blood pressure variability. Clin. Chim. Acta 2008, 395, 47–50. [Google Scholar] [CrossRef]
- Zarkesh-Esfahani, H.; Pockley, G.; Metcalfe, R.A.; Bidlingmaier, M.; Wu, Z.; Ajami, A.; Weetman, A.P.; Strasburger, C.J.; Ross, R.J. High-dose leptin activates human leukocytes via receptor expression on monocytes. J. Immunol. 2001, 167, 4593–4599. [Google Scholar] [CrossRef] [Green Version]
- Larrea, E.; Aldabe, R.; Gonzalez, I.; Segura, V.; Sarobe, P.; Echeverria, I.; Prieto, J. Oncostatin M enhances the antiviral effects of type I interferon and activates immunostimulatory functions in liver epithelial cells. J. Virol. 2009, 83, 3298–3311. [Google Scholar] [CrossRef] [Green Version]
- DeMarco, R.A.; Fink, M.P.; Lotze, M.T. Monocytes promote natural killer cell interferon gamma production in response to the endogenous danger signal HMGB1. Mol. Immunol. 2005, 42, 433–444. [Google Scholar] [CrossRef]
- Mita, Y.; Dobashi, K.; Endou, K.; Kawata, T.; Shimizu, Y.; Nakazawa, T.; Mori, M. Toll-like receptor 4 surface expression on human monocytes and B cells is modulated by IL-2 and IL-4. Immunol. Lett. 2002, 81, 71–75. [Google Scholar] [CrossRef]
- Owens, B.M.; Moore, J.W.; Kaye, P.M. IRF7 regulates TLR2-mediated activation of splenic CD11c(hi) dendritic cells. PLoS ONE 2012, 7, e41050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guiducci, C.; Ghirelli, C.; Marloie-Provost, M.A.; Matray, T.; Coffman, R.L.; Liu, Y.J.; Barrat, F.J.; Soumelis, V. PI3K is critical for the nuclear translocation of IRF-7 and type I IFN production by human plasmacytoid predendritic cells in response to TLR activation. J. Exp. Med. 2008, 205, 315–322. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Jagannath, C.; Liu, X.D.; Sharafkhaneh, A.; Kolodziejska, K.E.; Eissa, N.T. Toll-like receptor 4 is a sensor for autophagy associated with innate immunity. Immunity 2007, 27, 135–144. [Google Scholar] [CrossRef] [Green Version]
- Degterev, A.; Hitomi, J.; Germscheid, M.; Ch’en, I.L.; Korkina, O.; Teng, X.; Abbott, D.; Cuny, G.D.; Yuan, C.; Wagner, G.; et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat. Chem. Biol. 2008, 4, 313–321. [Google Scholar] [CrossRef] [Green Version]
- Hadadi, E.; Zhang, B.; Baidzajevas, K.; Yusof, N.; Puan, K.J.; Ong, S.M.; Yeap, W.H.; Rotzschke, O.; Kiss-Toth, E.; Wilson, H.; et al. Differential IL-1beta secretion by monocyte subsets is regulated by Hsp27 through modulating mRNA stability. Sci. Rep. 2016, 6, 39035. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, R.; Xia, C.Q.; Butfiloski, E.; Clare-Salzler, M. Effect of high glucose on cytokine production by human peripheral blood immune cells and type I interferon signaling in monocytes: Implications for the role of hyperglycemia in the diabetes inflammatory process and host defense against infection. Clin. Immunol. 2018, 195, 139–148. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, Y.; Herrera, M.T.; Soldevila, G.; Garcia-Garcia, L.; Fabian, G.; Perez-Armendariz, E.M.; Bobadilla, K.; Guzman-Beltran, S.; Sada, E.; Torres, M. High glucose concentrations induce TNF-alpha production through the down-regulation of CD33 in primary human monocytes. BMC Immunol. 2012, 13, 19. [Google Scholar] [CrossRef] [Green Version]
- Thewissen, M.M.; van de Gaar, J.; den Boer, A.T.; Munsters, M.J.; Blaak, E.E.; Duijvestijn, A. Monocytes, but not T cells, respond to insulin with Akt(S473) phosphorylation independent of the donor glucometabolic state. Diabetes Metab. Res. Rev. 2014, 30, 323–332. [Google Scholar] [CrossRef]
- Gonzalez, E.; Flier, E.; Molle, D.; Accili, D.; McGraw, T.E. Hyperinsulinemia leads to uncoupled insulin regulation of the GLUT4 glucose transporter and the FoxO1 transcription factor. Proc. Natl. Acad. Sci. USA 2011, 108, 10162–10167. [Google Scholar] [CrossRef] [Green Version]
- Cameron, A.R.; Morrison, V.L.; Levin, D.; Mohan, M.; Forteath, C.; Beall, C.; McNeilly, A.D.; Balfour, D.J.; Savinko, T.; Wong, A.K.; et al. Anti-Inflammatory Effects of Metformin Irrespective of Diabetes Status. Circ. Res. 2016, 119, 652–665. [Google Scholar] [CrossRef] [Green Version]
- Dandona, P.; Chaudhuri, A.; Mohanty, P.; Ghanim, H. Anti-inflammatory effects of insulin. Curr. Opin. Clin. Nutr. Metab. Care 2007, 10, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Katsiki, N.; Mikhailidis, D.P.; Banach, M. Leptin, cardiovascular diseases and type 2 diabetes mellitus. Acta Pharm. Sin. 2018, 39, 1176–1188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cohen, D.H.; LeRoith, D. Obesity, type 2 diabetes, and cancer: The insulin and IGF connection. Endocr. Relat. Cancer 2012, 19, F27–F45. [Google Scholar] [CrossRef]
- Lago, F.; Gomez, R.; Gomez-Reino, J.J.; Dieguez, C.; Gualillo, O. Adipokines as novel modulators of lipid metabolism. Trends Biochem. Sci. 2009, 34, 500–510. [Google Scholar] [CrossRef]
- Iikuni, N.; Lam, Q.L.; Lu, L.; Matarese, G.; La Cava, A. Leptin and Inflammation. Curr. Immunol. Rev. 2008, 4, 70–79. [Google Scholar] [CrossRef]
- Ray, A. Adipokine leptin in obesity-related pathology of breast cancer. J. Biosci. 2012, 37, 289–294. [Google Scholar] [CrossRef] [PubMed]
- Formica, V.; Luccchetti, J.; Cunningham, D.; Smyth, E.C.; Ferroni, P.; Nardecchia, A.; Tesauro, M.; Cereda, V.; Guadagni, F.; Roselli, M. Systemic inflammation, as measured by the neutrophil/lymphocyte ratio, may have differential prognostic impact before and during treatment with fluorouracil, irinotecan and bevacizumab in metastatic colorectal cancer patients. Med. Oncol. 2014, 31, 166. [Google Scholar] [CrossRef]
- Trayhurn, P.; Wood, I.S. Adipokines: Inflammation and the pleiotropic role of white adipose tissue. Br. J. Nutr. 2004, 92, 347–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanna, V.; Di Giacomo, A.; La Cava, A.; Lechler, R.I.; Fontana, S.; Zappacosta, S.; Matarese, G. Leptin surge precedes onset of autoimmune encephalomyelitis and correlates with development of pathogenic T cell responses. J. Clin. Investig. 2003, 111, 241–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Qiao, F.; Zhao, Y.; Wang, Y.; Liu, G. HMGB1 is activated in type 2 diabetes mellitus patients and in mesangial cells in response to high glucose. Int. J. Clin. Exp. Pathol. 2015, 8, 6683–6691. [Google Scholar] [PubMed]
- Coppola, A.; Arriga, R.; Lauro, D.; Del Principe, M.I.; Buccisano, F.; Maurillo, L.; Palomba, P.; Venditti, A.; Sconocchia, G. NK Cell Inflammation in the Clinical Outcome of Colorectal Carcinoma. Front. Med. 2015, 2, 33. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levitz, S.M. Activation of human peripheral blood mononuclear cells by interleukin-2 and granulocyte-macrophage colony-stimulating factor to inhibit Cryptococcus neoformans. Infect. Immun. 1991, 59, 3393–3397. [Google Scholar] [CrossRef] [Green Version]
- Bianchi, M.E. HMGB1 loves company. J. Leukoc. Biol. 2009, 86, 573–576. [Google Scholar] [CrossRef]
- Pereira, F.O.; Frode, T.S.; Medeiros, Y.S. Evaluation of tumour necrosis factor alpha, interleukin-2 soluble receptor, nitric oxide metabolites, and lipids as inflammatory markers in type 2 diabetes mellitus. Mediat. Inflamm. 2006, 2006, 39062. [Google Scholar] [CrossRef] [Green Version]
- Yang, Z.Z.; Grote, D.M.; Ziesmer, S.C.; Manske, M.K.; Witzig, T.E.; Novak, A.J.; Ansell, S.M. Soluble IL-2Ralpha facilitates IL-2-mediated immune responses and predicts reduced survival in follicular B-cell non-Hodgkin lymphoma. Blood 2011, 118, 2809–2820. [Google Scholar] [CrossRef]
- Ren, P.C.; Zhang, Y.; Zhang, X.D.; An, L.J.; Lv, H.G.; He, J.; Gao, C.J.; Sun, X.D. High-mobility group box 1 contributes to mechanical allodynia and spinal astrocytic activation in a mouse model of type 2 diabetes. Brain Res. Bull. 2012, 88, 332–337. [Google Scholar] [CrossRef]
- Sanchez-Infantes, D.; White, U.A.; Elks, C.M.; Morrison, R.F.; Gimble, J.M.; Considine, R.V.; Ferrante, A.W.; Ravussin, E.; Stephens, J.M. Oncostatin m is produced in adipose tissue and is regulated in conditions of obesity and type 2 diabetes. J. Clin. Endocrinol. Metab. 2014, 99, E217–E225. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGillicuddy, F.C.; Chiquoine, E.H.; Hinkle, C.C.; Kim, R.J.; Shah, R.; Roche, H.M.; Smyth, E.M.; Reilly, M.P. Interferon gamma attenuates insulin signaling, lipid storage, and differentiation in human adipocytes via activation of the JAK/STAT pathway. J. Biol. Chem. 2009, 284, 31936–31944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linsen, L.; Somers, V.; Stinissen, P. Immunoregulation of autoimmunity by natural killer T cells. Hum. Immunol. 2005, 66, 1193–1202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huebner, L.; Engeli, S.; Wrann, C.D.; Goudeva, L.; Laue, T.; Kielstein, H. Human NK cell subset functions are differentially affected by adipokines. PLoS ONE 2013, 8, e75703. [Google Scholar] [CrossRef] [Green Version]
- Piatkiewicz, P.; Milek, T.; Bernat-Karpinska, M.; Ohams, M.; Czech, A.; Ciostek, P. The dysfunction of NK cells in patients with type 2 diabetes and colon cancer. Arch. Immunol Ther. Exp. 2013, 61, 245–253. [Google Scholar] [CrossRef] [PubMed]
- Gajovic, N.; Jurisevic, M.; Pantic, J.; Radosavljevic, G.; Arsenijevic, N.; Lukic, M.L.; Jovanovic, I. Attenuation of NK cells facilitates mammary tumor growth in streptozotocin-induced diabetes in mice. Endocr. Relat. Cancer 2018, 25, 493–507. [Google Scholar] [CrossRef]
- Hornung, V.; Rothenfusser, S.; Britsch, S.; Krug, A.; Jahrsdorfer, B.; Giese, T.; Endres, S.; Hartmann, G. Quantitative expression of toll-like receptor 1-10 mRNA in cellular subsets of human peripheral blood mononuclear cells and sensitivity to CpG oligodeoxynucleotides. J. Immunol. 2002, 168, 4531–4537. [Google Scholar] [CrossRef] [Green Version]
- Gaidt, M.M.; Ebert, T.S.; Chauhan, D.; Schmidt, T.; Schmid-Burgk, J.L.; Rapino, F.; Robertson, A.A.; Cooper, M.A.; Graf, T.; Hornung, V. Human Monocytes Engage an Alternative Inflammasome Pathway. Immunity 2016, 44, 833–846. [Google Scholar] [CrossRef] [Green Version]
- Zhao, G.; Dharmadhikari, G.; Maedler, K.; Meyer-Hermann, M. Possible role of interleukin-1beta in type 2 diabetes onset and implications for anti-inflammatory therapy strategies. PLoS Comput. Biol. 2014, 10, e1003798. [Google Scholar] [CrossRef]
- Tsubai, T.; Noda, Y.; Ito, K.; Nakao, M.; Seino, Y.; Oiso, Y.; Hamada, Y. Insulin elevates leptin secretion and mRNA levels via cyclic AMP in 3T3-L1 adipocytes deprived of glucose. Heliyon 2016, 2, e00194. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, C.G.; Bryson, J.M.; Hancock, D.P.; Caterson, I.D. Leptin secretion is related to glucose-derived lipogenesis in isolated adipocytes. Int. J. Obes. 2007, 31, 723–729. [Google Scholar] [CrossRef] [Green Version]
- Miller, R.A.; Chu, Q.; Xie, J.; Foretz, M.; Viollet, B.; Birnbaum, M.J. Biguanides suppress hepatic glucagon signalling by decreasing production of cyclic AMP. Nature 2013, 494, 256–260. [Google Scholar] [CrossRef] [Green Version]
- Phillips, J.H.; Warner, N.L.; Lanier, L.L. Correlation of biophysical properties and cell surface antigenic profile of Percoll gradient-separated human natural killer cells. Nat. Immun Cell Growth Regul. 1983, 3, 73–86. [Google Scholar] [PubMed]
- Saidi, H.; Bras, M.; Formaglio, P.; Melki, M.T.; Charbit, B.; Herbeuval, J.P.; Gougeon, M.L. HMGB1 Is Involved in IFN-alpha Production and TRAIL Expression by HIV-1-Exposed Plasmacytoid Dendritic Cells: Impact of the Crosstalk with NK Cells. PLoS Pathog. 2016, 12, e1005407. [Google Scholar] [CrossRef] [PubMed]
- Matsunaga, N.; Tsuchimori, N.; Matsumoto, T.; Ii, M. TAK-242 (resatorvid), a small-molecule inhibitor of Toll-like receptor (TLR) 4 signaling, binds selectively to TLR4 and interferes with interactions between TLR4 and its adaptor molecules. Mol. Pharm. 2011, 79, 34–41. [Google Scholar] [CrossRef] [Green Version]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Coppola, A.; Capuani, B.; Pacifici, F.; Pastore, D.; Arriga, R.; Bellia, A.; Andreadi, A.; Di Daniele, N.; Lauro, R.; Della-Morte, D.; et al. Activation of Peripheral Blood Mononuclear Cells and Leptin Secretion: New Potential Role of Interleukin-2 and High Mobility Group Box (HMGB)1. Int. J. Mol. Sci. 2021, 22, 7988. https://doi.org/10.3390/ijms22157988
Coppola A, Capuani B, Pacifici F, Pastore D, Arriga R, Bellia A, Andreadi A, Di Daniele N, Lauro R, Della-Morte D, et al. Activation of Peripheral Blood Mononuclear Cells and Leptin Secretion: New Potential Role of Interleukin-2 and High Mobility Group Box (HMGB)1. International Journal of Molecular Sciences. 2021; 22(15):7988. https://doi.org/10.3390/ijms22157988
Chicago/Turabian StyleCoppola, Andrea, Barbara Capuani, Francesca Pacifici, Donatella Pastore, Roberto Arriga, Alfonso Bellia, Aikaterini Andreadi, Nicola Di Daniele, Renato Lauro, David Della-Morte, and et al. 2021. "Activation of Peripheral Blood Mononuclear Cells and Leptin Secretion: New Potential Role of Interleukin-2 and High Mobility Group Box (HMGB)1" International Journal of Molecular Sciences 22, no. 15: 7988. https://doi.org/10.3390/ijms22157988
APA StyleCoppola, A., Capuani, B., Pacifici, F., Pastore, D., Arriga, R., Bellia, A., Andreadi, A., Di Daniele, N., Lauro, R., Della-Morte, D., Sconocchia, G., & Lauro, D. (2021). Activation of Peripheral Blood Mononuclear Cells and Leptin Secretion: New Potential Role of Interleukin-2 and High Mobility Group Box (HMGB)1. International Journal of Molecular Sciences, 22(15), 7988. https://doi.org/10.3390/ijms22157988