
Osteoarthritis-Related Inflammation Blocks TGF-β’s Protective Effect on Chondrocyte Hypertrophy via (de)Phosphorylation of the SMAD2/3 Linker Region

 ,
,  and
and
Abstract
:1. Introduction
2. Results
2.1. IL-1β and OAS-cm Negate the Anti-Hypertrophic Function of TGF-β in Bovine Cartilage Explants
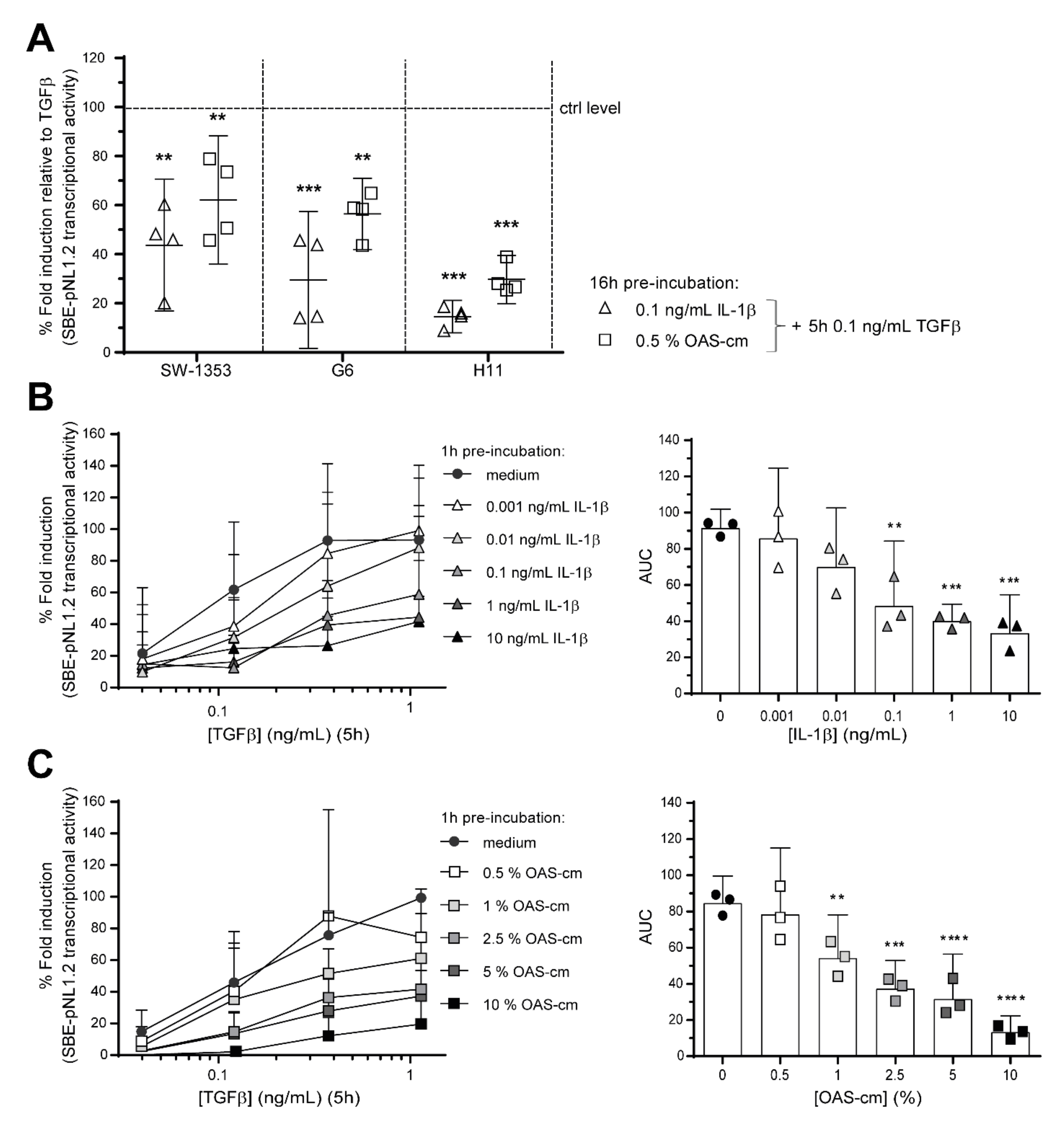
2.2. IL-1β and OAS-cm Inhibit TGF-β Transcriptional Activity in Different Chondrocyte Cell Lines
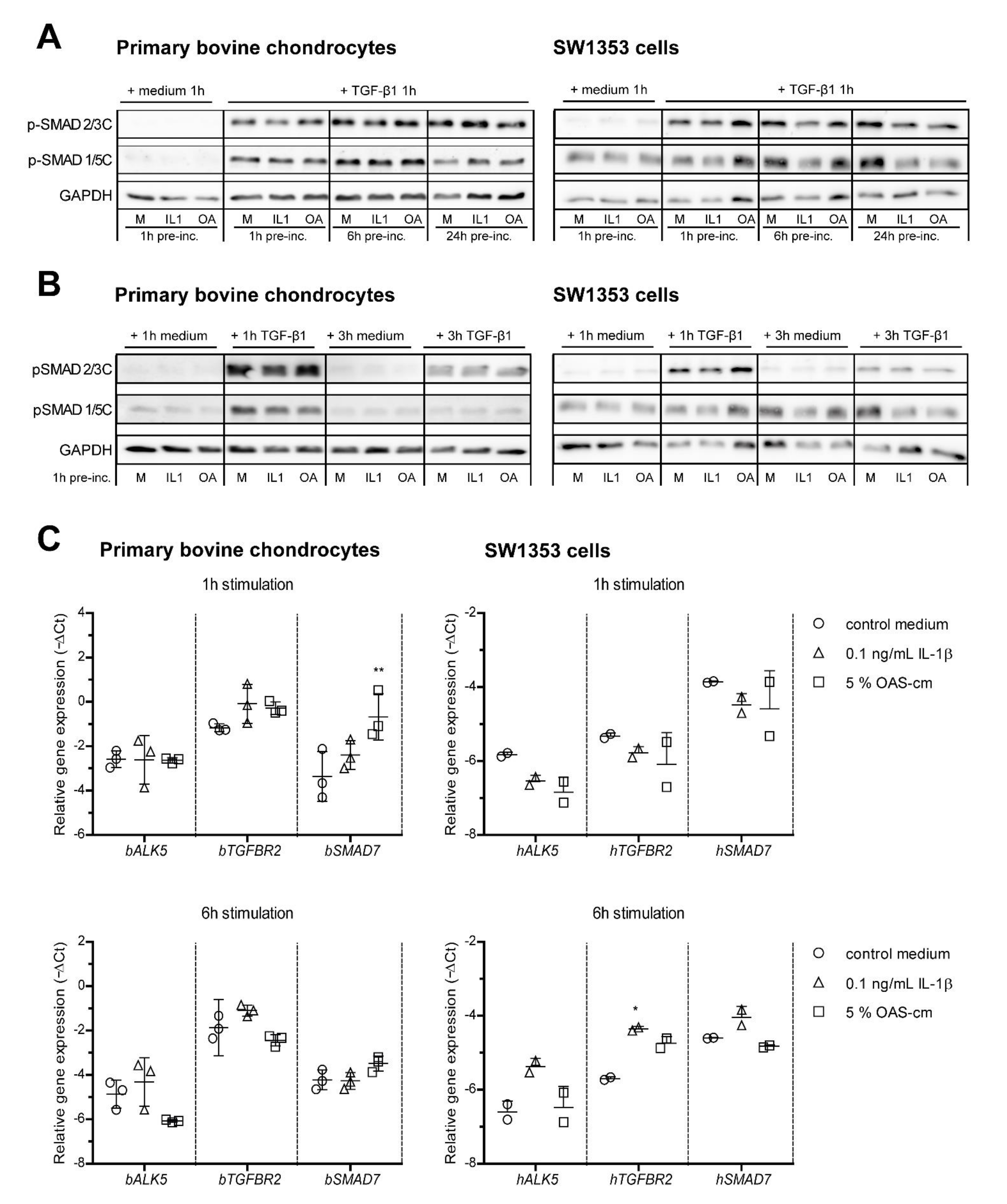
2.3. IL-1β and OAS-cm Do Not Inhibit C-Terminal Phosphorylation of SMAD2/3 and Do Not Regulate Receptor Level Expression
2.4. IL-1β and OAS-cm Inhibit TGF-β via(de-)Phosphorylation of the SMAD2/3 Linker Region
3. Discussion
4. Materials and Methods
4.1. Primary Cell Culture
4.2. Chondrocyte Cell Line Culture
4.3. Chondrocyte Stimulation
4.4. Plasmid DNA, Adenoviral Production and Transduction
4.5. SMAD-Luciferase Transcriptional Reporter Assay
4.6. Protein Isolation and Western Blot
4.7. RNA Isolation and Quantitative Real-Time PCR
4.8. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
Appendix A

Appendix B
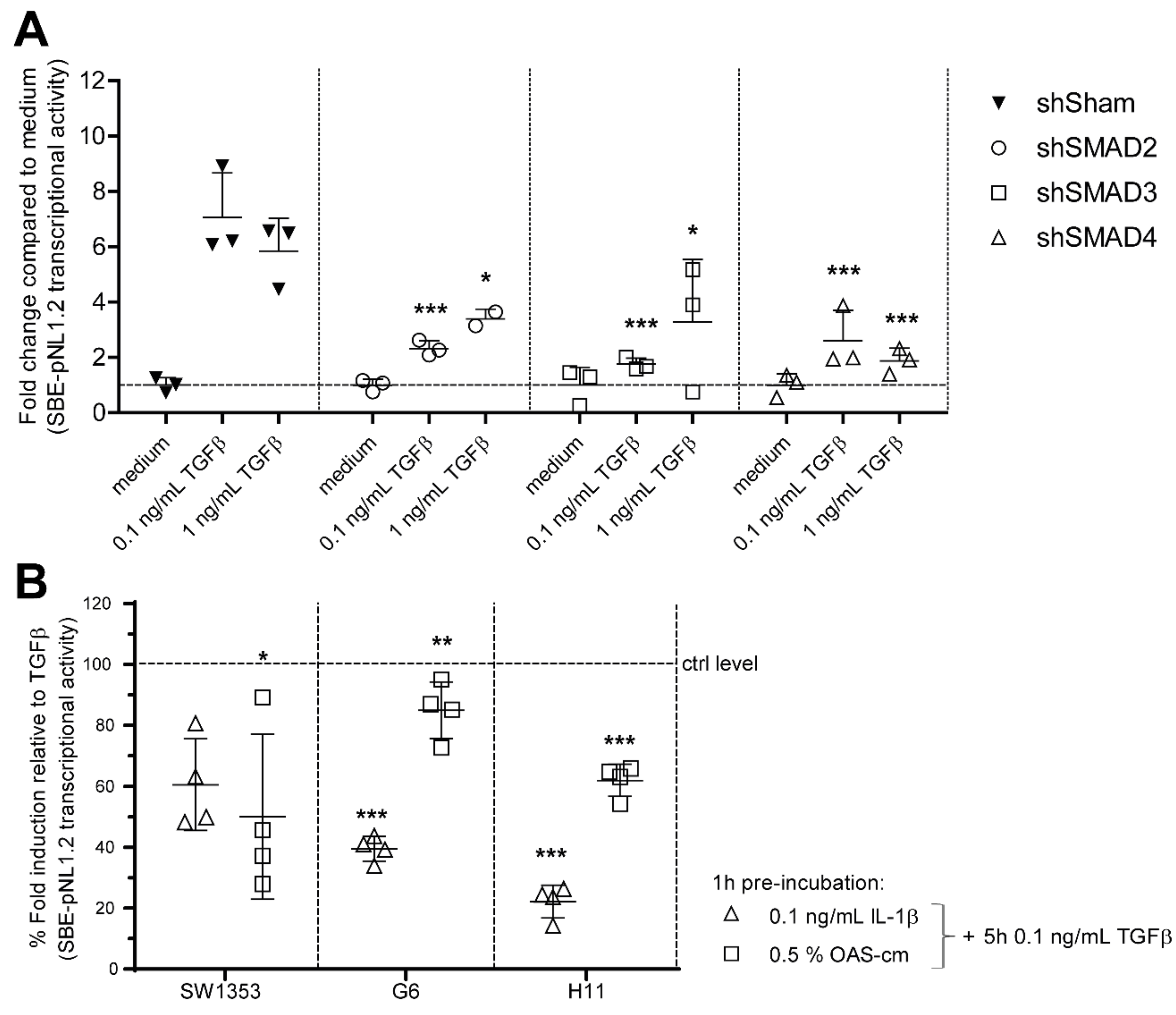
Appendix C
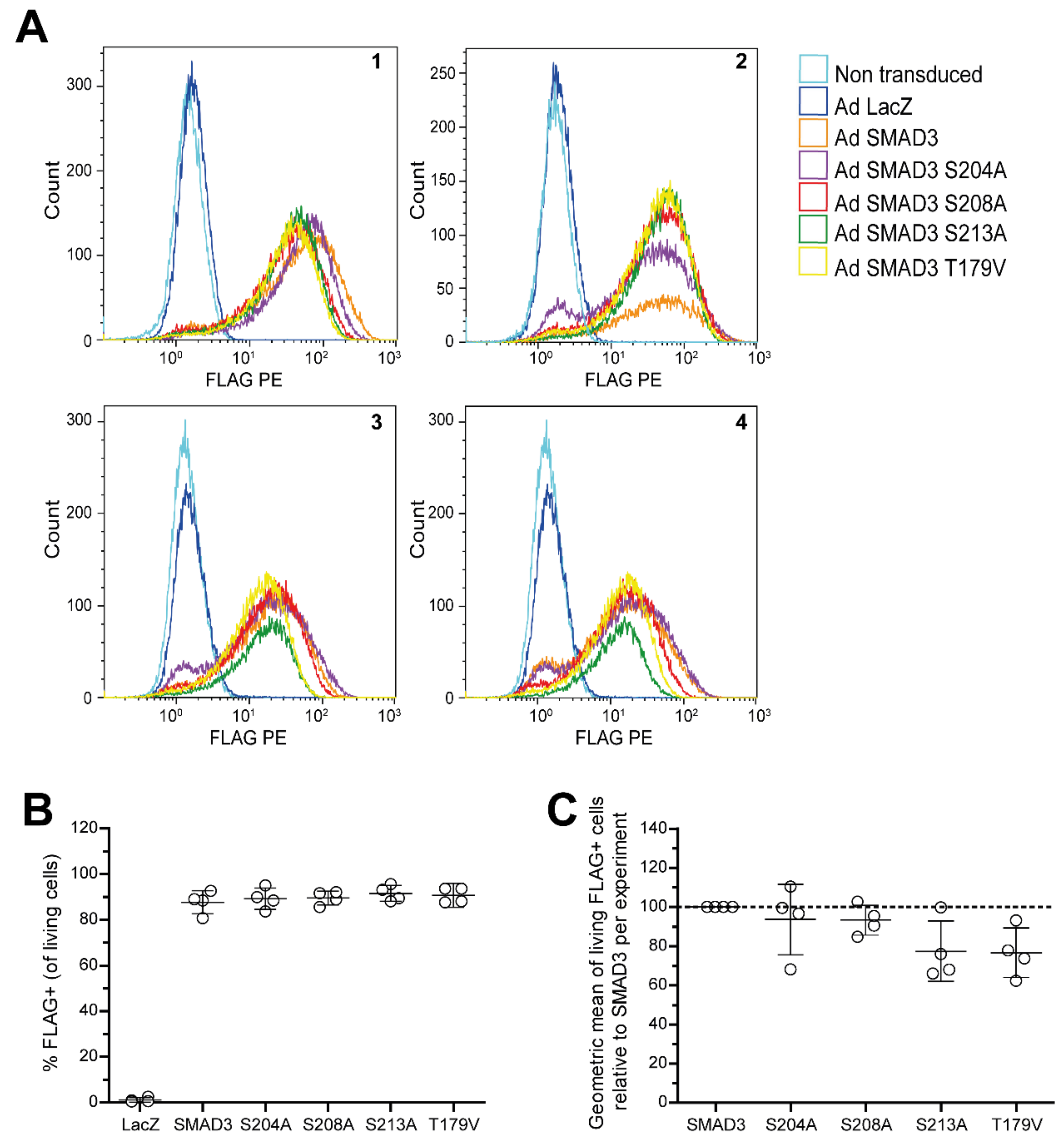
References
- Chow, Y.Y.; Chin, K.Y. The Role of Inflammation in the Pathogenesis of Osteoarthritis. Mediat. Inflamm. 2020, 2020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van den Bosch, M.H.J.; van Lent, P.; van der Kraan, P.M. Identifying effector molecules, cells, and cytokines of innate immunity in OA. Osteoarthr. Cartil. 2020, 28, 532–543. [Google Scholar] [CrossRef] [PubMed]
- Scanzello, C.R.; Goldring, S.R. The role of synovitis in osteoarthritis pathogenesis. Bone 2012, 51, 249–257. [Google Scholar] [CrossRef] [Green Version]
- D’Agostino1, M.A.; Conaghan, P.; le Bars, M.; Baron, G.; Grassi, W.; Martin-Mola, E.; Wakefield, R.; Brasseur, J.-L.; So, A.; Backhaus, M.; et al. EULAR report on the use of ultrasonography in painful knee osteoarthritis. Part 1: Prevalence of inflammation in osteoarthritis. Ann. Rheum. Dis 2005, 64, 1703–1709. [Google Scholar] [CrossRef] [PubMed]
- Roemer, F.W.; Guermazi, A.; Felson, D.T.; Niu, J.; Nevitt, M.C.; Crema, M.D.; Lynch, J.A.; Lewis, C.E.; Torner, J.; Zhalng, Y. Presence of MRI-detected joint effusion and synovitis increases the risk of cartilage loss in knees without osteoarthritis at 30-month follow-up: The MOST study. Ann. Rheum. Dis. 2011, 70, 1804–1809. [Google Scholar] [CrossRef]
- Kortekaas, M.C.; Kwok, W.Y.; Reijnierse, M.; Stijnen, T.; Kloppenburg, M. Brief Report: Association of Inflammation With Development of Erosions in Patients With Hand Osteoarthritis: A Prospective Ultrasonography Study. Arthritis Rheumatol. 2016, 68, 392–397. [Google Scholar] [CrossRef]
- Vincenti, M.P.; Brinckerhoff, C.E. Transcriptional regulation of collagenase (MMP-1, MMP-13) genes in arthritis: Integration of complex signaling pathways for the recruitment of gene-specific transcription factors. Arthritis Res. 2002, 4, 157–164. [Google Scholar] [CrossRef]
- Cortial, D.; Gouttenoire, J.; Rousseau, C.F.; Ronzieère, M.-C.; Piccardi, N.; Msika, P.; Herbage, D.; Malletin-Gerin, F.; Freyria, A.l.-M. Activation by IL-1 of bovine articular chondrocytes in culture within a 3D collagen-based scaffold. An in vitro model to address the effect of compounds with therapeutic potential in osteoarthritis. Osteoarthr. Cartil. 2006, 14, 631–640. [Google Scholar] [CrossRef] [Green Version]
- Bauge, C.; Attia, J.; Leclercq, S.; Pujol, J.P.; Galera, P.; Boumediene, K. Interleukin-1beta up-regulation of Smad7 via NF-kappaB activation in human chondrocytes. Arthritis Rheum. 2008, 58, 221–226. [Google Scholar] [CrossRef]
- Roman-Blas, J.A.; Stokes, D.G.; Jimenez, S.A. Modulation of TGF-beta signaling by proinflammatory cytokines in articular chondrocytes. Osteoarthr. Cartil. 2007, 15, 1367–1377. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thielen, N.G.M.; van der Kraan, P.M.; van Caam, A.P.M. TGFbeta/BMP Signaling Pathway in Cartilage Homeostasis. Cells 2019, 8, 969. [Google Scholar] [CrossRef] [Green Version]
- Chen, C.G.; Thuillier, D.; Chin, E.N.; Alliston, T. Chondrocyte-intrinsic Smad3 represses Runx2-inducible matrix metalloproteinase 13 expression to maintain articular cartilage and prevent osteoarthritis. Arthritis Rheum. 2012, 64, 3278–3289. [Google Scholar] [CrossRef] [Green Version]
- Van der Kraan, P.M.; Blaney Davidson, E.N.; Blom, A.; van den Berg, W.B. TGF-beta signaling in chondrocyte terminal differentiation and osteoarthritis: Modulation and integration of signaling pathways through receptor-Smads. Osteoarthr. Cartil. 2009, 17, 1539–1545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Kraan, P.M.; Blaney Davidson, E.N.; van den Berg, W.B. A role for age-related changes in TGFbeta signaling in aberrant chondrocyte differentiation and osteoarthritis. Arthritis Res. Ther. 2010, 12, 201. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bauge, C.; Legendre, F.; Leclercq, S.; Elissalde, J.M.; Pujol, J.P.; Galera, P.; Boumediene, K. Interleukin-1beta impairment of transforming growth factor beta1 signaling by down-regulation of transforming growth factor beta receptor type II and up-regulation of Smad7 in human articular chondrocytes. Arthritis Rheum. 2007, 56, 3020–3032. [Google Scholar] [CrossRef]
- Matsuzaki, K. Smad phospho-isoforms direct context-dependent TGF-beta signaling. Cytokine Growth Factor Rev. 2013, 24, 385–399. [Google Scholar] [CrossRef] [PubMed]
- Kamato, D.; Burch, M.L.; Piva, T.J.; Rezaei, H.B.; Rostam, M.A.; Xu, S.; Zheng, W.; Little, P.J.; Osman, N. Transforming growth factor-beta signalling: Role and consequences of Smad linker region phosphorylation. Cell. Signal. 2013, 25, 2017–2024. [Google Scholar] [CrossRef] [PubMed]
- Massague, J. TGF-beta signal transduction. Annu. Rev. Biochem. 1998, 67, 753–791. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y. Structural insights on Smad function in TGFbeta signaling. Bioessays 2001, 23, 223–232. [Google Scholar] [CrossRef]
- Hill, C.S. Nucleocytoplasmic shuttling of Smad proteins. Cell Res. 2009, 19, 36–46. [Google Scholar] [CrossRef] [Green Version]
- Sapkota, G.; Knockaert, M.; Alarcon, C.; Montalvo, E.; Brivanlou, A.H.; Massague, J. Dephosphorylation of the linker regions of Smad1 and Smad2/3 by small C-terminal domain phosphatases has distinct outcomes for bone morphogenetic protein and transforming growth factor-beta pathways. J. Biol. Chem. 2006, 281, 40412–40419. [Google Scholar] [CrossRef] [Green Version]
- Matsuzaki, K. Smad phosphoisoform signaling specificity: The right place at the right time. Carcinogenesis 2011, 32, 1578–1588. [Google Scholar] [CrossRef]
- Millet, C.; Yamashita, M.; Heller, M.; Yu, L.R.; Veenstra, T.D.; Zhang, Y.E. A negative feedback control of transforming growth factor-beta signaling by glycogen synthase kinase 3-mediated Smad3 linker phosphorylation at Ser-204. J. Biol. Chem. 2009, 284, 19808–19816. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van der Kraan, P.M.; van den Berg, W.B. Chondrocyte hypertrophy and osteoarthritis: Role in initiation and progression of cartilage degeneration? Osteoarthr. Cartil. 2012, 20, 223–232. [Google Scholar] [CrossRef] [Green Version]
- Li, T.F.; O’Keefe, R.J.; Chen, D. TGF-beta signaling in chondrocytes. Front. Biosci. 2005, 10, 681–688. [Google Scholar] [CrossRef]
- Zhao, W.; Wang, T.; Luo, Q.; Chen, Y.; Leung, V.Y.L.; Wen, C.; Shah, M.F.; Pan, H.; Chiu, K.; Cao, X.; et al. Cartilage degeneration and excessive subchondral bone formation in spontaneous osteoarthritis involves altered TGF-beta signaling. J. Orthop. Res. 2016, 34, 763–770. [Google Scholar] [CrossRef] [Green Version]
- Van Caam, A.; Madej, W.; Thijssen, E.; Garcia de Vinuesa, A.; van den Berg, W.; Goumans, M.J.; ten Diike, P.; Davidson, E.B.; van der Kraan, P.M. Expression of TGFbeta-family signalling components in ageing cartilage: Age-related loss of TGFbeta and BMP receptors. Osteoarthr. Cartil. 2016, 24, 1235–1245. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hata, A.; Chen, Y.G. TGF-beta Signaling from Receptors to Smads. Cold Spring Harb. Perspect. Biol. 2016, 8, a022061. [Google Scholar] [CrossRef] [PubMed]
- Bae, E.; Kim, S.J.; Hong, S.; Liu, F.; Ooshima, A. Smad3 linker phosphorylation attenuates Smad3 transcriptional activity and TGF-beta1/Smad3-induced epithelial-mesenchymal transition in renal epithelial cells. Biochem. Biophys. Res. Commun. 2012, 427, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Bae, E.; Sato, M.; Kim, R.-J.; Kwak, M.-K.; Naka, K.; Gim, J.; Kadota, M.; Tang, B.; Flanders, K.C.; Kim, T.-A.; et al. Definition of smad3 phosphorylation events that affect malignant and metastatic behaviors in breast cancer cells. Cancer Res. 2014, 74, 6139–6149. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burch, M.L.; Zheng, W.; Little, P.J. Smad linker region phosphorylation in the regulation of extracellular matrix synthesis. Cell. Mol. Life Sci. 2011, 68, 97–107. [Google Scholar] [CrossRef]
- Li, F.; Zeng, B.; Chai, Y.; Cai, P.; Fan, C.; Cheng, T. The linker region of Smad2 mediates TGF-beta-dependent ERK2-induced collagen synthesis. Biochem. Biophys. Res. Commun. 2009, 386, 289–293. [Google Scholar] [CrossRef]
- Melchiorri, C.; Meliconi, R.; Frizziero, L.; Silvestri, T.; Pulsatelli, L.; Mazzetti, I.; Borzì, R.M.; Uguccioni, M.; Facchini, A. Enhanced and coordinated in vivo expression of inflammatory cytokines and nitric oxide synthase by chondrocytes from patients with osteoarthritis. Arthritis Rheum. 1998, 41, 2165–2174. [Google Scholar] [CrossRef]
- Martel-Pelletier, J.; McCollum, R.; DiBattista, J.; Faure, M.P.; Chin, J.A.; Fournier, S.; Sarfati, M.; Pelletier, J.P. The interleukin-1 receptor in normal and osteoarthritic human articular chondrocytes. Identification as the type I receptor and analysis of binding kinetics and biologic function. Arthritis Rheum. 1992, 35, 530–540. [Google Scholar] [CrossRef] [PubMed]
- Radons, J.; Bosserhoff, A.K.; Grassel, S.; Falk, W.; Schubert, T.E. p38MAPK mediates IL-1-induced down-regulation of aggrecan gene expression in human chondrocytes. Int. J. Mol. Med. 2006, 17, 661–668. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fernandes, J.C.; Martel-Pelletier, J.; Pelletier, J.P. The role of cytokines in osteoarthritis pathophysiology. Biorheology 2002, 39, 237–246. [Google Scholar] [PubMed]
- Nasi, S.; Ea, H.K.; So, A.; Busso, N. Revisiting the Role of Interleukin-1 Pathway in Osteoarthritis: Interleukin-1alpha and -1beta, and NLRP3 Inflammasome Are Not Involved in the Pathological Features of the Murine Menisectomy Model of Osteoarthritis. Front. Pharmacol. 2017, 8, 282. [Google Scholar] [CrossRef] [PubMed]
- Van Dalen, S.C.; Blom, A.B.; Sloetjes, A.W.; Helsen, M.M.; Roth, J.; Vogl, T.; van de Loo, F.A.J.; Koenders, M.I.; van der Kraan, P.M.; van den Berg, W.B.; et al. Interleukin-1 is not involved in synovial inflammation and cartilage destruction in collagenase-induced osteoarthritis. Osteoarthr. Cartil. 2017, 25, 385–396. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapoor, M.; Martel-Pelletier, J.; Lajeunesse, D.; Pelletier, J.P.; Fahmi, H. Role of proinflammatory cytokines in the pathophysiology of osteoarthritis. Nat. Rev. Rheumatol. 2011, 7, 33–42. [Google Scholar] [CrossRef] [PubMed]
- Liacini, A.; Sylvester, J.; Li, W.Q.; Huang, W.; Dehnade, F.; Ahmad, M.; Zafarullah, M. Induction of matrix metalloproteinase-13 gene expression by TNF-alpha is mediated by MAP kinases, AP-1, and NF-kappaB transcription factors in articular chondrocytes. Exp. Cell Res. 2003, 288, 208–217. [Google Scholar] [CrossRef]
- Laavola, M.; Leppanen, T.; Hamalainen, M.; Vuolteenaho, K.; Moilanen, T.; Nieminen, R.; Moilanen, E. IL-6 in Osteoarthritis: Effects of Pine Stilbenoids. Molecules 2018, 24, 109. [Google Scholar] [CrossRef] [Green Version]
- Van Lent, P.L.; Blom, A.B.; Schelbergen, R.F.; Sloetjes, A.; Lafeber, F.P.; Lems, W.F.; Cats, H.; Vogl, T.; Roth, J.; van den Berg, W.B. Active involvement of alarmins S100A8 and S100A9 in the regulation of synovial activation and joint destruction during mouse and human osteoarthritis. Arthritis Rheum. 2012, 64, 1466–1476. [Google Scholar] [CrossRef] [PubMed]
- Hulin-Curtis, S.L.; Bidwell, J.L.; Perry, M.J. Association between CCL2 haplotypes and knee osteoarthritis. Int. J. Immunogenet. 2013, 40, 280–283. [Google Scholar] [CrossRef]
- Heldens, G.T.; Blaney Davidson, E.N.; Vitters, E.L.; Schreurs, B.W.; Piek, E.; van den Berg, W.B.; van der Kraan, P.M. Catabolic factors and osteoarthritis-conditioned medium inhibit chondrogenesis of human mesenchymal stem cells. Tissue Eng. Part A 2012, 18, 45–54. [Google Scholar] [CrossRef]
- Cecil, D.L.; Johnson, K.; Rediske, J.; Lotz, M.; Schmidt, A.M.; Terkeltaub, R. Inflammation-induced chondrocyte hypertrophy is driven by receptor for advanced glycation end products. J. Immunol. 2005, 175, 8296–8302. [Google Scholar] [CrossRef] [Green Version]
- Merz, D.; Liu, R.; Johnson, K.; Terkeltaub, R. IL-8/CXCL8 and growth-related oncogene alpha/CXCL1 induce chondrocyte hypertrophic differentiation. J. Immunol. 2003, 171, 4406–4415. [Google Scholar] [CrossRef] [Green Version]
- Morita, K.; Miyamoto, T.; Fujita, N.; Kubota, Y.; Ito, K.; Takubo, K.; Miyamoto, K.; Ninomiya, K.; Suzuki, T.; Iwasaki, R.; et al. Reactive oxygen species induce chondrocyte hypertrophy in endochondral ossification. J. Exp. Med. 2007, 204, 1613–1623. [Google Scholar] [CrossRef]
- Van Beuningen, H.M.; de Vries-van Melle, M.L.; Vitters, E.L.; Schreurs, W.; van den Berg, W.B.; van Osch, G.J.; van der Kraan, P.M. Inhibition of TAK1 and/or JAK can rescue impaired chondrogenic differentiation of human mesenchymal stem cells in osteoarthritis-like conditions. Tissue Eng. Part A 2014, 20, 2243–2252. [Google Scholar] [CrossRef] [Green Version]
- Van der Kraan, P.M. Differential Role of Transforming Growth Factor-beta in an Osteoarthritic or a Healthy Joint. J. Bone Metab. 2018, 25, 65–72. [Google Scholar] [CrossRef]
- Madej, W.; Buma, P.; van der Kraan, P. Inflammatory conditions partly impair the mechanically mediated activation of Smad2/3 signaling in articular cartilage. Arthritis Res. Ther. 2016, 18, 146. [Google Scholar] [CrossRef] [Green Version]
- Elshaier, A.M.; Hakimiyan, A.A.; Rappoport, L.; Rueger, D.C.; Chubinskaya, S. Effect of interleukin-1beta on osteogenic protein 1-induced signaling in adult human articular chondrocytes. Arthritis Rheum. 2009, 60, 143–154. [Google Scholar] [CrossRef] [Green Version]
- Murata, M.; Yoshida, K.; Yamaguchi, T.; Matsuzaki, K. Linker phosphorylation of Smad3 promotes fibro-carcinogenesis in chronic viral hepatitis of hepatocellular carcinoma. World J. Gastroenterol. 2014, 20, 15018–15027. [Google Scholar] [CrossRef] [PubMed]
- Rostam, M.A.; Kamato, D.; Piva, T.J.; Zheng, W.; Little, P.J.; Osman, N. The role of specific Smad linker region phosphorylation in TGF-beta mediated expression of glycosaminoglycan synthesizing enzymes in vascular smooth muscle. Cell. Signal. 2016, 28, 956–966. [Google Scholar] [CrossRef]
- Burch, M.L.; Yang, S.N.; Ballinger, M.L.; Getachew, R.; Osman, N.; Little, P.J. TGF-beta stimulates biglycan synthesis via p38 and ERK phosphorylation of the linker region of Smad2. Cell. Mol. Life Sci. 2010, 67, 2077–2090. [Google Scholar] [CrossRef]
- Yu, J.S.; Ramasamy, T.S.; Murphy, N.; Holt, M.K.; Czapiewski, R.; Wei, S.K.; Cui, W. PI3K/mTORC2 regulates TGF-beta/Activin signalling by modulating Smad2/3 activity via linker phosphorylation. Nat. Commun. 2015, 6, 7212. [Google Scholar] [CrossRef] [Green Version]
- Bruce, D.L.; Sapkota, G.P. Phosphatases in SMAD regulation. FEBS Lett. 2012, 586, 1897–1905. [Google Scholar] [CrossRef] [Green Version]
- Djouad, F.; Rackwitz, L.; Song, Y.; Janjanin, S.; Tuan, R.S. ERK1/2 activation induced by inflammatory cytokines compromises effective host tissue integration of engineered cartilage. Tissue Eng. Part A 2009, 15, 2825–2835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, Y.; Valbracht, J.; Lotz, M. Selective activation of the mitogen-activated protein kinase subgroups c-Jun NH2 terminal kinase and p38 by IL-1 and TNF in human articular chondrocytes. J. Clin. Investig. 1996, 98, 2425–2430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Manley, G.C.A.; Stokes, C.A.; Marsh, E.K.; Sabroe, I.; Parker, L.C. DUSP10 Negatively Regulates the Inflammatory Response to Rhinovirus through Interleukin-1beta Signaling. J. Virol. 2019, 93, e01659-18. [Google Scholar] [CrossRef] [Green Version]
- Van den Akker, G.G.; van Beuningen, H.M.; Vitters, E.L.; Koenders, M.I.; van de Loo, F.A.; van Lent, P.L.; Davidson, E.N.B.; van der Kraan, P.M. Interleukin 1 beta-induced SMAD2/3 linker modifications are TAK1 dependent and delay TGFbeta signaling in primary human mesenchymal stem cells. Cell. Signal. 2017, 40, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Kretzschmar, M.; Doody, J.; Timokhina, I.; Massague, J. A mechanism of repression of TGFbeta/ Smad signaling by oncogenic Ras. Genes Dev. 1999, 13, 804–816. [Google Scholar] [CrossRef]
- Park, S.; Yang, K.M.; Park, Y.; Hong, E.; Hong, C.P.; Park, J.; Pang, K.; Lee, J.; Park, B.; Lee, S. Identification of Epithelial-Mesenchymal Transition-related Target Genes Induced by the Mutation of Smad3 Linker Phosphorylation. J. Cancer Prev. 2018, 23, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Neefjes, M.; Housmans, B.A.C.; van den Akker, G.G.H.; van Rhijn, L.W.; Welting, T.J.M.; van der Kraan, P.M. Reporter gene comparison demonstrates interference of complex body fluids with secreted luciferase activity. Sci. Rep. 2021, 11, 1359. [Google Scholar] [CrossRef]
- Sekimoto, G.; Matsuzaki, K.; Yoshida, K.; Mori, S.; Murata, M.; Seki, T.; Matsui, H.; Fujisawa, J.; Okazaki, K. Reversible Smad-dependent signaling between tumor suppression and oncogenesis. Cancer Res. 2007, 67, 5090–5096. [Google Scholar] [CrossRef] [Green Version]
- Tarasewicz, E.; Jeruss, J.S. Phospho-specific Smad3 signaling: Impact on breast oncogenesis. Cell Cycle 2012, 11, 2443–2451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Browne, J.A.; Liu, X.; Schnaper, H.W.; Hayashida, T. Serine-204 in the linker region of Smad3 mediates the collagen-I response to TGF-beta in a cell phenotype-specific manner. Exp. Cell Res. 2013, 319, 2928–2937. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gao, S.; Alarcon, C.; Sapkota, G.; Rahman, S.; Chen, P.Y.; Goerner, N.; Macias, M.J.; Erdjument-Bromage, H.; Tempst, P.; Massagué, J. Ubiquitin ligase Nedd4L targets activated Smad2/3 to limit TGF-beta signaling. Mol. Cell. 2009, 36, 457–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heldin, C.H.; Moustakas, A. Role of Smads in TGFbeta signaling. Cell Tissue Res. 2012, 347, 21–36. [Google Scholar] [CrossRef]
- Kamaraju, A.K.; Roberts, A.B. Role of Rho/ROCK and p38 MAP kinase pathways in transforming growth factor-beta-mediated Smad-dependent growth inhibition of human breast carcinoma cells in vivo. J. Biol. Chem. 2005, 280, 1024–1036. [Google Scholar] [CrossRef] [Green Version]
- Matsuura, I.; Denissova, N.G.; Wang, G.; He, D.; Long, J.; Liu, F. Cyclin-dependent kinases regulate the antiproliferative function of Smads. Nature 2004, 430, 226–231. [Google Scholar] [CrossRef]
- Wrighton, K.H.; Willis, D.; Long, J.; Liu, F.; Lin, X.; Feng, X.H. Small C-terminal domain phosphatases dephosphorylate the regulatory linker regions of Smad2 and Smad3 to enhance transforming growth factor-beta signaling. J. Biol. Chem. 2006, 281, 38365–38375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wrighton, K.H.; Lin, X.; Feng, X.H. Phospho-control of TGF-beta superfamily signaling. Cell Res. 2009, 19, 8–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffmann, A.; Preobrazhenska, O.; Wodarczyk, C.; Medler, Y.; Winkel, A.; Shahab, S.; Huylebroeck, D.; Gross, G.; Verschueren, K. Transforming growth factor-beta-activated kinase-1 (TAK1), a MAP3K, interacts with Smad proteins and interferes with osteogenesis in murine mesenchymal progenitors. J. Biol. Chem. 2005, 280, 27271–27283. [Google Scholar] [CrossRef] [Green Version]
- Benus, G.F.J.D.; Wierenga, A.T.J.; de Gorter, D.J.J.; Schuringa, J.J.; van Bennekum, A.M.; Drenth-Diephuis, L.; Vellenga, E.; Eggen, B.J.L. Inhibition of the transforming growth factor beta (TGFbeta) pathway by interleukin-1beta is mediated through TGFbeta-activated kinase 1 phosphorylation of SMAD3. Mol. Biol. Cell 2005, 16, 3501–3510. [Google Scholar] [CrossRef] [Green Version]
- Nagata, H.; Hatano, E.; Tada, M.; Murata, M.; Kitamura, K.; Asechi, H.; Narita, M.; Yanagida, A.; Tamaki, N.; Yagi, S.; et al. Inhibition of c-Jun NH2-terminal kinase switches Smad3 signaling from oncogenesis to tumor- suppression in rat hepatocellular carcinoma. Hepatology 2009, 49, 1944–1953. [Google Scholar] [CrossRef]
- Chen, D.; Lin, Q.; Box, N.; Roop, D.; Ishii, S.; Matsuzaki, K.; Fan, T.; Hornyak, T.J.; Reed, J.A.; Stavnezer, E.; et al. SKI knockdown inhibits human melanoma tumor growth in vivo. Pigment Cell Melanoma Res. 2009, 22, 761–772. [Google Scholar] [CrossRef] [PubMed]
- Hamajima, H.; Ozaki, I.; Zhang, H.; Iwane, S.; Kawaguchi, Y.; Eguchi, Y.; Matsuhashi, S.; Mizuta, T.; Matsuzaki, K.; Fujimoto, K. Modulation of the transforming growth factor-beta1-induced Smad phosphorylation by the extracellular matrix receptor beta1-integrin. Int. J. Oncol. 2009, 35, 1441–1447. [Google Scholar] [PubMed] [Green Version]
- Liu, T.; Feng, X.H. Regulation of TGF-beta signalling by protein phosphatases. Biochem. J. 2010, 430, 191–198. [Google Scholar] [CrossRef] [Green Version]
- Knockaert, M.; Sapkota, G.; Alarcon, C.; Massague, J.; Brivanlou, A.H. Unique players in the BMP pathway: Small C-terminal domain phosphatases dephosphorylate Smad1 to attenuate BMP signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 11940–11945. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Cho, E.J.; Burstein, G.; Siegel, D.; Zhang, Y. Selective inactivation of a human neuronal silencing phosphatase by a small molecule inhibitor. ACS Chem. Biol. 2011, 6, 511–519. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Otsubo, K.; Yoneda, T.; Kaneko, A.; Yagi, S.; Furukawa, K.; Chuman, Y. Development of a Substrate Identification Method for Human Scp1 Phosphatase Using Phosphorylation Mimic Phage Display. Protein Pept. Lett. 2018, 25, 76–83. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Matsuzaki, K.; Yoshida, K.; Furukawa, F.; Tahashi, Y.; Yamagata, H.; Sekimoto, G.; Seki, T.; Matsui, H.; Nishizawa, M.; et al. TGF-beta and HGF transmit the signals through JNK-dependent Smad2/3 phosphorylation at the linker regions. Oncogene 2004, 23, 7416–7429. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Neefjes, M.; van Caam, A.P.M.; van der Kraan, P.M. Transcription Factors in Cartilage Homeostasis and Osteoarthritis. Biology 2020, 9, 290. [Google Scholar] [CrossRef]
- Dreher, S.I.; Fischer, J.; Walker, T.; Diederichs, S.; Richter, W. Significance of MEF2C and RUNX3 Regulation for Endochondral Differentiation of Human Mesenchymal Progenitor Cells. Front. Cell Dev. Biol. 2020, 8, 81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kamato, D.; Do, B.H.; Osman, N.; Ross, B.P.; Mohamed, R.; Xu, S.; Little, P.J. Smad linker region phosphorylation is a signalling pathway in its own right and not only a modulator of canonical TGF-beta signalling. Cell. Mol. Life Sci. 2020, 77, 243–251. [Google Scholar] [CrossRef]
- Van de Loo, F.A.; Veenbergen, S.; van den Brand, B.; Bennink, M.B.; Blaney-Davidson, E.; Arntz, O.J.; van Beuningen, H.M.; van der Kraan, P.M.; van den Berg, W.B. Enhanced suppressor of cytokine signaling 3 in arthritic cartilage dysregulates human chondrocyte function. Arthritis Rheum. 2012, 64, 3313–3323. [Google Scholar] [CrossRef]

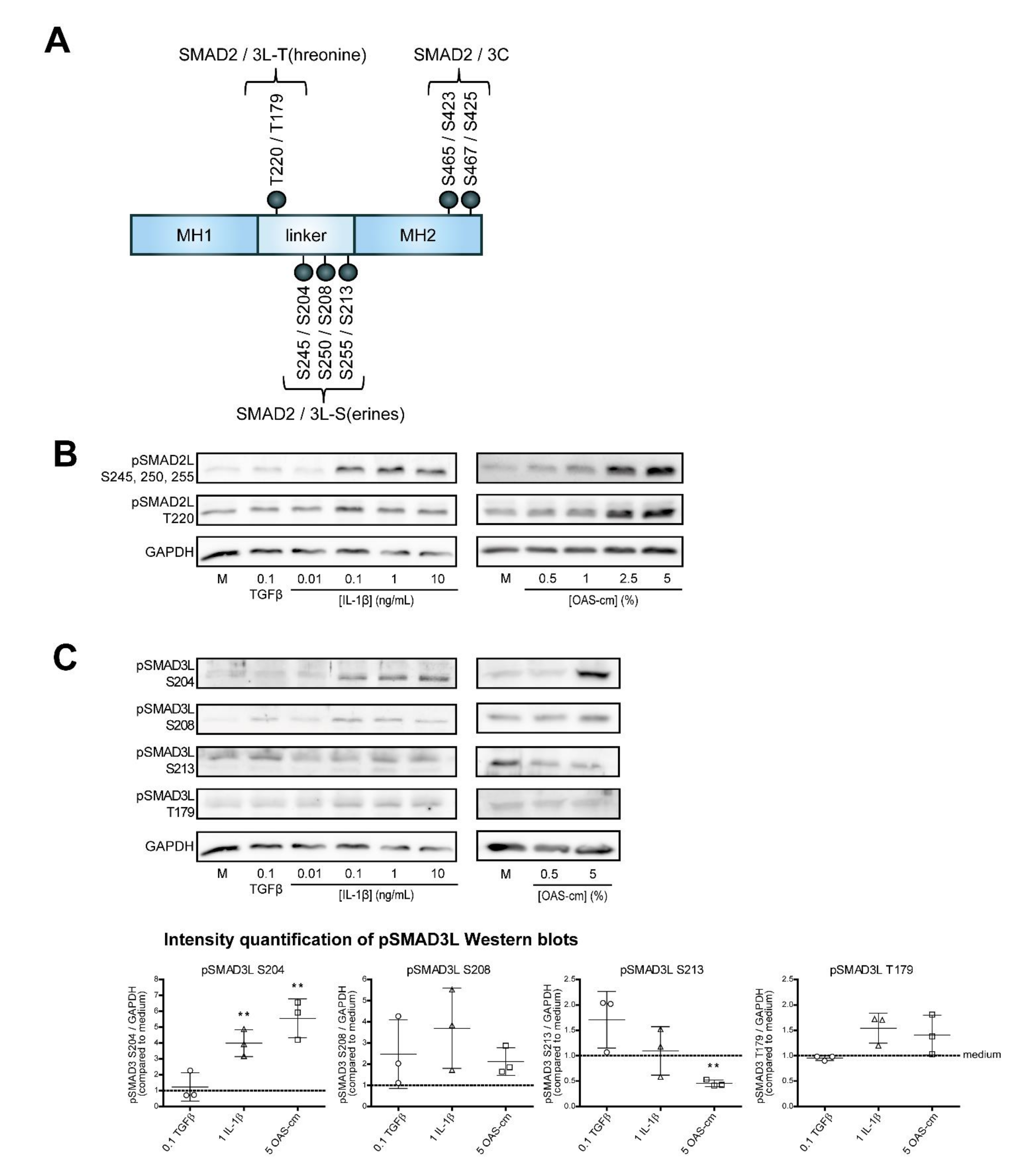
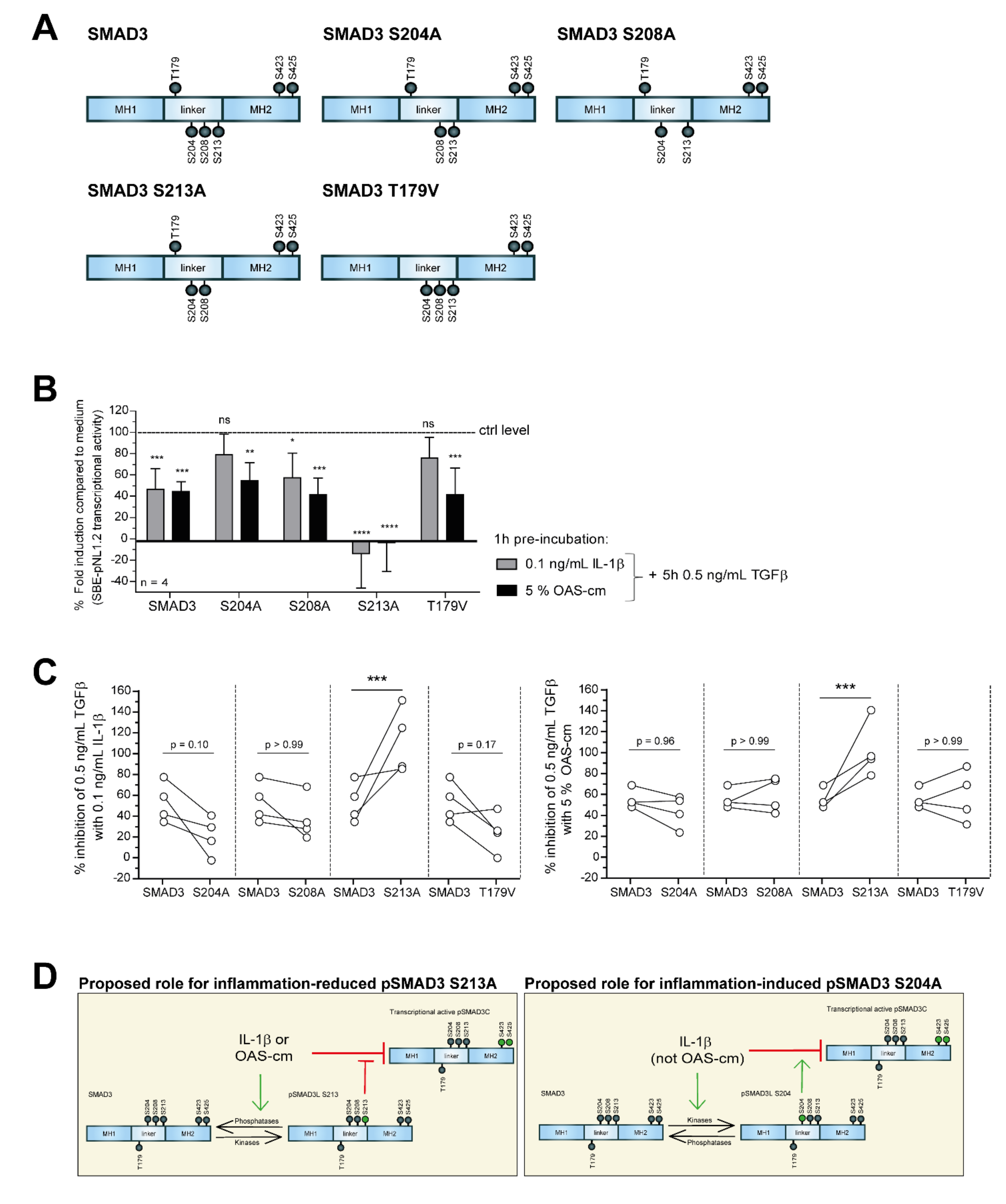

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene and Species | Forward Sequence (5′ → 3′) | Reverse Sequence (5′ → 3′) |
|---|---|---|
| bGAPDH | CACCCACGGCAAGTTCAAC | TCTCGCTCCTGGAAGATGGT |
| bRPS14 | CATCACTGCCCTCCACATCA | TTCCAATCCGCCCAATCTTCA |
| bRPL22 | GTTCGCTCACCTCCCTTTCTG | GCAGCATCCATGATTCCATCT |
| bCOL10A1 | CCATCCAACACCAAGACACAGT | TGCTCTCCTCTCAGTGATACACCTT |
| bMMP3 | AAACTCACCTCACGTACAGAATTG | TCCCAGACCGTCAGAGCTTT |
| bSMAD7 | GGGCTTTCAGATTCCCAACTT | CTCCCAGTATGCCACCACG |
| bTGFBR2 | GGCTGTCTGGAGGAAGAATGA | GTCTCTCCGGACCCCTTTCT |
| bALK5 | CAGGACCACTGCAATAAAATAGAACTT | TGCCAGTTCAACAGGACCAA |
| hGAPDH | ATCTTCTTTTGCGTCGCCAG | TTCCCCATGGTGTCTGAGC |
| hRPL22 | TCGCTCACCTCCCTTTCTAA | TCACGGTGATCTTGCTCTTG |
| hRPS27A | TGGCTGTCCTGAAATATTATAAGGT | CCCCAGCACCACATTCATCA |
| hSMAD7 | CCTTAGCCGACTCTGCGAACTA | CCAGATAATTCGTTCCCCCTGT |
| hTGFBR2 | CTGGTGCTCTGGGAAATGACA | TCGCCCTCGATCTCTCAACA |
| hALK5 | CGACGGCGTTACAGTGTTTCT | CCCATCTGTCACACAAGTAAA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Thielen, N.; Neefjes, M.; Wiegertjes, R.; van den Akker, G.; Vitters, E.; van Beuningen, H.; Blaney Davidson, E.; Koenders, M.; van Lent, P.; van de Loo, F.; et al. Osteoarthritis-Related Inflammation Blocks TGF-β’s Protective Effect on Chondrocyte Hypertrophy via (de)Phosphorylation of the SMAD2/3 Linker Region. Int. J. Mol. Sci. 2021, 22, 8124. https://doi.org/10.3390/ijms22158124
Thielen N, Neefjes M, Wiegertjes R, van den Akker G, Vitters E, van Beuningen H, Blaney Davidson E, Koenders M, van Lent P, van de Loo F, et al. Osteoarthritis-Related Inflammation Blocks TGF-β’s Protective Effect on Chondrocyte Hypertrophy via (de)Phosphorylation of the SMAD2/3 Linker Region. International Journal of Molecular Sciences. 2021; 22(15):8124. https://doi.org/10.3390/ijms22158124
Chicago/Turabian StyleThielen, Nathalie, Margot Neefjes, Renske Wiegertjes, Guus van den Akker, Elly Vitters, Henk van Beuningen, Esmeralda Blaney Davidson, Marije Koenders, Peter van Lent, Fons van de Loo, and et al. 2021. "Osteoarthritis-Related Inflammation Blocks TGF-β’s Protective Effect on Chondrocyte Hypertrophy via (de)Phosphorylation of the SMAD2/3 Linker Region" International Journal of Molecular Sciences 22, no. 15: 8124. https://doi.org/10.3390/ijms22158124