
Effect of Microbiome on Non-Alcoholic Fatty Liver Disease and the Role of Probiotics, Prebiotics, and Biogenics


{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Relationship between Hepatic Immune Cells and the Pathogenesis of NAFLD and NASH
2.1. Macrophages and Kupffer Cellsn
2.2. Chemokines and Monocytes
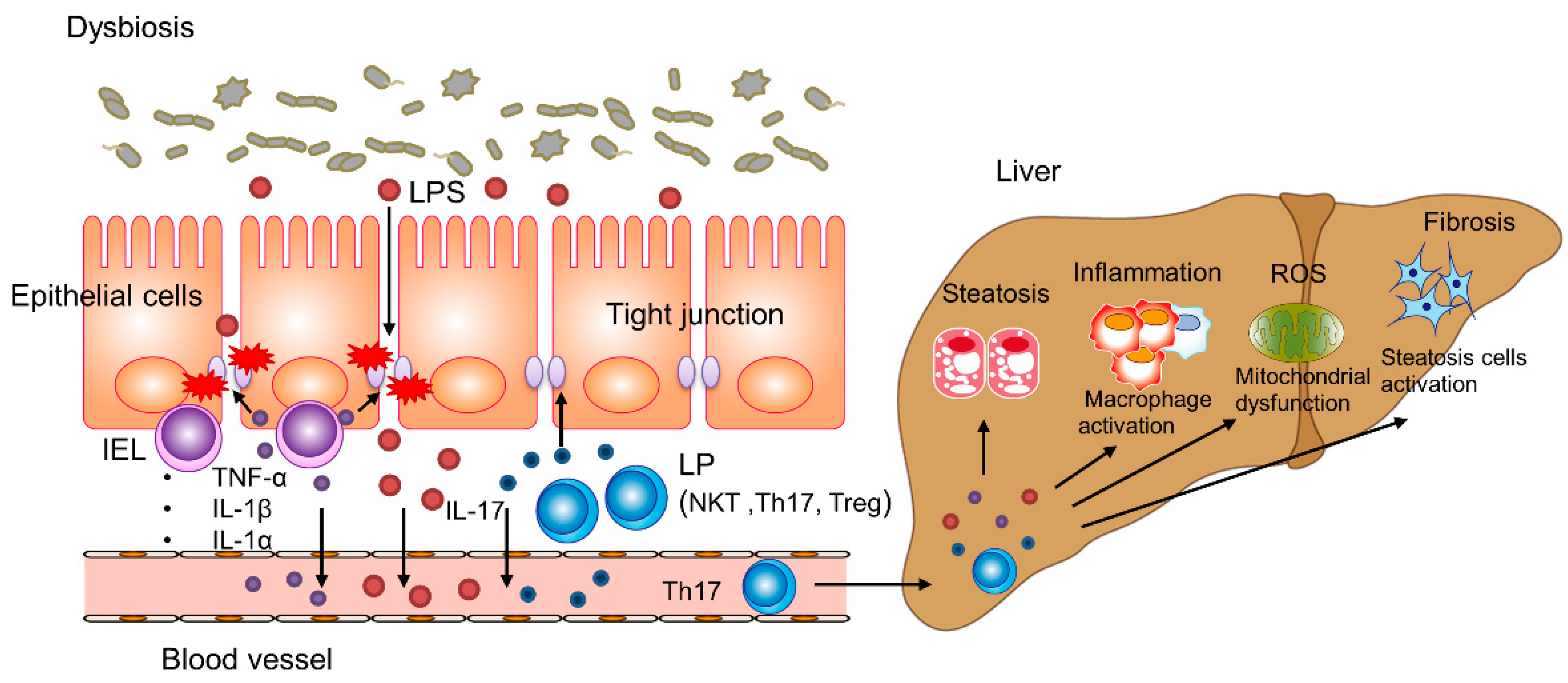
3. The Gut–Liver Axis in NAFLD and NASH
3.1. Intestinal Permeability and the Microbiota
3.2. Intestinal Immune System
4. Treatment of NAFLD and NASH with Probiotics, Prebiotics, and Biogenics
4.1. Lactic Fermentation and Control of Gut Microbiome
4.2. Therapeutic Effects of Probiotics, Prebiotics, and Biogenics on NAFLD and NASH
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Tilg, H.; Moschen, A.R.; Roden, M. NAFLD and diabetes mellitus. Nat. Rev. Gastroenterol. Hepatol. 2017, 14, 32–42. [Google Scholar] [CrossRef]
- Italian Association for the Study of the Liver (AISF). AISF position paper on nonalcoholic fatty liver disease (NAFLD): Updates and future directions. Dig. Liver Dis. 2017, 49, 471–483. [Google Scholar] [CrossRef]
- Sheka, A.C.; Adeyi, O.; Thompson, J.; Hameed, B.; Crawford, P.A.; Ikramuddin, S. Nonalcoholic Steatohepatitis: A Review. JAMA 2020, 323, 1175–1183. [Google Scholar] [CrossRef]
- Benedict, M.; Zhang, X. Non-alcoholic fatty liver disease: An expanded review. World J. Hepatol. 2017, 9, 715–732. [Google Scholar] [CrossRef]
- Younossi, Z.M.; Loomba, R.; Anstee, Q.M.; Rinella, M.E.; Bugianesi, E.; Marchesini, G.; Neuschwander-Tetri, B.A.; Serfaty, L.; Negro, F.; Caldwell, S.H.; et al. Diagnostic modalities for nonalcoholic fatty liver disease, nonalcoholic steatohepatitis, and associated fibrosis. Hepatology 2018, 68, 349–360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marchesini, G.; Brizi, M.; Bianchi, G.; Tomassetti, S.; Bugianesi, E.; Lenzi, M.; McCullough, A.J.; Natale, S.; Forlani, G.; Melchionda, N. Nonalcoholic fatty liver disease: A feature of the metabolic syndrome. Diabetes 2001, 50, 1844–1850. [Google Scholar] [CrossRef] [Green Version]
- Hebbard, L.; George, J. Animal models of nonalcoholic fatty liver disease. Nat. Rev. Gastroenterol. Hepatol. 2011, 8, 35–44. [Google Scholar] [CrossRef]
- Matsumoto, M.; Hada, N.; Sakamaki, Y.; Uno, A.; Shiga, T.; Tanaka, C.; Ito, T.; Katsume, A.; Sudoh, M. An improved mouse model that rapidly develops fibrosis in non-alcoholic steatohepatitis. Int. J. Exp. Pathol. 2013, 94, 93–103. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wada, T.; Kenmochi, H.; Miyashita, Y.; Sasaki, M.; Ojima, M.; Sasahara, M.; Koya, D.; Tsuneki, H.; Sasaoka, T. Spironolactone improves glucose and lipid metabolism by ameliorating hepatic steatosis and inflammation and suppressing enhanced gluconeogenesis induced by high-fat and high-fructose diet. Endocrinology 2010, 151, 2040–2049. [Google Scholar] [CrossRef] [Green Version]
- Matsuzawa, N.; Takamura, T.; Kurita, S.; Misu, H.; Ota, T.; Ando, H.; Yokoyama, M.; Honda, M.; Zen, Y.; Nakanuma, Y.; et al. Lipid-induced oxidative stress causes steatohepatitis in mice fed an atherogenic diet. Hepatology 2007, 46, 1392–1403. [Google Scholar] [CrossRef] [PubMed]
- Kwok, R.; Choi, K.C.; Wong, G.L.; Zhang, Y.; Chan, H.L.; Luk, A.O.; Shu, S.S.; Chan, A.W.; Yeung, M.W.; Chan, J.C.; et al. Screening diabetic patients for non-alcoholic fatty liver disease with controlled attenuation parameter and liver stiffness measurements: A prospective cohort study. Gut 2016, 65, 1359–1368. [Google Scholar] [CrossRef] [PubMed]
- Younossi, Z.M.; Koenig, A.B.; Abdelatif, D.; Fazel, Y.; Henry, L.; Wymer, M. Global epidemiology of nonalcoholic fatty liver disease-Meta-analytic assessment of prevalence, incidence, and outcomes. Hepatology 2016, 64, 73–84. [Google Scholar] [CrossRef] [Green Version]
- Williams, C.D.; Stengel, J.; Asike, M.I.; Torres, D.M.; Shaw, J.; Contreras, M.; Landt, C.L.; Harrison, S.A. Prevalence of nonalcoholic fatty liver disease and nonalcoholic steatohepatitis among a largely middle-aged population utilizing ultrasound and liver biopsy: A prospective study. Gastroenterology 2011, 140, 124–131. [Google Scholar] [CrossRef]
- Loomba, R.; Abraham, M.; Unalp, A.; Wilson, L.; Lavine, J.; Doo, E.; Bass, N.M. Association between diabetes, family history of diabetes, and risk of nonalcoholic steatohepatitis and fibrosis. Hepatology 2012, 56, 943–951. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lonardo, A.; Nascimbeni, F.; Mantovani, A.; Targher, G. Hypertension, diabetes, atherosclerosis and NASH: Cause or consequence? J. Hepatol. 2018, 68, 335–352. [Google Scholar] [CrossRef]
- Ota, T.; Takamura, T.; Kurita, S.; Matsuzawa, N.; Kita, Y.; Uno, M.; Akahori, H.; Misu, H.; Sakurai, M.; Zen, Y.; et al. Insulin resistance accelerates a dietary rat model of nonalcoholic steatohepatitis. Gastroenterology 2007, 132, 282–293. [Google Scholar] [CrossRef]
- Takaki, A.; Kawai, D.; Yamamoto, K. Multiple hits, including oxidative stress, as pathogenesis and treatment target in non-alcoholic steatohepatitis (NASH). Int. J. Mol. Sci. 2013, 14, 20704–20728. [Google Scholar] [CrossRef] [Green Version]
- Konig, J.; Wells, J.; Cani, P.D.; Garcia-Rodenas, C.L.; MacDonald, T.; Mercenier, A.; Whyte, J.; Troost, F.; Brummer, R.J. Human Intestinal Barrier Function in Health and Disease. Clin. Transl. Gastroenterol. 2016, 7, e196. [Google Scholar] [CrossRef]
- Parthasarathy, G.; Revelo, X.; Malhi, H. Pathogenesis of Nonalcoholic Steatohepatitis: An Overview. Hepatol. Commun. 2020, 4, 478–492. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.; Mehal, W.; Nagy, L.E.; Rotman, Y. Immunological mechanisms and therapeutic targets of fatty liver diseases. Cell. Mol. Immunol. 2021, 18, 73–91. [Google Scholar] [CrossRef]
- Ortiz, M.; Soto-Alarcon, S.A.; Orellana, P.; Espinosa, A.; Campos, C.; Lopez-Arana, S.; Rincon, M.A.; Illesca, P.; Valenzuela, R.; Videla, L.A. Suppression of high-fat diet-induced obesity-associated liver mitochondrial dysfunction by docosahexaenoic acid and hydroxytyrosol co-administration. Dig. Liver Dis. 2020, 52, 895–904. [Google Scholar] [CrossRef]
- Sica, A.; Mantovani, A. Macrophage plasticity and polarization: In vivo veritas. J. Clin. Investig. 2012, 122, 787–795. [Google Scholar] [CrossRef] [PubMed]
- Wan, J.; Benkdane, M.; Teixeira-Clerc, F.; Bonnafous, S.; Louvet, A.; Lafdil, F.; Pecker, F.; Tran, A.; Gual, P.; Mallat, A.; et al. M2 Kupffer cells promote M1 Kupffer cell apoptosis: A protective mechanism against alcoholic and nonalcoholic fatty liver disease. Hepatology 2014, 59, 130–142. [Google Scholar] [CrossRef] [PubMed]
- Xue, J.; Sharma, V.; Hsieh, M.H.; Chawla, A.; Murali, R.; Pandol, S.J.; Habtezion, A. Alternatively activated macrophages promote pancreatic fibrosis in chronic pancreatitis. Nat. Commun. 2015, 6, 7158. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valenzuela, R.; Illesca, P.; Echeverria, F.; Espinosa, A.; Rincon-Cervera, M.A.; Ortiz, M.; Hernandez-Rodas, M.C.; Valenzuela, A.; Videla, L.A. Molecular adaptations underlying the beneficial effects of hydroxytyrosol in the pathogenic alterations induced by a high-fat diet in mouse liver: PPAR-alpha and Nrf2 activation, and NF-kappaB down-regulation. Food Funct. 2017, 8, 1526–1537. [Google Scholar] [CrossRef]
- Paradies, G.; Paradies, V.; Ruggiero, F.M.; Petrosillo, G. Oxidative stress, cardiolipin and mitochondrial dysfunction in nonalcoholic fatty liver disease. World J. Gastroenterol. WJG 2014, 20, 14205–14218. [Google Scholar] [CrossRef]
- Suzuki, T.; Hara, H. Dietary fat and bile juice, but not obesity, are responsible for the increase in small intestinal permeability induced through the suppression of tight junction protein expression in LETO and OLETF rats. Nutr. Metab. 2010, 7, 19. [Google Scholar] [CrossRef] [Green Version]
- Meng, J.; Banerjee, S.; Li, D.; Sindberg, G.M.; Wang, F.; Ma, J.; Roy, S. Opioid Exacerbation of Gram-positive sepsis, induced by Gut Microbial Modulation, is Rescued by IL-17A Neutralization. Sci. Rep. 2015, 5, 10918. [Google Scholar] [CrossRef] [Green Version]
- Zhou, D.; Pan, Q.; Liu, X.L.; Yang, R.X.; Chen, Y.W.; Liu, C.; Fan, J.G. Clostridium butyricum B1 alleviates high-fat diet-induced steatohepatitis in mice via enterohepatic immunoregulation. J. Gastroenterol. Hepatol. 2017, 32, 1640–1648. [Google Scholar] [CrossRef]
- Mouries, J.; Brescia, P.; Silvestri, A.; Spadoni, I.; Sorribas, M.; Wiest, R.; Mileti, E.; Galbiati, M.; Invernizzi, P.; Adorini, L.; et al. Microbiota-driven gut vascular barrier disruption is a prerequisite for non-alcoholic steatohepatitis development. J. Hepatol. 2019, 71, 1216–1228. [Google Scholar] [CrossRef] [Green Version]
- Sutti, S.; Albano, E. Adaptive immunity: An emerging player in the progression of NAFLD. Nat. Rev. Gastroenterol. Hepatol. 2020, 17, 81–92. [Google Scholar] [CrossRef]
- Echeverria, F.; Valenzuela, R.; Espinosa, A.; Bustamante, A.; Alvarez, D.; Gonzalez-Manan, D.; Ortiz, M.; Soto-Alarcon, S.A.; Videla, L.A. Reduction of high-fat diet-induced liver proinflammatory state by eicosapentaenoic acid plus hydroxytyrosol supplementation: Involvement of resolvins RvE1/2 and RvD1/2. J. Nutr. Biochem. 2019, 63, 35–43. [Google Scholar] [CrossRef]
- Korbecki, J.; Bajdak-Rusinek, K. The effect of palmitic acid on inflammatory response in macrophages: An overview of molecular mechanisms. Inflamm. Res. Off. J. Eur. Histamine Res. Soc. 2019, 68, 915–932. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kazankov, K.; Jorgensen, S.M.D.; Thomsen, K.L.; Moller, H.J.; Vilstrup, H.; George, J.; Schuppan, D.; Gronbaek, H. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat. Rev. Gastroenterol. Hepatol. 2019, 16, 145–159. [Google Scholar] [CrossRef]
- Gomez Perdiguero, E.; Klapproth, K.; Schulz, C.; Busch, K.; Azzoni, E.; Crozet, L.; Garner, H.; Trouillet, C.; de Bruijn, M.F.; Geissmann, F.; et al. Tissue-resident macrophages originate from yolk-sac-derived erythro-myeloid progenitors. Nature 2015, 518, 547–551. [Google Scholar] [CrossRef]
- Krenkel, O.; Tacke, F. Liver macrophages in tissue homeostasis and disease. Nat. Rev. Immunol. 2017, 17, 306–321. [Google Scholar] [CrossRef]
- Mass, E.; Ballesteros, I.; Farlik, M.; Halbritter, F.; Gunther, P.; Crozet, L.; Jacome-Galarza, C.E.; Handler, K.; Klughammer, J.; Kobayashi, Y.; et al. Specification of tissue-resident macrophages during organogenesis. Science 2016, 353, aaf4238. [Google Scholar] [CrossRef] [Green Version]
- Scott, C.L.; Zheng, F.; De Baetselier, P.; Martens, L.; Saeys, Y.; De Prijck, S.; Lippens, S.; Abels, C.; Schoonooghe, S.; Raes, G.; et al. Bone marrow-derived monocytes give rise to self-renewing and fully differentiated Kupffer cells. Nat. Commun. 2016, 7, 10321. [Google Scholar] [CrossRef]
- Ni, Y.; Zhuge, F.; Nagashimada, M.; Ota, T. Novel Action of Carotenoids on Non-Alcoholic Fatty Liver Disease: Macrophage Polarization and Liver Homeostasis. Nutrients 2016, 8, 391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heymann, F.; Tacke, F. Immunology in the liver--from homeostasis to disease. Nat. Rev. Gastroenterol. Hepatol. 2016, 13, 88–110. [Google Scholar] [CrossRef] [PubMed]
- Ramachandran, P.; Pellicoro, A.; Vernon, M.A.; Boulter, L.; Aucott, R.L.; Ali, A.; Hartland, S.N.; Snowdon, V.K.; Cappon, A.; Gordon-Walker, T.T.; et al. Differential Ly-6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc. Natl. Acad. Sci. USA 2012, 109, E3186–E3195. [Google Scholar] [CrossRef] [Green Version]
- Jager, J.; Aparicio-Vergara, M.; Aouadi, M. Liver innate immune cells and insulin resistance: The multiple facets of Kupffer cells. J. Intern. Med. 2016, 280, 209–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Odegaard, J.I.; Ricardo-Gonzalez, R.R.; Red Eagle, A.; Vats, D.; Morel, C.R.; Goforth, M.H.; Subramanian, V.; Mukundan, L.; Ferrante, A.W.; Chawla, A. Alternative M2 activation of Kupffer cells by PPARdelta ameliorates obesity-induced insulin resistance. Cell Metab. 2008, 7, 496–507. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, S.; Martinez, F.O. Alternative activation of macrophages: Mechanism and functions. Immunity 2010, 32, 593–604. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murray, P.J.; Allen, J.E.; Biswas, S.K.; Fisher, E.A.; Gilroy, D.W.; Goerdt, S.; Gordon, S.; Hamilton, J.A.; Ivashkiv, L.B.; Lawrence, T.; et al. Macrophage activation and polarization: Nomenclature and experimental guidelines. Immunity 2014, 41, 14–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinez, F.O.; Gordon, S. The M1 and M2 paradigm of macrophage activation: Time for reassessment. F1000prime Rep. 2014, 6, 13. [Google Scholar] [CrossRef] [Green Version]
- Sica, A.; Invernizzi, P.; Mantovani, A. Macrophage plasticity and polarization in liver homeostasis and pathology. Hepatology 2014, 59, 2034–2042. [Google Scholar] [CrossRef] [PubMed]
- Kanda, H.; Tateya, S.; Tamori, Y.; Kotani, K.; Hiasa, K.; Kitazawa, R.; Kitazawa, S.; Miyachi, H.; Maeda, S.; Egashira, K.; et al. MCP-1 contributes to macrophage infiltration into adipose tissue, insulin resistance, and hepatic steatosis in obesity. J. Clin. Investig. 2006, 116, 1494–1505. [Google Scholar] [CrossRef]
- Obstfeld, A.E.; Sugaru, E.; Thearle, M.; Francisco, A.M.; Gayet, C.; Ginsberg, H.N.; Ables, E.V.; Ferrante, A.W., Jr. C-C chemokine receptor 2 (CCR2) regulates the hepatic recruitment of myeloid cells that promote obesity-induced hepatic steatosis. Diabetes 2010, 59, 916–925. [Google Scholar] [CrossRef] [Green Version]
- Inouye, K.E.; Shi, H.; Howard, J.K.; Daly, C.H.; Lord, G.M.; Rollins, B.J.; Flier, J.S. Absence of CC chemokine ligand 2 does not limit obesity-associated infiltration of macrophages into adipose tissue. Diabetes 2007, 56, 2242–2250. [Google Scholar] [CrossRef] [Green Version]
- Kirk, E.A.; Sagawa, Z.K.; McDonald, T.O.; O’Brien, K.D.; Heinecke, J.W. Monocyte chemoattractant protein deficiency fails to restrain macrophage infiltration into adipose tissue [corrected]. Diabetes 2008, 57, 1254–1261. [Google Scholar] [CrossRef] [Green Version]
- Kitade, H.; Sawamoto, K.; Nagashimada, M.; Inoue, H.; Yamamoto, Y.; Sai, Y.; Takamura, T.; Yamamoto, H.; Miyamoto, K.; Ginsberg, H.N.; et al. CCR5 plays a critical role in obesity-induced adipose tissue inflammation and insulin resistance by regulating both macrophage recruitment and M1/M2 status. Diabetes 2012, 61, 1680–1690. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.; Zhang, L.; Yu, C.; Yang, X.F.; Wang, H. Monocyte and macrophage differentiation: Circulation inflammatory monocyte as biomarker for inflammatory diseases. Biomark. Res. 2014, 2, 1. [Google Scholar] [CrossRef] [Green Version]
- Lumeng, C.N.; DelProposto, J.B.; Westcott, D.J.; Saltiel, A.R. Phenotypic switching of adipose tissue macrophages with obesity is generated by spatiotemporal differences in macrophage subtypes. Diabetes 2008, 57, 3239–3246. [Google Scholar] [CrossRef] [Green Version]
- Nagashimada, M.; Sawamoto, K.; Ni, Y.; Kitade, H.; Nagata, N.; Xu, L.; Kobori, M.; Mukaida, N.; Yamashita, T.; Kaneko, S.; et al. CX3CL1-CX3CR1 signalling deficiency exacerbates obesity-induced inflammation and insulin resistance in male mice. Endocrinology 2021, 162, bqab064. [Google Scholar] [CrossRef] [PubMed]
- Morris, D.L.; Oatmen, K.E.; Wang, T.; DelProposto, J.L.; Lumeng, C.N. CX3CR1 deficiency does not influence trafficking of adipose tissue macrophages in mice with diet-induced obesity. Obesity 2012, 20, 1189–1199. [Google Scholar] [CrossRef] [Green Version]
- Lee, M.; Lee, Y.; Song, J.; Lee, J.; Chang, S.Y. Tissue-specific Role of CX3CR1 Expressing Immune Cells and Their Relationships with Human Disease. Immune Netw. 2018, 18, e5. [Google Scholar] [CrossRef]
- Ni, Y.; Ni, L.; Zhuge, F.; Xu, L.; Fu, Z.; Ota, T. Adipose Tissue Macrophage Phenotypes and Characteristics: The Key to Insulin Resistance in Obesity and Metabolic Disorders. Obesity 2020, 28, 225–234. [Google Scholar] [CrossRef]
- Karlmark, K.R.; Zimmermann, H.W.; Roderburg, C.; Gassler, N.; Wasmuth, H.E.; Luedde, T.; Trautwein, C.; Tacke, F. The fractalkine receptor CX3CR1 protects against liver fibrosis by controlling differentiation and survival of infiltrating hepatic monocytes. Hepatology 2010, 52, 1769–1782. [Google Scholar] [CrossRef]
- Schneider, K.M.; Bieghs, V.; Heymann, F.; Hu, W.; Dreymueller, D.; Liao, L.; Frissen, M.; Ludwig, A.; Gassler, N.; Pabst, O.; et al. CX3CR1 is a gatekeeper for intestinal barrier integrity in mice: Limiting steatohepatitis by maintaining intestinal homeostasis. Hepatology 2015, 62, 1405–1416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tappy, L.; Le, K.A.; Tran, C.; Paquot, N. Fructose and metabolic diseases: New findings, new questions. Nutrition 2010, 26, 1044–1049. [Google Scholar] [CrossRef]
- Abid, A.; Taha, O.; Nseir, W.; Farah, R.; Grosovski, M.; Assy, N. Soft drink consumption is associated with fatty liver disease independent of metabolic syndrome. J. Hepatol. 2009, 51, 918–924. [Google Scholar] [CrossRef]
- Hernandez-Rodas, M.C.; Valenzuela, R.; Videla, L.A. Relevant Aspects of Nutritional and Dietary Interventions in Non-Alcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2015, 16, 25168–25198. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kirpich, I.A.; Marsano, L.S.; McClain, C.J. Gut-liver axis, nutrition, and non-alcoholic fatty liver disease. Clin. Biochem. 2015, 48, 923–930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Miele, L.; Valenza, V.; La Torre, G.; Montalto, M.; Cammarota, G.; Ricci, R.; Masciana, R.; Forgione, A.; Gabrieli, M.L.; Perotti, G.; et al. Increased intestinal permeability and tight junction alterations in nonalcoholic fatty liver disease. Hepatology 2009, 49, 1877–1887. [Google Scholar] [CrossRef] [PubMed]
- Albillos, A.; de Gottardi, A.; Rescigno, M. The gut-liver axis in liver disease: Pathophysiological basis for therapy. J. Hepatol. 2020, 72, 558–577. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tilg, H.; Moschen, A.R.; Szabo, G. Interleukin-1 and inflammasomes in alcoholic liver disease/acute alcoholic hepatitis and nonalcoholic fatty liver disease/nonalcoholic steatohepatitis. Hepatology 2016, 64, 955–965. [Google Scholar] [CrossRef]
- Gonzalez-Mariscal, L.; Betanzos, A.; Nava, P.; Jaramillo, B.E. Tight junction proteins. Prog. Biophys. Mol. Biol. 2003, 81, 1–44. [Google Scholar] [CrossRef]
- Wieland, A.; Frank, D.N.; Harnke, B.; Bambha, K. Systematic review: Microbial dysbiosis and nonalcoholic fatty liver disease. Aliment. Pharmacol. Ther. 2015, 42, 1051–1063. [Google Scholar] [CrossRef]
- Loomba, R.; Seguritan, V.; Li, W.; Long, T.; Klitgord, N.; Bhatt, A.; Dulai, P.S.; Caussy, C.; Bettencourt, R.; Highlander, S.K.; et al. Gut Microbiome-Based Metagenomic Signature for Non-invasive Detection of Advanced Fibrosis in Human Nonalcoholic Fatty Liver Disease. Cell Metab. 2017, 25, 1054–1062.e5. [Google Scholar] [CrossRef] [PubMed]
- Zhu, L.; Baker, S.S.; Gill, C.; Liu, W.; Alkhouri, R.; Baker, R.D.; Gill, S.R. Characterization of gut microbiomes in nonalcoholic steatohepatitis (NASH) patients: A connection between endogenous alcohol and NASH. Hepatology 2013, 57, 601–609. [Google Scholar] [CrossRef] [PubMed]
- Boursier, J.; Mueller, O.; Barret, M.; Machado, M.; Fizanne, L.; Araujo-Perez, F.; Guy, C.D.; Seed, P.C.; Rawls, J.F.; David, L.A.; et al. The severity of nonalcoholic fatty liver disease is associated with gut dysbiosis and shift in the metabolic function of the gut microbiota. Hepatology 2016, 63, 764–775. [Google Scholar] [CrossRef] [Green Version]
- Gophna, U. Microbiology. The guts of dietary habits. Science 2011, 334, 45–46. [Google Scholar] [CrossRef]
- Olivares-Villagomez, D.; Van Kaer, L. Intestinal Intraepithelial Lymphocytes: Sentinels of the Mucosal Barrier. Trends Immunol. 2018, 39, 264–275. [Google Scholar] [CrossRef]
- McDonald, B.D.; Jabri, B.; Bendelac, A. Diverse developmental pathways of intestinal intraepithelial lymphocytes. Nat. Rev. Immunol. 2018, 18, 514–525. [Google Scholar] [CrossRef]
- Middendorp, S.; Nieuwenhuis, E.E. NKT cells in mucosal immunity. Mucosal Immunol. 2009, 2, 393–402. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Hao, G.L.; Wang, B.Y.; Gao, C.C.; Wang, Y.X.; Li, L.S.; Xu, J.D. Function and dysfunction of plasma cells in intestine. Cell Biosci. 2019, 9, 26. [Google Scholar] [CrossRef]
- Blaschitz, C.; Raffatellu, M. Th17 cytokines and the gut mucosal barrier. J. Clin. Immunol. 2010, 30, 196–203. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.S.; Tato, C.M.; Joyce-Shaikh, B.; Gulen, M.F.; Cayatte, C.; Chen, Y.; Blumenschein, W.M.; Judo, M.; Ayanoglu, G.; McClanahan, T.K.; et al. Interleukin-23-Independent IL-17 Production Regulates Intestinal Epithelial Permeability. Immunity 2015, 43, 727–738. [Google Scholar] [CrossRef] [Green Version]
- Hong, C.P.; Park, A.; Yang, B.G.; Yun, C.H.; Kwak, M.J.; Lee, G.W.; Kim, J.H.; Jang, M.S.; Lee, E.J.; Jeun, E.J.; et al. Gut-Specific Delivery of T-Helper 17 Cells Reduces Obesity and Insulin Resistance in Mice. Gastroenterology 2017, 152, 1998–2010. [Google Scholar] [CrossRef]
- Rau, M.; Schilling, A.K.; Meertens, J.; Hering, I.; Weiss, J.; Jurowich, C.; Kudlich, T.; Hermanns, H.M.; Bantel, H.; Beyersdorf, N.; et al. Progression from Nonalcoholic Fatty Liver to Nonalcoholic Steatohepatitis Is Marked by a Higher Frequency of Th17 Cells in the Liver and an Increased Th17/Resting Regulatory T Cell Ratio in Peripheral Blood and in the Liver. J. Immunol. 2016, 196, 97–105. [Google Scholar] [CrossRef]
- Saltzman, E.T.; Palacios, T.; Thomsen, M.; Vitetta, L. Intestinal Microbiome Shifts, Dysbiosis, Inflammation, and Non-alcoholic Fatty Liver Disease. Front. Microbiol. 2018, 9, 61. [Google Scholar] [CrossRef]
- Chen, G.; Ni, Y.; Nagata, N.; Xu, L.; Ota, T. Micronutrient Antioxidants and Nonalcoholic Fatty Liver Disease. Int. J. Mol. Sci. 2016, 17, 1379. [Google Scholar] [CrossRef] [Green Version]
- Nagashimada, M.; Ota, T. Role of vitamin E in nonalcoholic fatty liver disease. IUBMB Life 2019, 71, 516–522. [Google Scholar] [CrossRef]
- Sanyal, A.J.; Chalasani, N.; Kowdley, K.V.; McCullough, A.; Diehl, A.M.; Bass, N.M.; Neuschwander-Tetri, B.A.; Lavine, J.E.; Tonascia, J.; Unalp, A.; et al. Pioglitazone, vitamin E, or placebo for nonalcoholic steatohepatitis. N. Engl. J. Med. 2010, 362, 1675–1685. [Google Scholar] [CrossRef] [Green Version]
- Lavine, J.E.; Schwimmer, J.B.; Van Natta, M.L.; Molleston, J.P.; Murray, K.F.; Rosenthal, P.; Abrams, S.H.; Scheimann, A.O.; Sanyal, A.J.; Chalasani, N.; et al. Effect of vitamin E or metformin for treatment of nonalcoholic fatty liver disease in children and adolescents: The TONIC randomized controlled trial. JAMA 2011, 305, 1659–1668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mitsuoka, T. Development of functional foods. Biosci. Microbiota Food Health 2014, 33, 117–128. [Google Scholar] [CrossRef] [Green Version]
- Meroni, M.; Longo, M.; Dongiovanni, P. The Role of Probiotics in Nonalcoholic Fatty Liver Disease: A New Insight into Therapeutic Strategies. Nutrients 2019, 11, 2642. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gibson, G.R.; Roberfroid, M.B. Dietary modulation of the human colonic microbiota: Introducing the concept of prebiotics. J. Nutr. 1995, 125, 1401–1412. [Google Scholar] [CrossRef] [PubMed]
- Mitsuoka, T. Significance of dietary modulation of intestinal flora and intestinal environment. Biosci. Microflora 2000, 19, 15–25. [Google Scholar] [CrossRef] [Green Version]
- Chiba, Y.; Shida, K.; Nagata, S.; Wada, M.; Bian, L.; Wang, C.; Shimizu, T.; Yamashiro, Y.; Kiyoshima-Shibata, J.; Nanno, M.; et al. Well-controlled proinflammatory cytokine responses of Peyer’s patch cells to probiotic Lactobacillus casei. Immunology 2010, 130, 352–362. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Hua, J.; Li, Z. Probiotics improve high fat diet-induced hepatic steatosis and insulin resistance by increasing hepatic NKT cells. J. Hepatol. 2008, 49, 821–830. [Google Scholar] [CrossRef] [Green Version]
- Li, Z.; Yang, S.; Lin, H.; Huang, J.; Watkins, P.A.; Moser, A.B.; Desimone, C.; Song, X.Y.; Diehl, A.M. Probiotics and antibodies to TNF inhibit inflammatory activity and improve nonalcoholic fatty liver disease. Hepatology 2003, 37, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Velayudham, A.; Dolganiuc, A.; Ellis, M.; Petrasek, J.; Kodys, K.; Mandrekar, P.; Szabo, G. VSL#3 probiotic treatment attenuates fibrosis without changes in steatohepatitis in a diet-induced nonalcoholic steatohepatitis model in mice. Hepatology 2009, 49, 989–997. [Google Scholar]
- Xue, L.; He, J.; Gao, N.; Lu, X.; Li, M.; Wu, X.; Liu, Z.; Jin, Y.; Liu, J.; Xu, J.; et al. Probiotics may delay the progression of nonalcoholic fatty liver disease by restoring the gut microbiota structure and improving intestinal endotoxemia. Sci. Rep. 2017, 7, 45176. [Google Scholar] [CrossRef]
- Briskey, D.; Heritage, M.; Jaskowski, L.A.; Peake, J.; Gobe, G.; Subramaniam, V.N.; Crawford, D.; Campbell, C.; Vitetta, L. Probiotics modify tight-junction proteins in an animal model of nonalcoholic fatty liver disease. Ther. Adv. Gastroenterol. 2016, 9, 463–472. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, Z.; Wang, C.; Zhang, L.; Zhao, Y.; Duan, C.; Zhang, X.; Gao, L.; Li, S. Lactobacillus plantarum NA136 improves the non-alcoholic fatty liver disease by modulating the AMPK/Nrf2 pathway. Appl. Microbiol. Biotechnol. 2019, 103, 5843–5850. [Google Scholar] [CrossRef]
- Sohn, W.; Jun, D.W.; Lee, K.N.; Lee, H.L.; Lee, O.Y.; Choi, H.S.; Yoon, B.C. Lactobacillus paracasei Induces M2-Dominant Kupffer Cell Polarization in a Mouse Model of Nonalcoholic Steatohepatitis. Dig. Dis. Sci. 2015, 60, 3340–3350. [Google Scholar] [CrossRef]
- Moens, F.; Van den Abbeele, P.; Basit, A.W.; Dodoo, C.; Chatterjee, R.; Smith, B.; Gaisford, S. A four-strain probiotic exerts positive immunomodulatory effects by enhancing colonic butyrate production in vitro. Int. J. Pharm. 2019, 555, 1–10. [Google Scholar] [CrossRef]
- Ma, Y.Y.; Li, L.; Yu, C.H.; Shen, Z.; Chen, L.H.; Li, Y.M. Effects of probiotics on nonalcoholic fatty liver disease: A meta-analysis. World J. Gastroenterol. WJG 2013, 19, 6911–6918. [Google Scholar] [CrossRef]
- Cani, P.D.; Possemiers, S.; Van de Wiele, T.; Guiot, Y.; Everard, A.; Rottier, O.; Geurts, L.; Naslain, D.; Neyrinck, A.; Lambert, D.M.; et al. Changes in gut microbiota control inflammation in obese mice through a mechanism involving GLP-2-driven improvement of gut permeability. Gut 2009, 58, 1091–1103. [Google Scholar] [CrossRef] [Green Version]
- Parnell, J.A.; Reimer, R.A. Prebiotic fibres dose-dependently increase satiety hormones and alter Bacteroidetes and Firmicutes in lean and obese JCR:LA-cp rats. Br. J. Nutr. 2012, 107, 601–613. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parnell, J.A.; Reimer, R.A. Weight loss during oligofructose supplementation is associated with decreased ghrelin and increased peptide YY in overweight and obese adults. Am. J. Clin. Nutr. 2009, 89, 1751–1759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loman, B.R.; Hernandez-Saavedra, D.; An, R.; Rector, R.S. Prebiotic and probiotic treatment of nonalcoholic fatty liver disease: A systematic review and meta-analysis. Nutr. Rev. 2018, 76, 822–839. [Google Scholar] [CrossRef]
- Ting, W.J.; Kuo, W.W.; Hsieh, D.J.; Yeh, Y.L.; Day, C.H.; Chen, Y.H.; Chen, R.J.; Padma, V.V.; Huang, C.Y. Heat Killed Lactobacillus reuteri GMNL-263 Reduces Fibrosis Effects on the Liver and Heart in High Fat Diet-Hamsters via TGF-beta Suppression. Int. J. Mol. Sci. 2015, 16, 25881–25896. [Google Scholar] [CrossRef] [Green Version]
- Hsieh, F.C.; Lee, C.L.; Chai, C.Y.; Chen, W.T.; Lu, Y.C.; Wu, C.S. Oral administration of Lactobacillus reuteri GMNL-263 improves insulin resistance and ameliorates hepatic steatosis in high fructose-fed rats. Nutr. Metab. 2013, 10, 35. [Google Scholar] [CrossRef] [Green Version]
- Uchinaka, A.; Azuma, N.; Mizumoto, H.; Nakano, S.; Minamiya, M.; Yoneda, M.; Aoyama, K.; Komatsu, Y.; Yamada, Y.; Murohara, T.; et al. Anti-inflammatory effects of heat-killed Lactobacillus plantarum L-137 on cardiac and adipose tissue in rats with metabolic syndrome. Sci. Rep. 2018, 8, 8156. [Google Scholar] [CrossRef]
- Izumo, T.; Ida, M.; Maekawa, T.; Furukawa, Y.; Kitagawa, Y.; Kiso, Y. Comparison of the Immunomodulatory Effects of Live and Heat-killed Lactobacillus pentosus S-PT84. J. Health Sci. 2011, 57, 304–310. [Google Scholar] [CrossRef] [Green Version]
- Nonaka, Y.; Izumo, T.; Izumi, F.; Maekawa, T.; Shibata, H.; Nakano, A.; Kishi, A.; Akatani, K.; Kiso, Y. Antiallergic effects of Lactobacillus pentosus strain S-PT84 mediated by modulation of Th1/Th2 immunobalance and induction of IL-10 production. Int. Arch. Allergy Immunol. 2008, 145, 249–257. [Google Scholar] [CrossRef]
- Sakai, Y.; Arie, H.; Ni, Y.; Zhuge, F.; Xu, L.; Chen, G.; Nagata, N.; Suzuki, T.; Kaneko, S.; Ota, T.; et al. Lactobacillus pentosus strain S-PT84 improves steatohepatitis by maintaining gut permeability. J. Endocrinol. 2020, 247, 169–181. [Google Scholar] [CrossRef]
- Zhao, X.; Higashikawa, F.; Noda, M.; Kawamura, Y.; Matoba, Y.; Kumagai, T.; Sugiyama, M. The obesity and fatty liver are reduced by plant-derived Pediococcus pentosaceus LP28 in high fat diet-induced obese mice. PLoS ONE 2012, 7, e30696. [Google Scholar] [CrossRef] [Green Version]
- McNaught, C.E.; Woodcock, N.P.; MacFie, J.; Mitchell, C.J. A prospective randomised study of the probiotic Lactobacillus plantarum 299V on indices of gut barrier function in elective surgical patients. Gut 2002, 51, 827–831. [Google Scholar] [CrossRef] [PubMed]
- Khan, A.; Ding, Z.; Ishaq, M.; Bacha, A.S.; Khan, I.; Hanif, A.; Li, W.; Guo, X. Understanding the Effects of Gut Microbiota Dysbiosis on Nonalcoholic Fatty Liver Disease and the Possible Probiotics Role: Recent Updates. Int. J. Biol. Sci. 2021, 17, 818–833. [Google Scholar] [CrossRef]
- Gou, S.; Yang, Z.; Liu, T.; Wu, H.; Wang, C. Use of probiotics in the treatment of severe acute pancreatitis: A systematic review and meta-analysis of randomized controlled trials. Crit. Care 2014, 18, R57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilkins, T.; Sequoia, J. Probiotics for Gastrointestinal Conditions: A Summary of the Evidence. Am. Fam. Phys. 2017, 96, 170–178. [Google Scholar]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nagashimada, M.; Honda, M. Effect of Microbiome on Non-Alcoholic Fatty Liver Disease and the Role of Probiotics, Prebiotics, and Biogenics. Int. J. Mol. Sci. 2021, 22, 8008. https://doi.org/10.3390/ijms22158008
Nagashimada M, Honda M. Effect of Microbiome on Non-Alcoholic Fatty Liver Disease and the Role of Probiotics, Prebiotics, and Biogenics. International Journal of Molecular Sciences. 2021; 22(15):8008. https://doi.org/10.3390/ijms22158008
Chicago/Turabian StyleNagashimada, Mayumi, and Masao Honda. 2021. "Effect of Microbiome on Non-Alcoholic Fatty Liver Disease and the Role of Probiotics, Prebiotics, and Biogenics" International Journal of Molecular Sciences 22, no. 15: 8008. https://doi.org/10.3390/ijms22158008
APA StyleNagashimada, M., & Honda, M. (2021). Effect of Microbiome on Non-Alcoholic Fatty Liver Disease and the Role of Probiotics, Prebiotics, and Biogenics. International Journal of Molecular Sciences, 22(15), 8008. https://doi.org/10.3390/ijms22158008