
Early Life Febrile Seizures Impair Hippocampal Synaptic Plasticity in Young Rats

 , ,
, ,
Abstract
:1. Introduction
2. Results
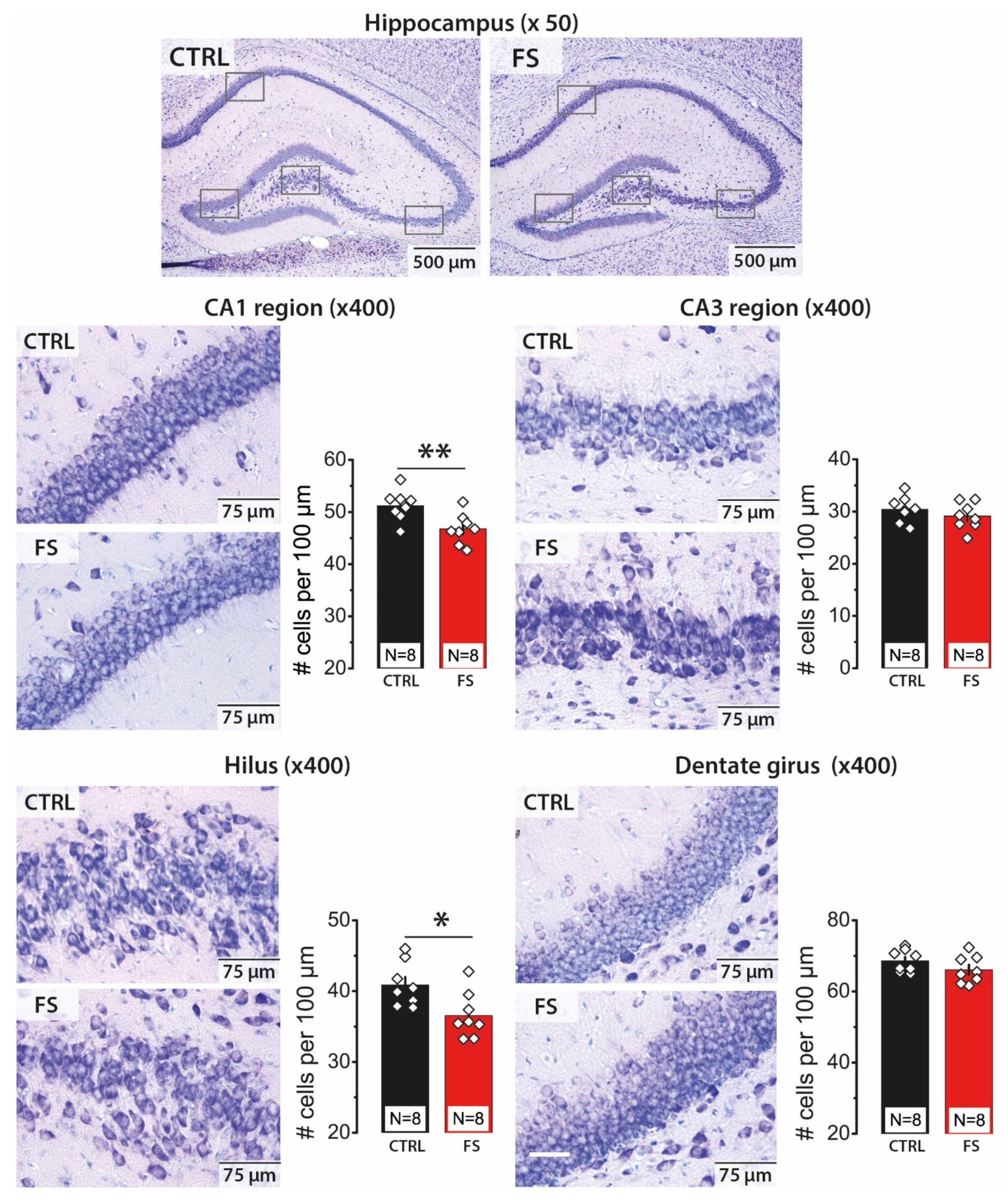
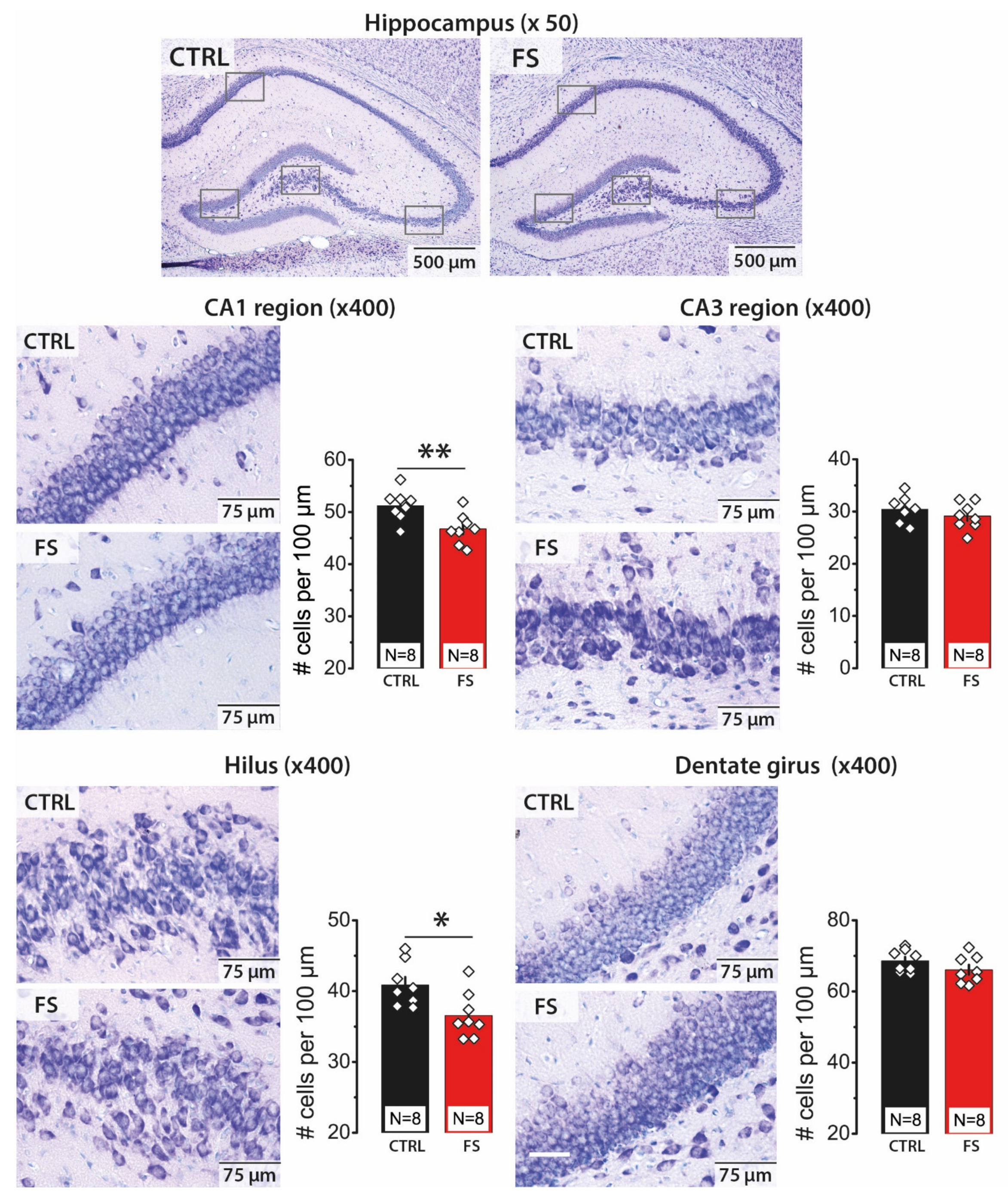
2.1. Febrile Seizures Led to a Decrease in the Number of Neurons in the CA1 Area and Hilus of the Hippocampus
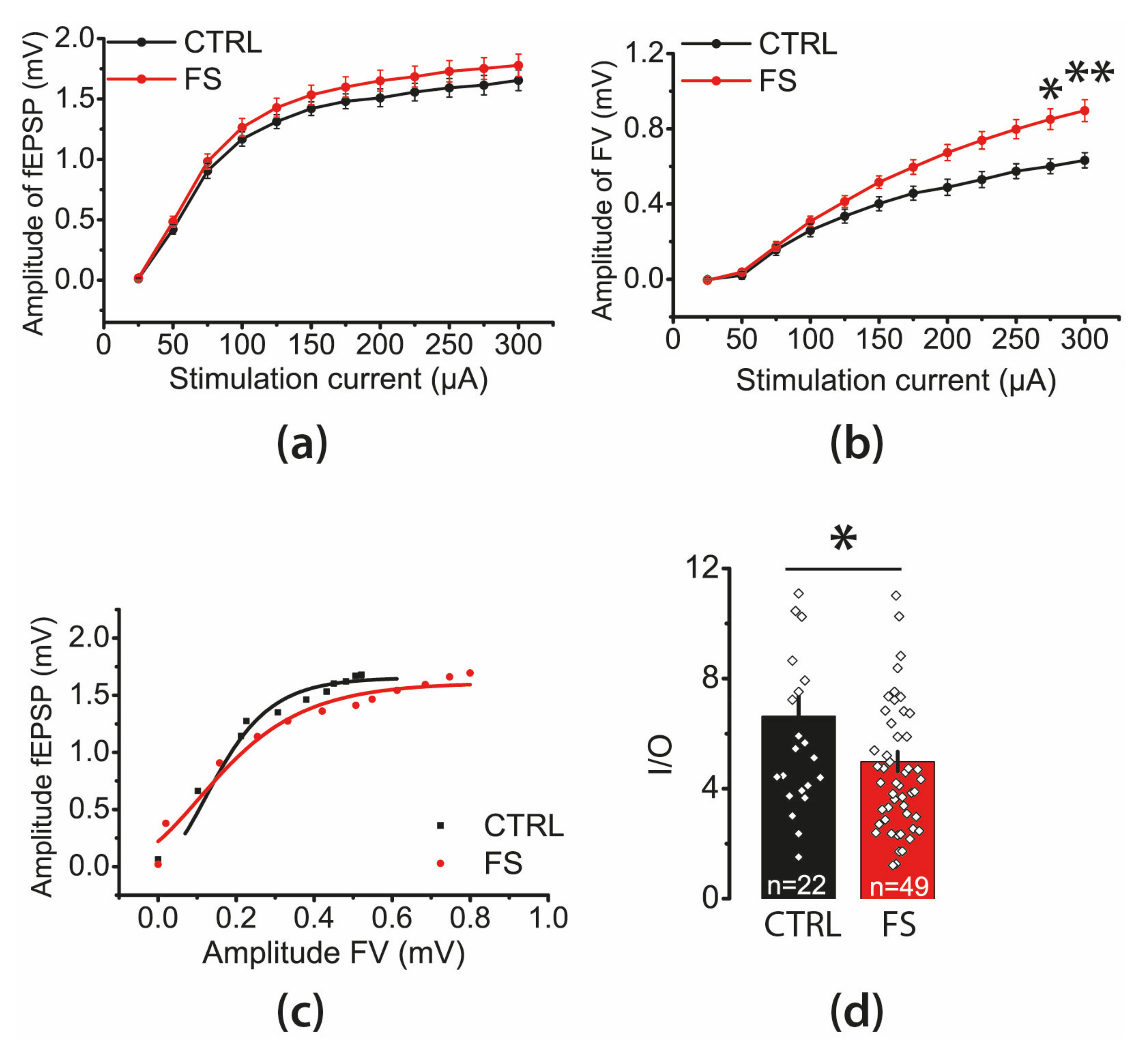
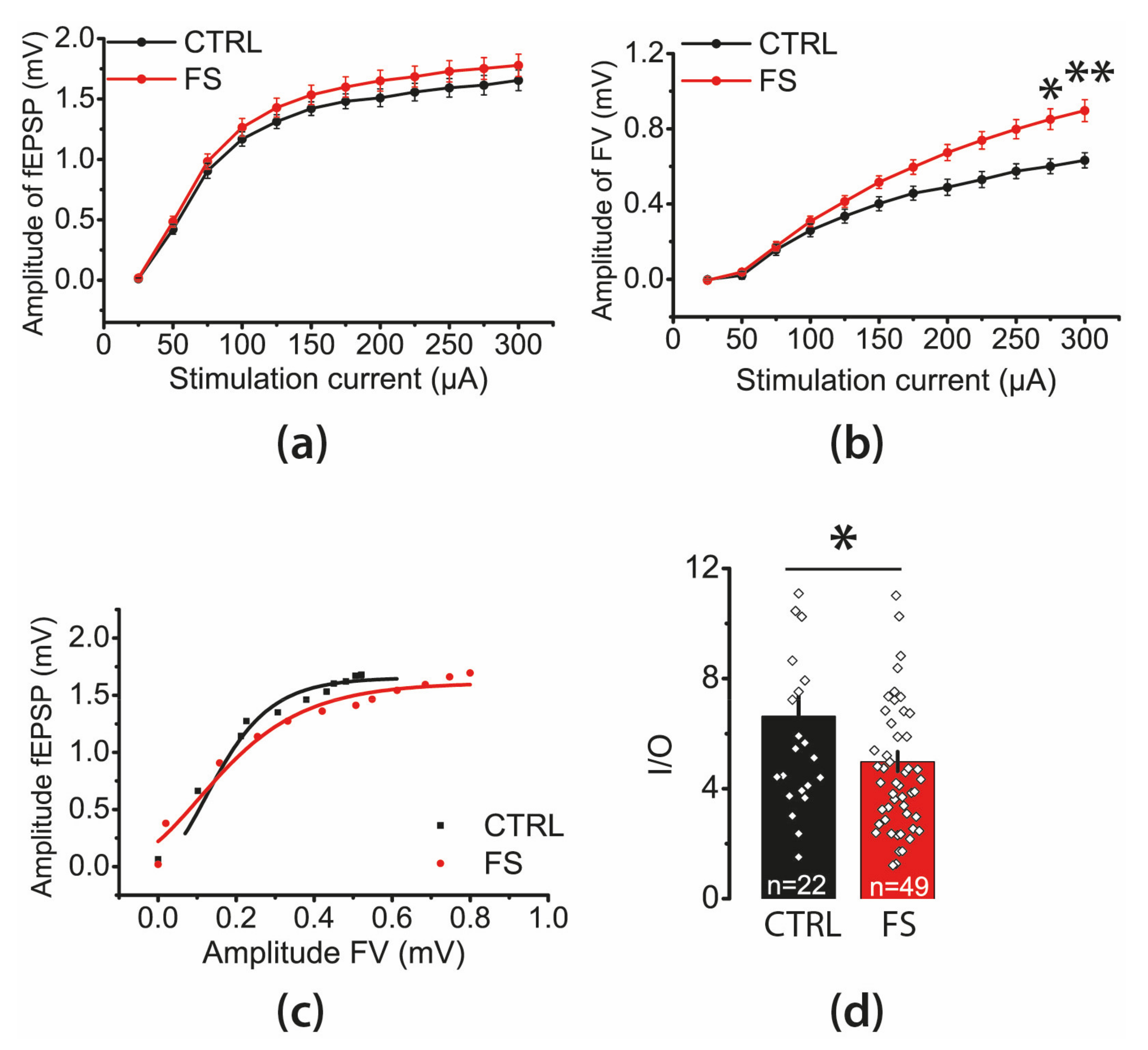
2.2. The Efficacy of Synaptic Neurotransmission at CA3-CA1 Is Reduced after FS
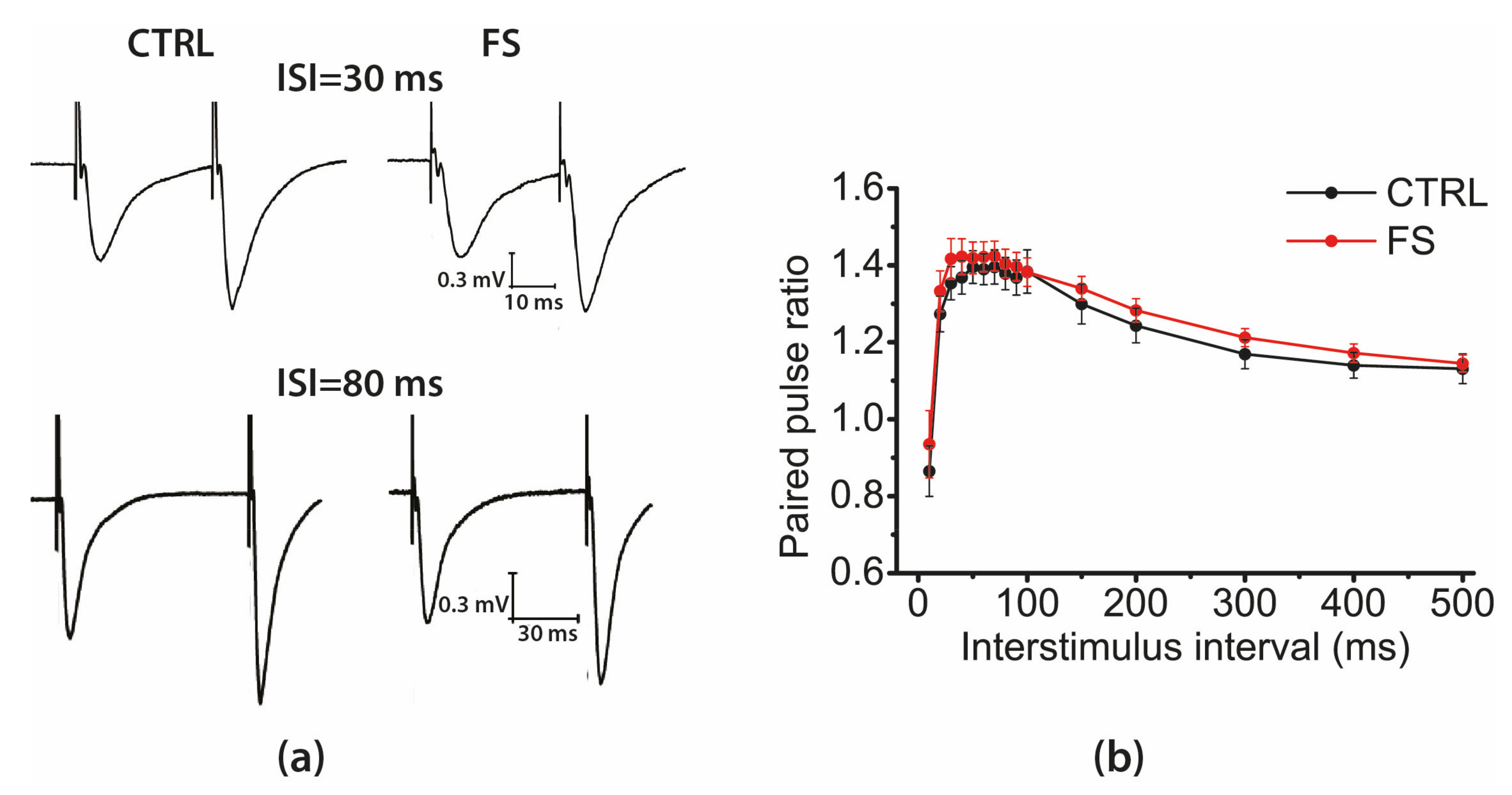
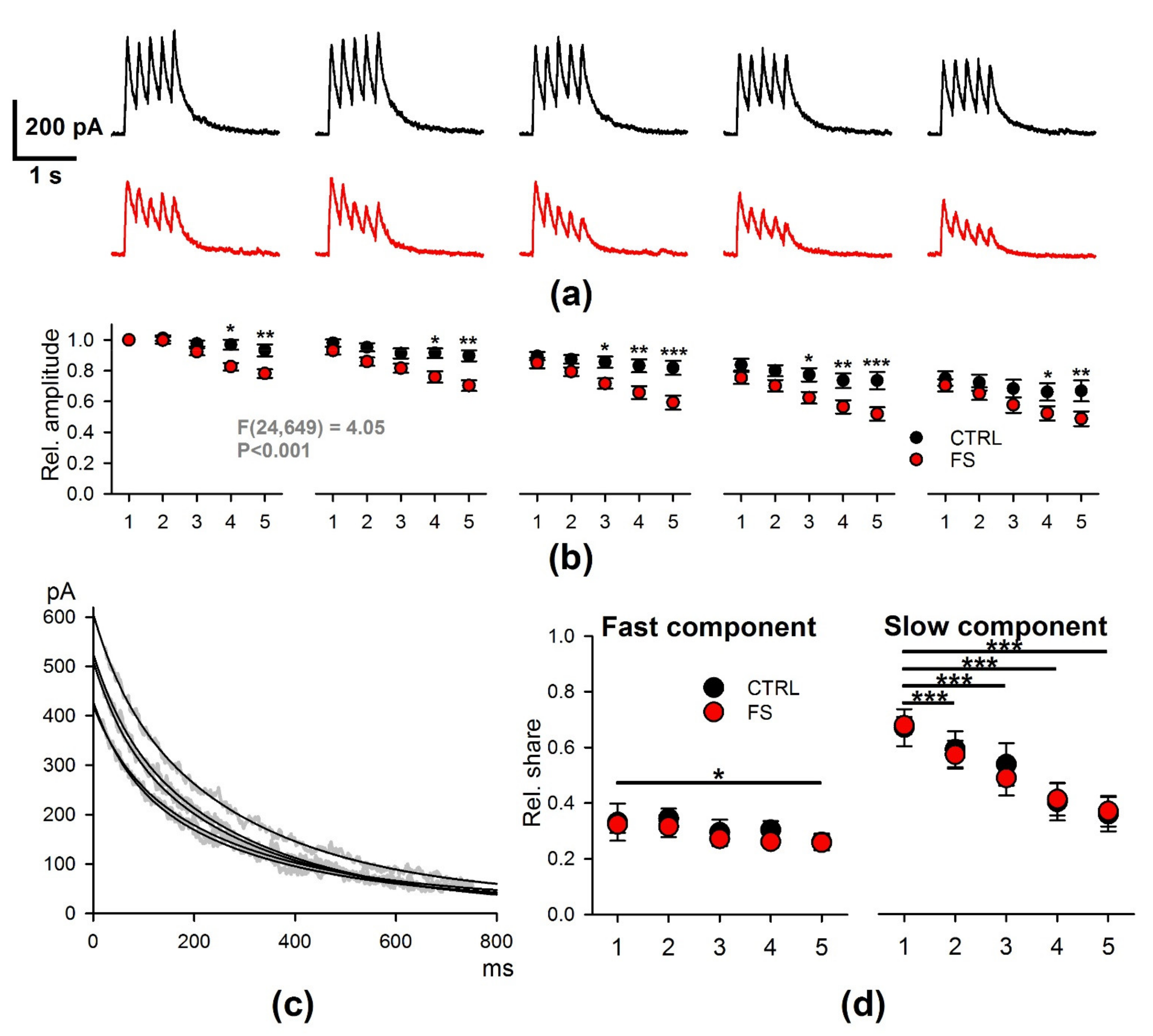
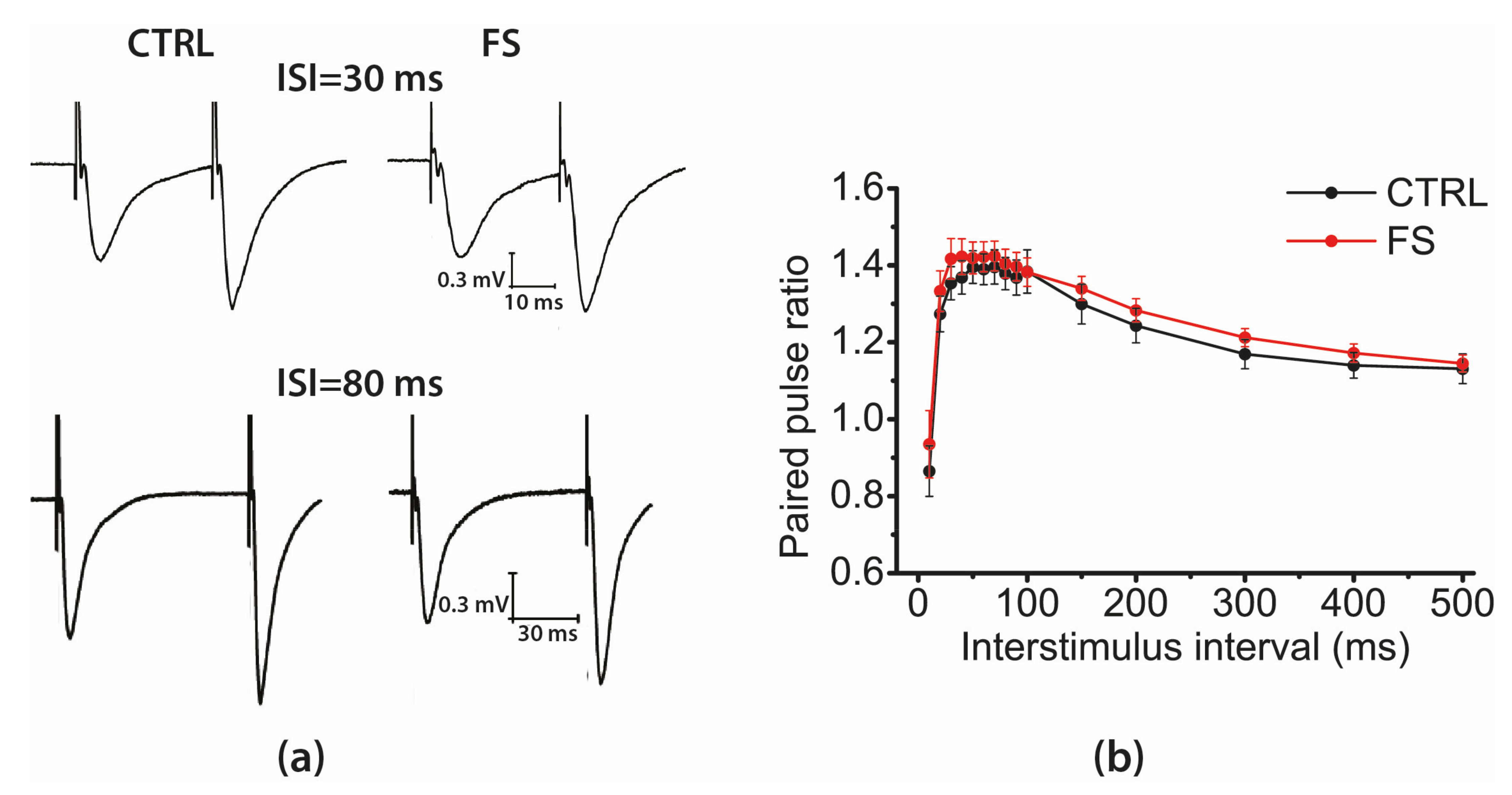
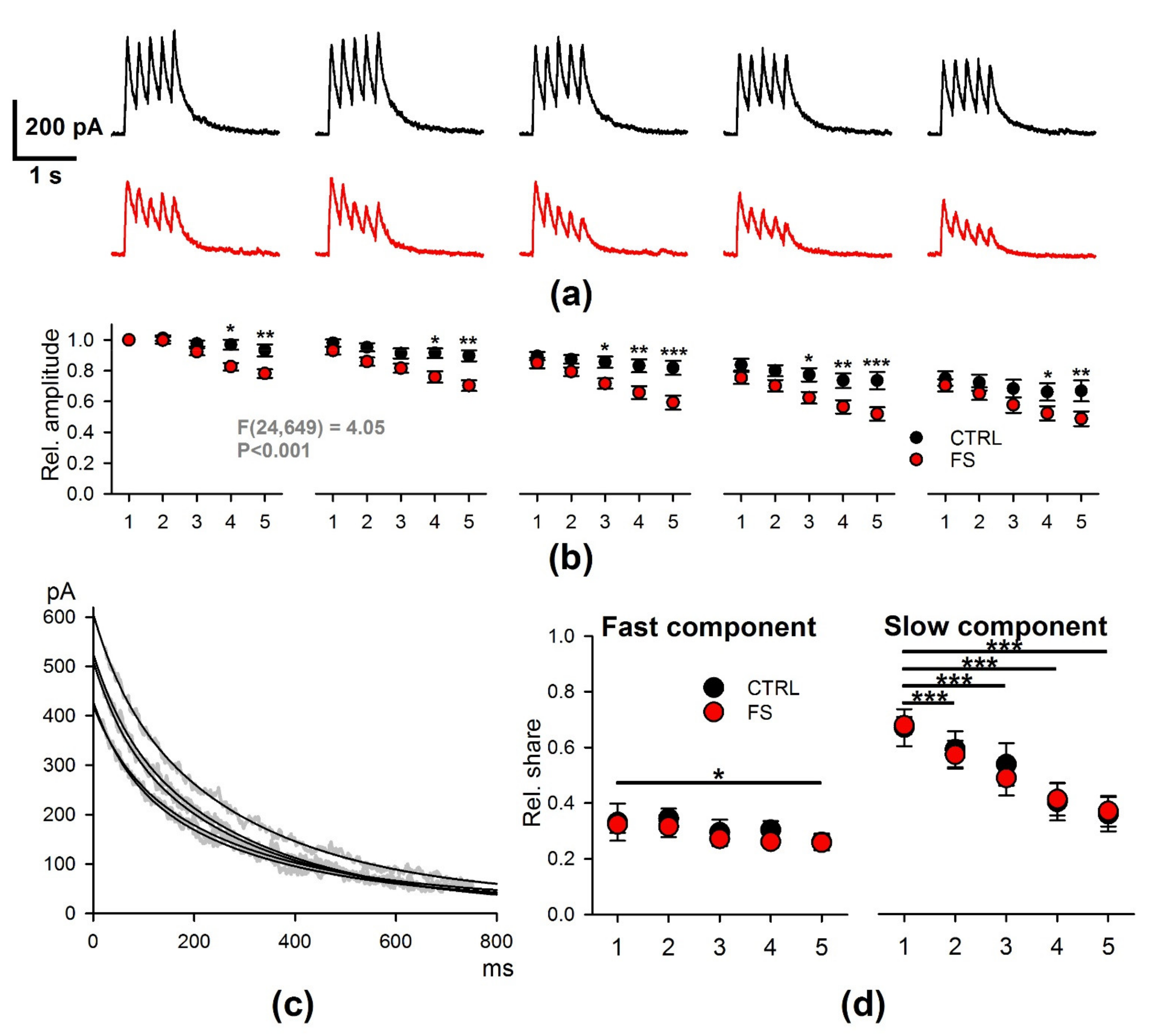
2.3. Short-Term Synaptic Plasticity of Hippocampal Neurons Does not Change in Rats that Underwent FS in Early Postnatal Ontogenesis
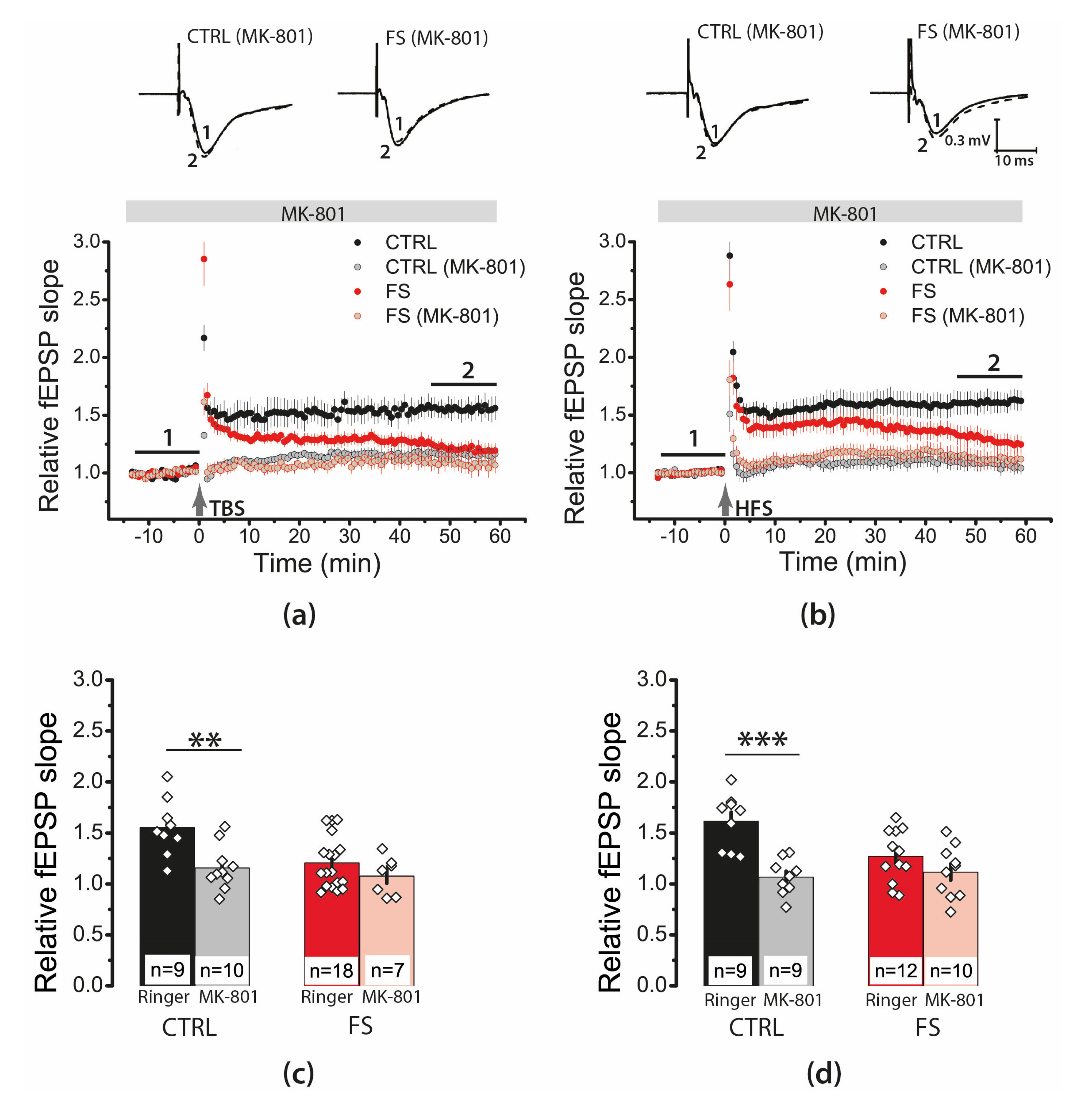
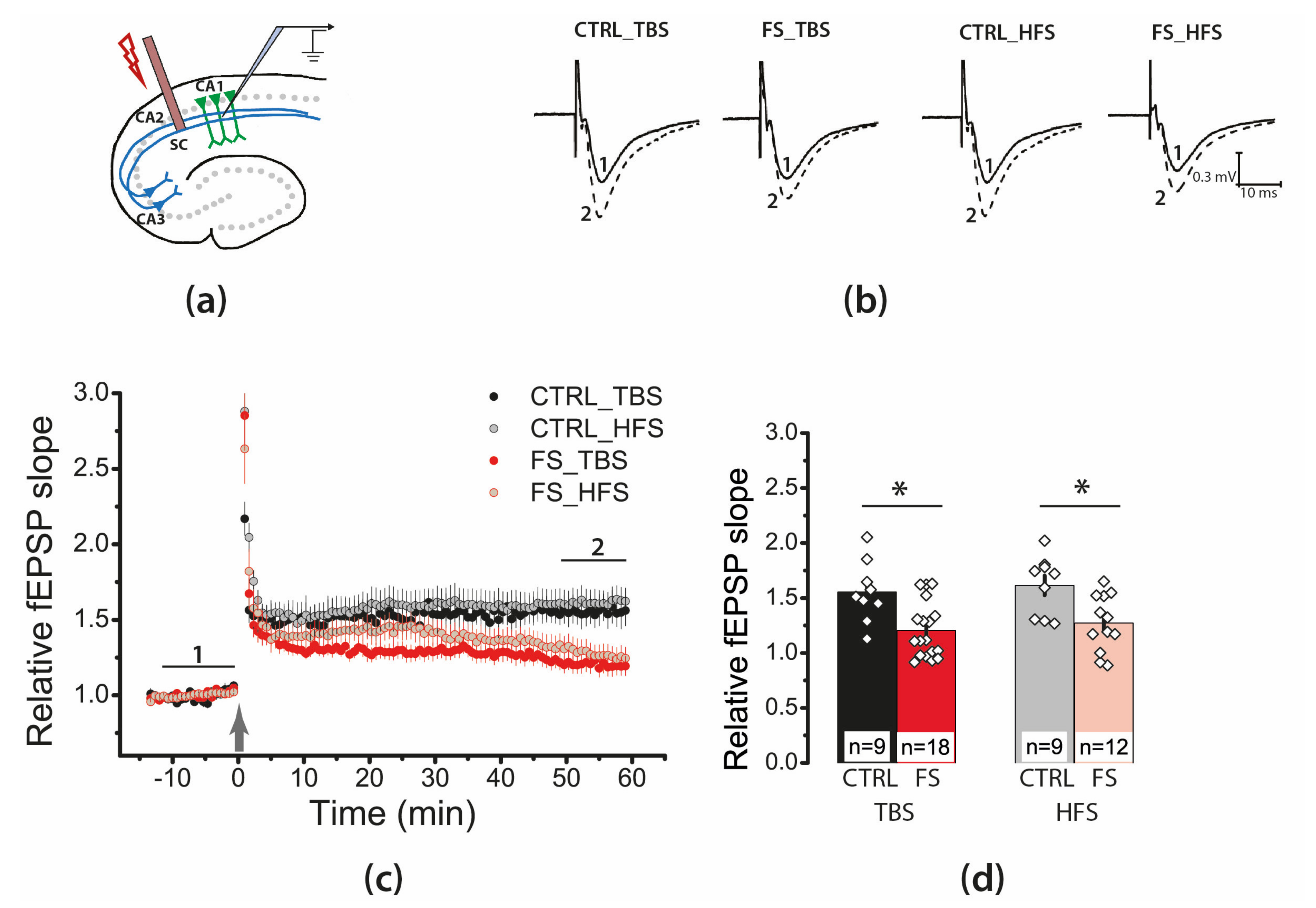
2.4. Febrile Seizures Impair Long-Term Synaptic Plasticity and Synaptic Transmission in the Rat Hippocampus
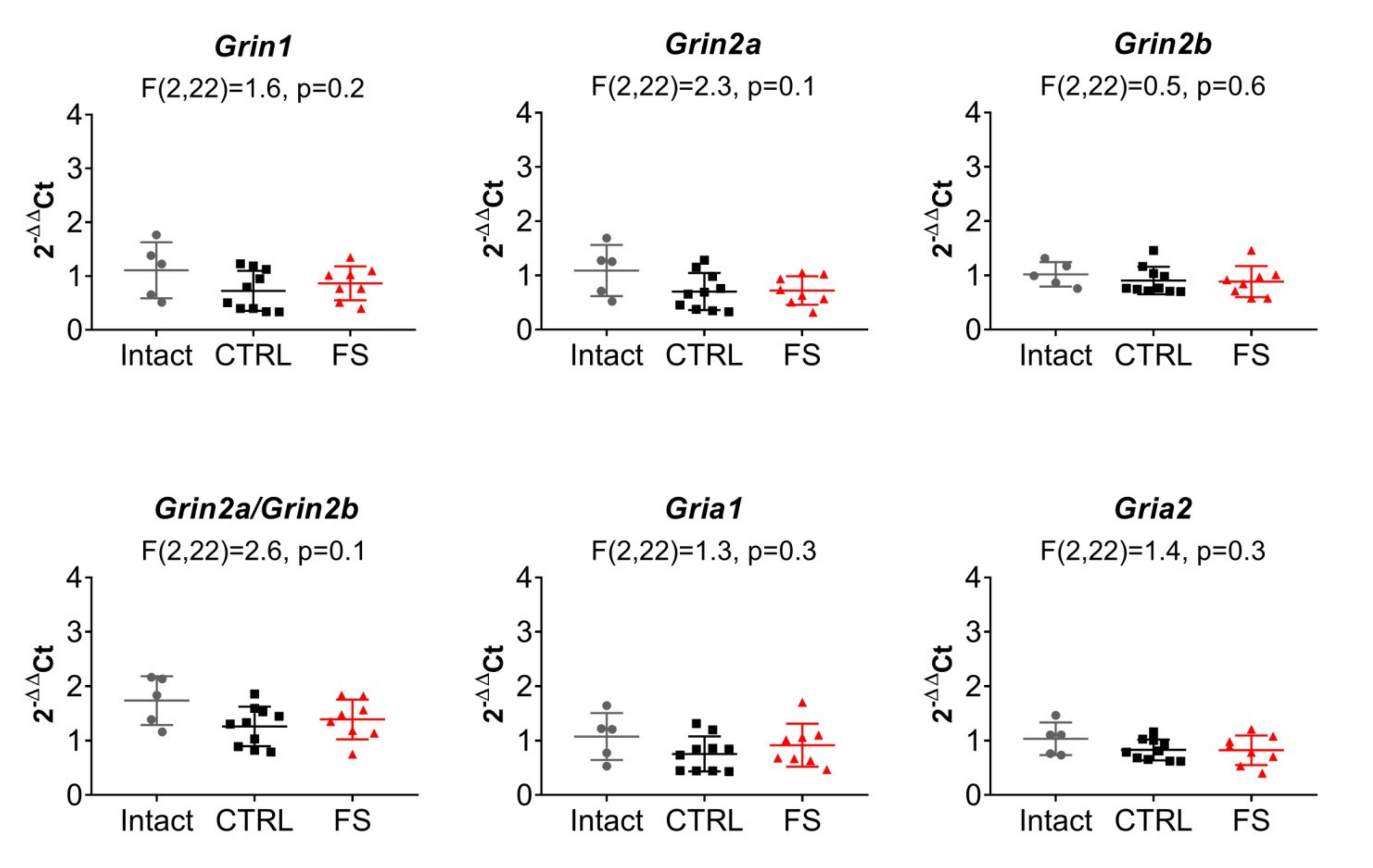
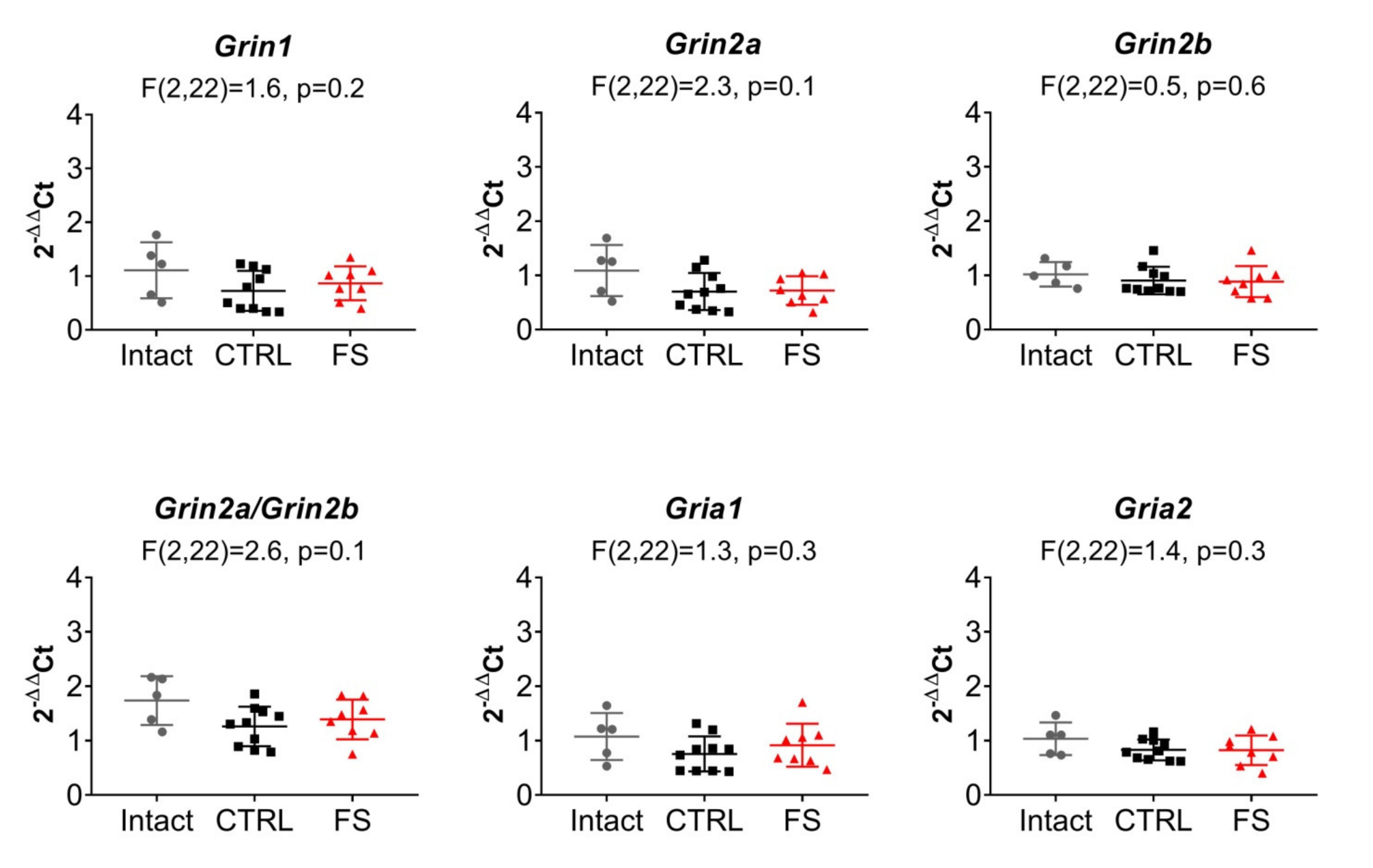
2.5. The Relative Expression of NMDAR and AMPAR Subunit Genes Does not Change after FS
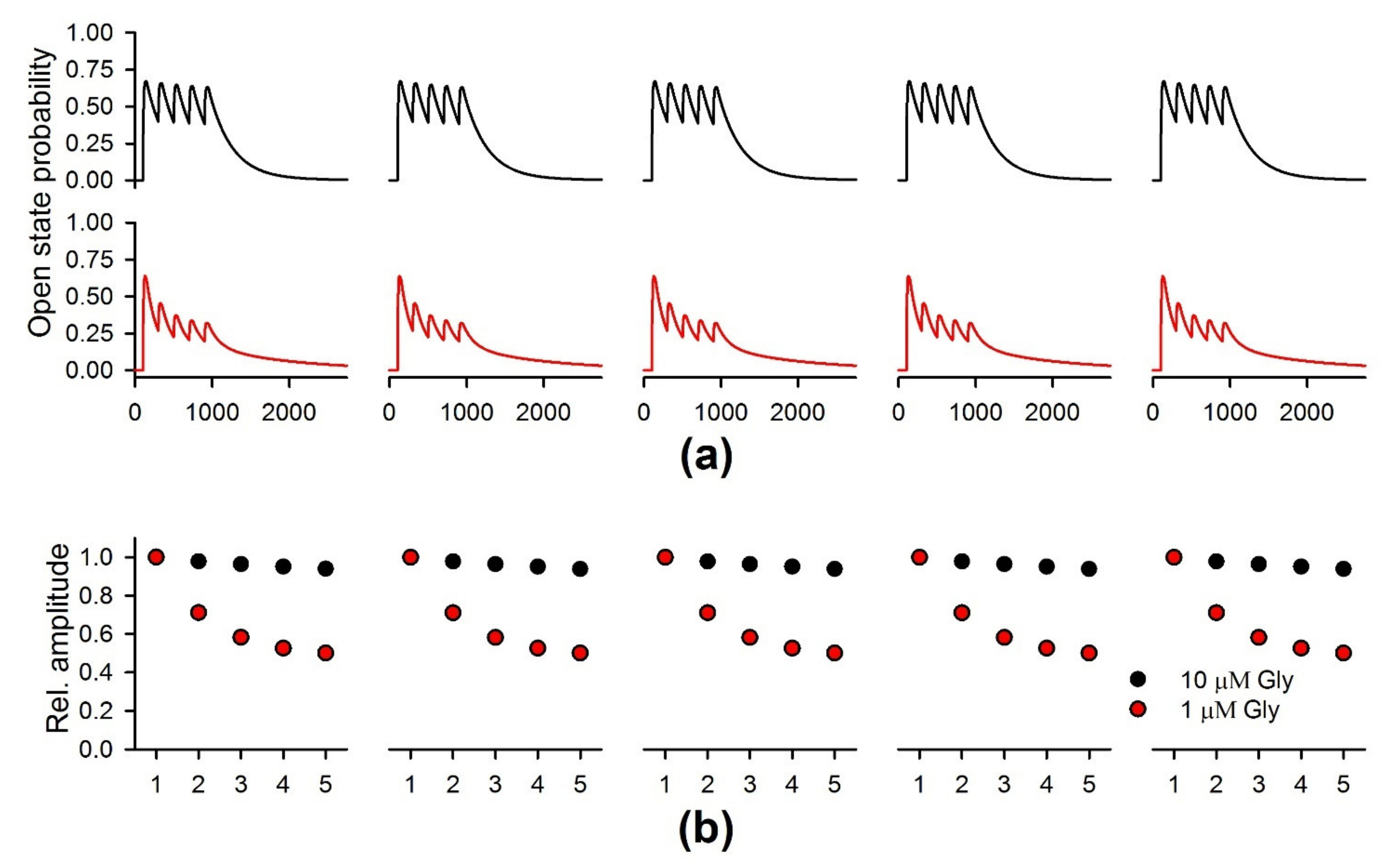
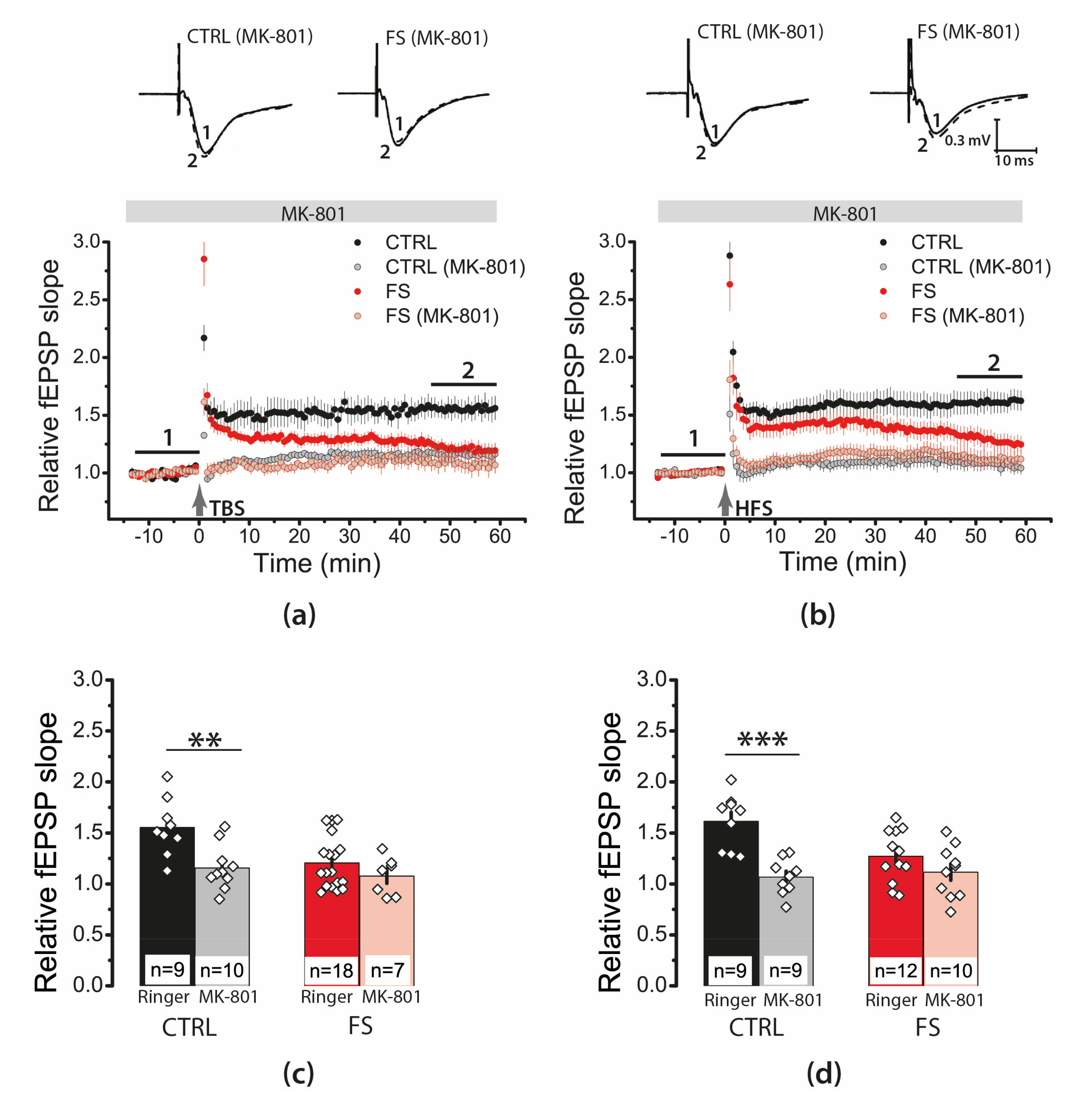
2.6. Febrile Seizures Affect the Properties of NMDAR-Mediated Synaptic Currents Evoked by TBS
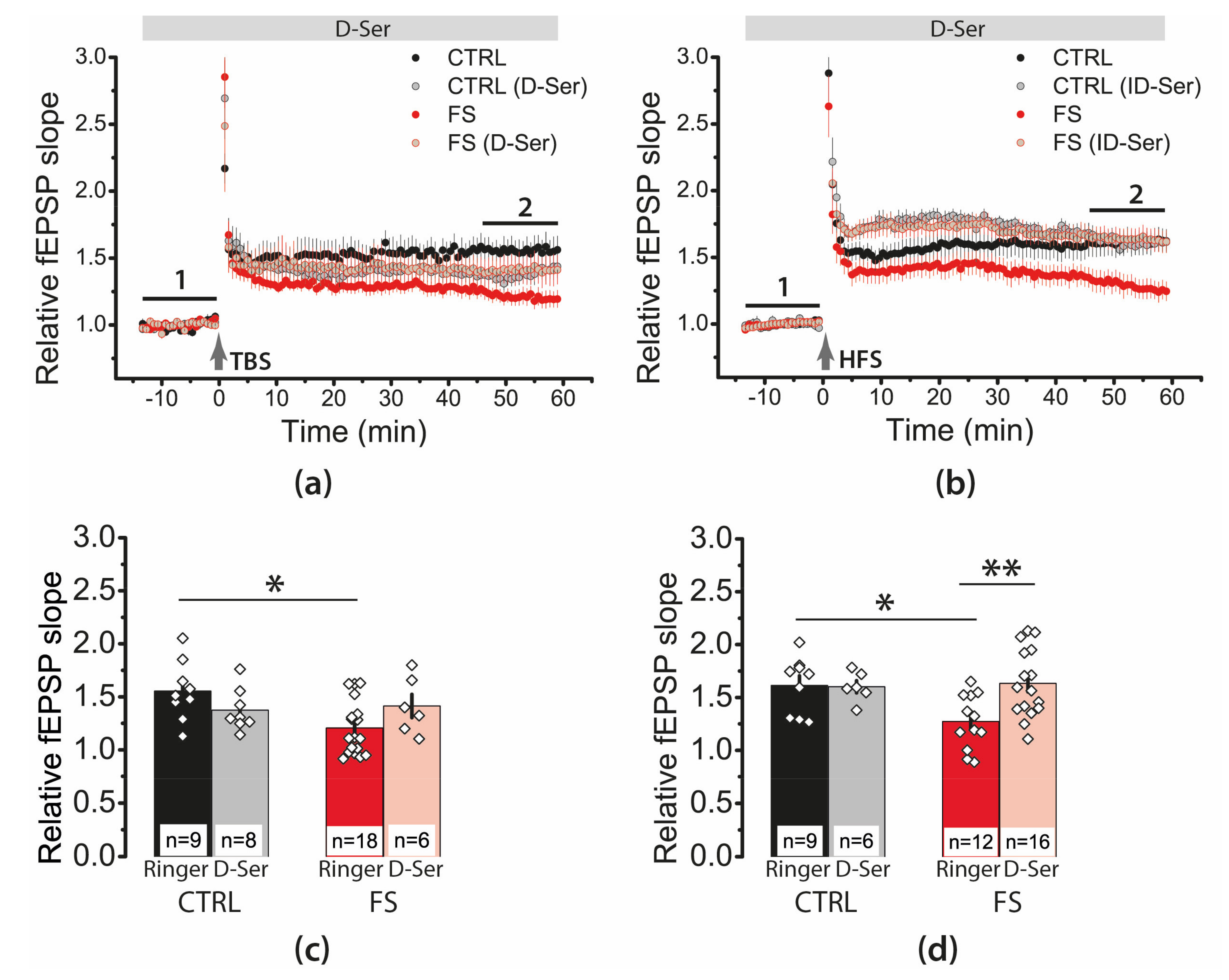
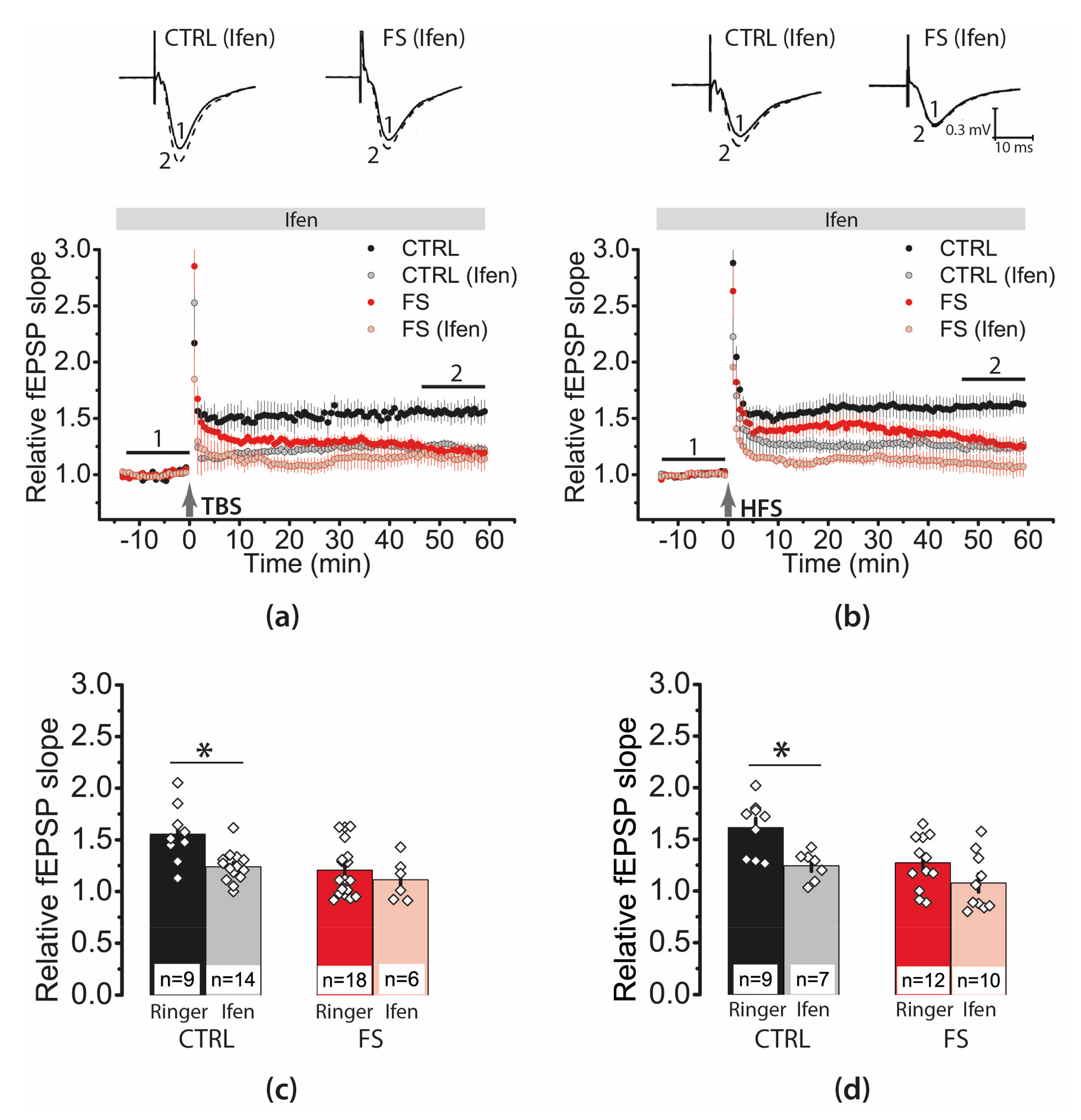
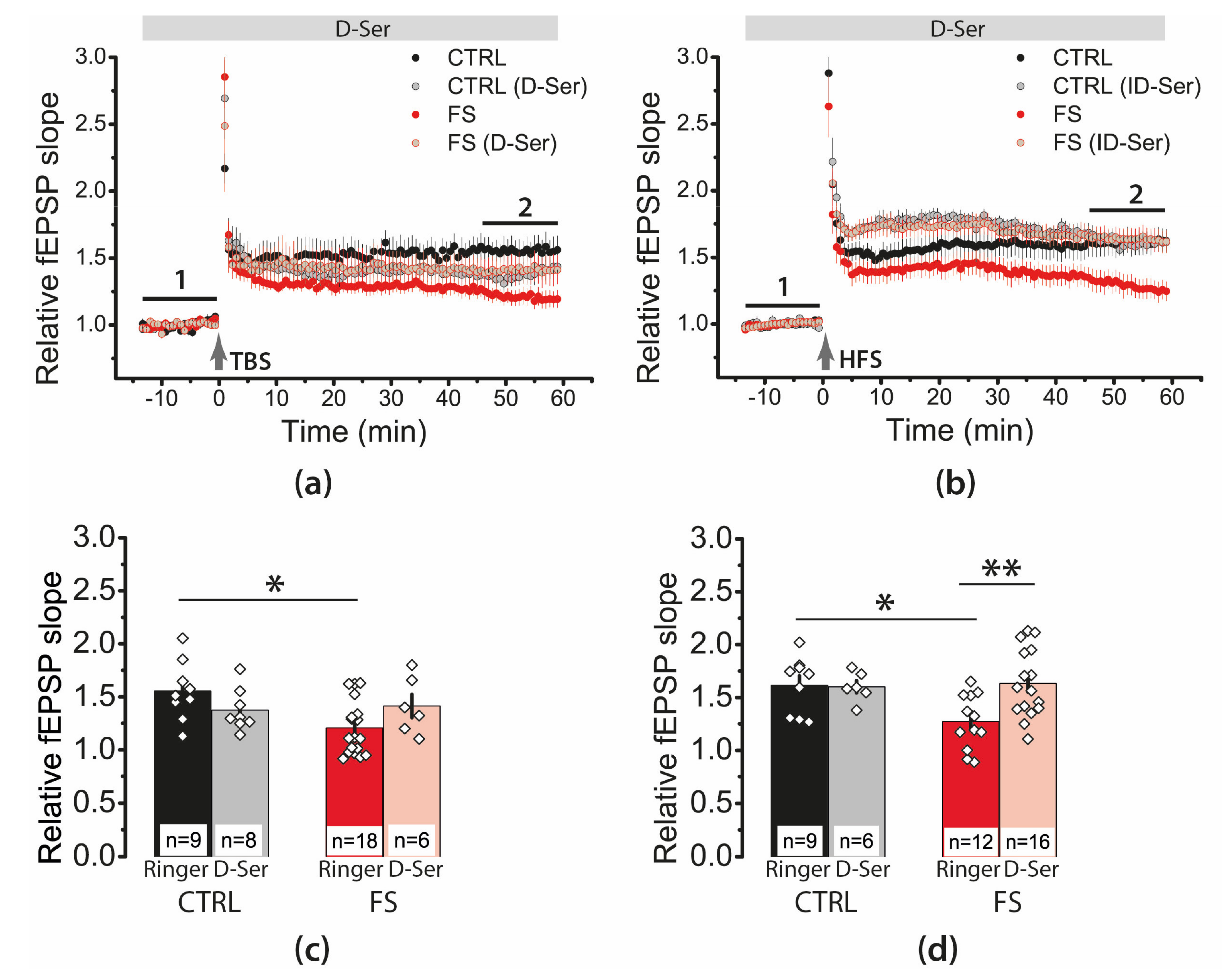
2.7. D-Serine Partially Restore LTP in the FS-Rats
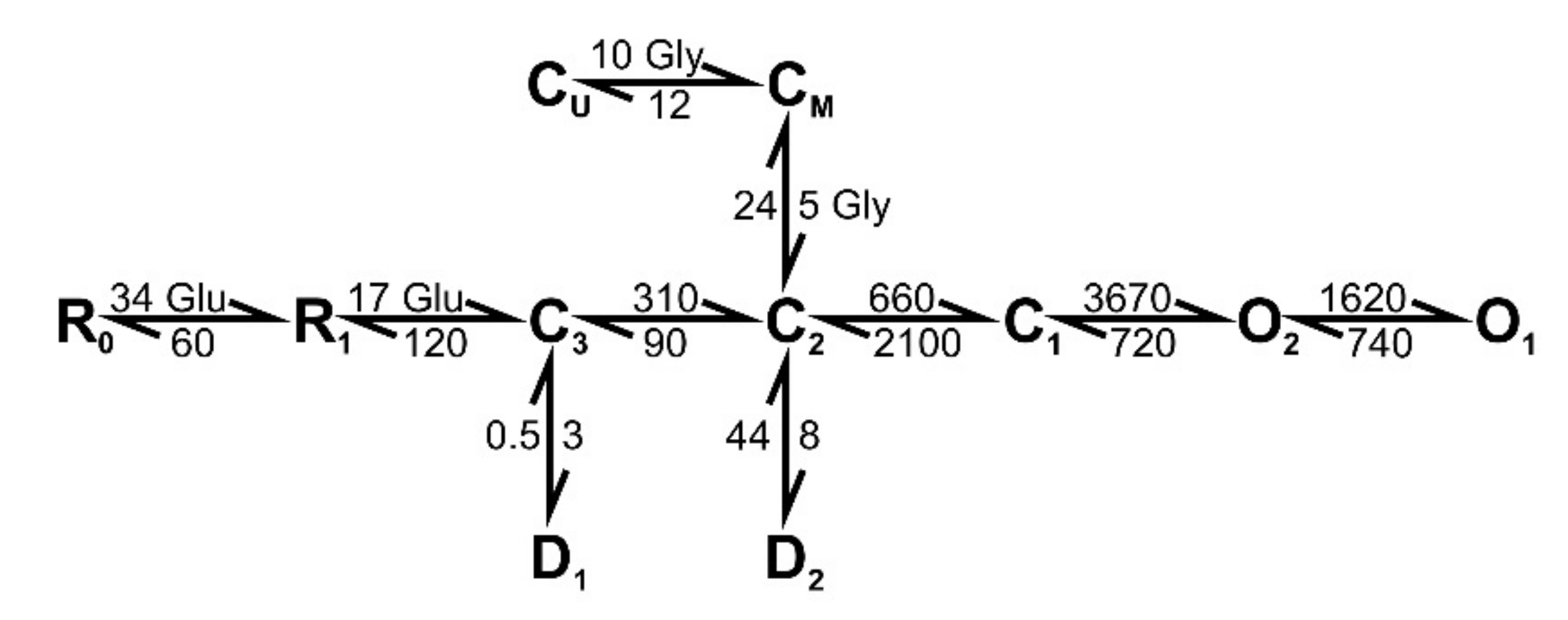
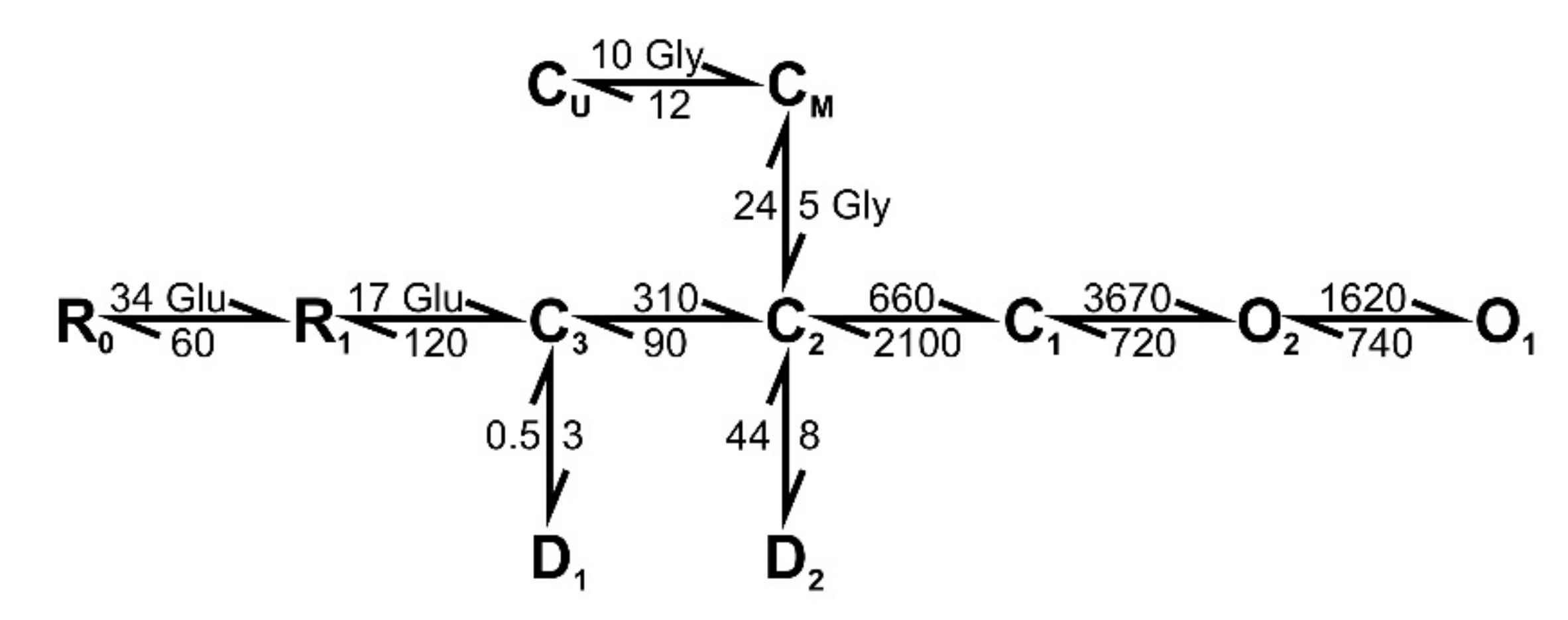
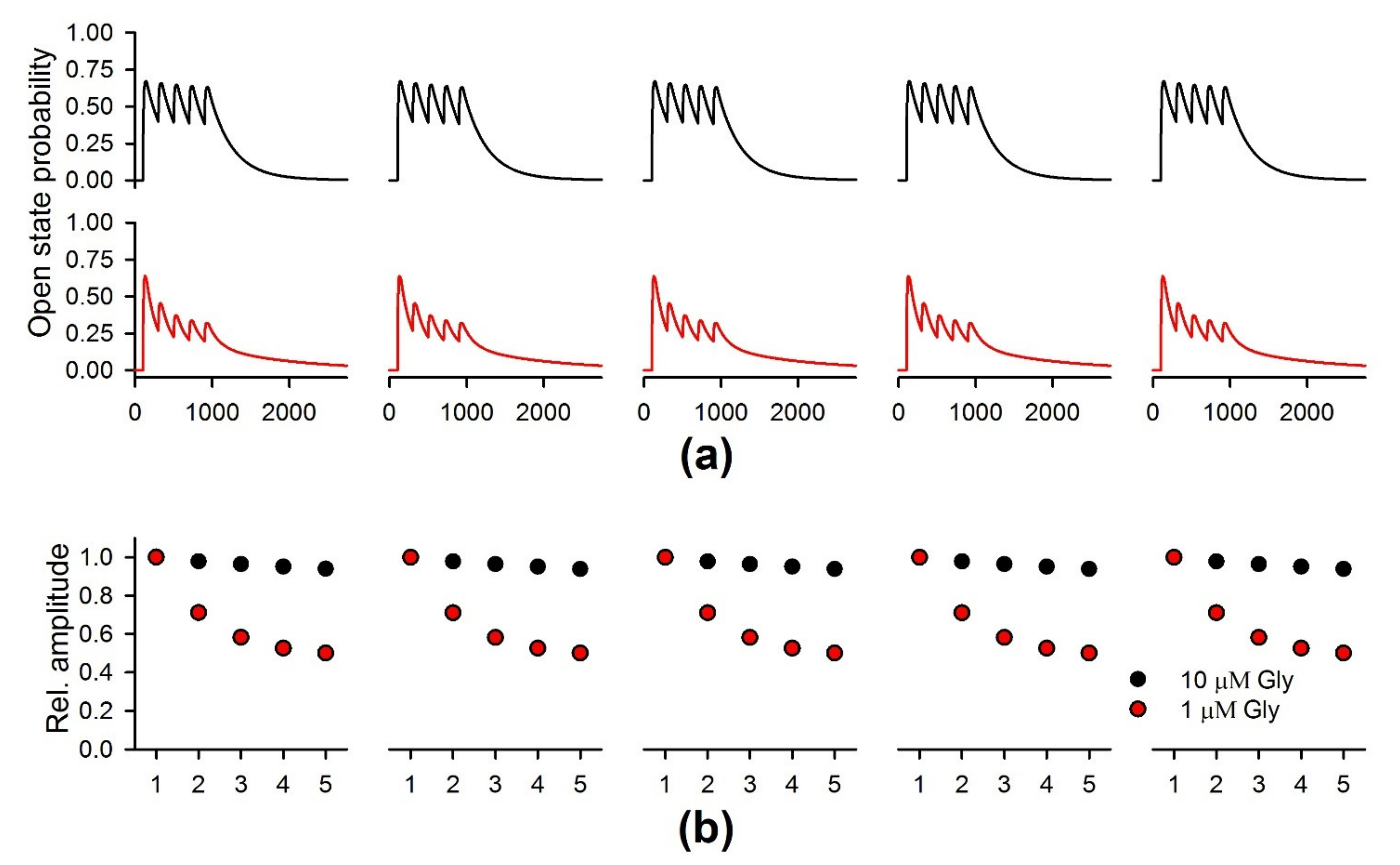
2.8. The Simulation of the NMDAR-Mediated Response to TBS
3. Discussion
4. Materials and Methods
4.1. Animals and FS Model
4.2. Brain Slices Preparation
4.3. Field Potential Recordings
4.4. The Whole-Cell Patch-Clamp Recordings
4.5. Quantitative PCR (qRT-PCR)
4.6. Histology
4.7. Simulations
4.8. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Chiu, S.S.; Tse, C.Y.; Lau, Y.L.; Peiris, M. Influenza A infection is an important cause of febrile seizures. Pediatrics 2001, 108, E63. [Google Scholar] [CrossRef] [Green Version]
- Francis, J.R.; Richmond, P.; Robins, C.; Lindsay, K.; Levy, A.; Effler, P.V.; Borland, M.; Blyth, C.C. An observational study of febrile seizures: The importance of viral infection and immunization. BMC Pediatr. 2016, 16, 202. [Google Scholar] [CrossRef] [Green Version]
- Feng, B.; Chen, Z. Generation of Febrile Seizures and Subsequent Epileptogenesis. Neurosci. Bull. 2016, 32, 481–492. [Google Scholar] [CrossRef] [Green Version]
- Millichap, J.G.J.; Millichap, J.G.J. Role of viral infections in the etiology of febrile seizures. Pediatr. Neurol. 2006, 35, 165–172. [Google Scholar] [CrossRef] [PubMed]
- Mewasingh, L.D.; Chin, R.F.M.; Scott, R.C. Current understanding of febrile seizures and their long-term outcomes. Dev. Med. Child Neurol. 2020, 62, 1245–1249. [Google Scholar] [CrossRef] [PubMed]
- Knudsen, F.U. Febrile seizures: Treatment and prognosis. Epilepsia 2000, 41, 2–9. [Google Scholar] [CrossRef] [PubMed]
- Dubé, C.M.; Ravizza, T.; Hamamura, M.; Zha, Q.; Keebaugh, A.; Fok, K.; Andres, A.L.; Nalcioglu, O.; Obenaus, A.; Vezzani, A.; et al. Epileptogenesis provoked by prolonged experimental febrile seizures: Mechanisms and biomarkers. J. Neurosci. 2010, 30, 7484–7494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lewis, D.V.; Shinnar, S.; Hesdorffer, D.C.; Bagiella, E.; Bello, J.A.; Chan, S.; Xu, Y.; MacFall, J.; Gomes, W.A.; Moshé, S.L.; et al. Hippocampal sclerosis after febrile status epilepticus: The FEBSTAT study. Ann. Neurol. 2014, 75, 178–185. [Google Scholar] [CrossRef] [Green Version]
- Cendes, F. Febrile seizures and mesial temporal sclerosis. Curr. Opin. Neurol. 2004, 17, 161–164. [Google Scholar] [CrossRef] [PubMed]
- Avishai-Eliner, S.; Brunson, K.L.; Sandman, C.A.; Baram, T.Z. Stressed-out, or in (utero)? Trends Neurosci. 2002, 25, 518–524. [Google Scholar] [CrossRef]
- Kipp, K.H.; Opitz, B.; Becker, M.; Hofmann, J.; Krick, C.; Gortner, L.; Mecklinger, A. Neural correlates of recognition memory in children with febrile seizures: Evidence from functional magnetic resonance imaging. Front. Hum. Neurosci. 2012, 6, 17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kipp, K.H.; Mecklinger, A.; Becker, M.; Reith, W.; Gortner, L. Infant febrile seizures: Changes in declarative memory as revealed by event-related potentials. Clin. Neurophysiol. Off. J. Int. Fed. Clin. Neurophysiol. 2010, 121, 2007–2016. [Google Scholar] [CrossRef]
- Martinos, M.M.; Pujar, S.; O’Reilly, H.; de Haan, M.; Neville, B.G.R.; Scott, R.C.; Chin, R.F.M. Intelligence and memory outcomes within 10 years of childhood convulsive status epilepticus. Epilepsy Behav. 2019, 95, 18–25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martinos, M.M.; Yoong, M.; Patil, S.; Chong, W.K.; Mardari, R.; Chin, R.F.M.; Neville, B.G.R.; de Haan, M.; Scott, R.C. Early developmental outcomes in children following convulsive status epilepticus: A longitudinal study. Epilepsia 2013, 54, 1012–1019. [Google Scholar] [CrossRef] [PubMed]
- Baram, T.Z.; Gerth, A.; Schultz, L. Febrile seizures: An appropriate-aged model suitable for long-term studies. Brain Res. Dev. Brain Res. 1997, 98, 265–270. [Google Scholar] [CrossRef] [Green Version]
- Bender, R.A.; Baram, T.Z. Epileptogenesis in the developing brain: What can we learn from animal models? Epilepsia 2007, 48 (Suppl. S5), 2–6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walker, M.C. Pathophysiology of status epilepticus. Neurosci. Lett. 2018, 667, 84–91. [Google Scholar] [CrossRef]
- Mihály, A. The Reactive Plasticity of Hippocampal Ionotropic Glutamate Receptors in Animal Epilepsies. Int. J. Mol. Sci. 2019, 20, 1030. [Google Scholar] [CrossRef] [Green Version]
- Toth, Z.; Yan, X.X.; Haftoglou, S.; Ribak, C.E.; Baram, T.Z. Seizure-induced neuronal injury: Vulnerability to febrile seizures in an immature rat model. J. Neurosci. 1998, 18, 4285–4294. [Google Scholar] [CrossRef] [Green Version]
- Bender, R.A.; Dubé, C.; Gonzalez-Vega, R.; Mina, E.W.; Baram, T.Z. Mossy fiber plasticity and enhanced hippocampal excitability, without hippocampal cell loss or altered neurogenesis, in an animal model of prolonged febrile seizures. Hippocampus 2003, 13, 399–412. [Google Scholar] [CrossRef] [Green Version]
- Brewster, A.; Bender, R.A.; Chen, Y.; Dube, C.; Eghbal-Ahmadi, M.; Baram, T.Z. Developmental febrile seizures modulate hippocampal gene expression of hyperpolarization-activated channels in an isoform- and cell-specific manner. J. Neurosci. 2002, 22, 4591–4599. [Google Scholar] [CrossRef]
- Dube, C.; Chen, K.; Eghbal-Ahmadi, M.; Brunson, K.; Soltesz, I.; Baram, T.Z.; Eghbal-Ahmadi, M.; Brunson, K.; Soltesz, I.; Baram, T.Z.; et al. Prolonged febrile seizures in the immature rat model enhance hippocampal excitability long term. Ann. Neurol. 2000, 47, 336–344. [Google Scholar] [CrossRef]
- Dubé, C.; Richichi, C.; Bender, R.A.; Chung, G.; Litt, B.; Baram, T.Z. Temporal lobe epilepsy after experimental prolonged febrile seizures: Prospective analysis. Brain 2006, 129, 911–922. [Google Scholar] [CrossRef] [PubMed]
- Chang, Y.-C.; Kuo, Y.-M.; Huang, A.-M.; Huang, C.-C. Repetitive febrile seizures in rat pups cause long-lasting deficits in synaptic plasticity and NR2A tyrosine phosphorylation. Neurobiol. Dis. 2005, 18, 466–475. [Google Scholar] [CrossRef] [PubMed]
- Notenboom, R.G.E.; Ramakers, G.M.J.; Kamal, A.; Spruijt, B.M.; De Graan, P.N.E. Long-lasting modulation of synaptic plasticity in rat hippocampus after early-life complex febrile seizures. Eur. J. Neurosci. 2010, 32. [Google Scholar] [CrossRef] [PubMed]
- Karnam, H.B.; Zhao, Q.; Shatskikh, T.; Holmes, G.L. Effect of age on cognitive sequelae following early life seizures in rats. Epilepsy Res. 2009, 85, 221–230. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mikati, M.A.; Zeinieh, M.P.; Kurdi, R.M.; Harb, S.A.; El Hokayem, J.A.; Daderian, R.H.; Shamseddine, A.; Obeid, M.; Bitar, F.F.; El Sabban, M. Long-term effects of acute and of chronic hypoxia on behavior and on hippocampal histology in the developing brain. Brain Res. Dev. Brain Res. 2005, 157, 98–102. [Google Scholar] [CrossRef]
- Huang, L.-T.; Yang, S.N.; Liou, C.-W.; Hung, P.-L.; Lai, M.-C.; Wang, C.-L.; Wang, T.-J. Pentylenetetrazol-induced recurrent seizures in rat pups: Time course on spatial learning and long-term effects. Epilepsia 2002, 43, 567–573. [Google Scholar] [CrossRef]
- Holmes, G.L.; Ben-Ari, Y. A single episode of neonatal seizures permanently alters glutamatergic synapses. Ann. Neurol. 2007, 61, 379–381. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.-N.; Huang, C.-B.; Yang, C.-H.; Lai, M.-C.; Chen, W.-F.; Wang, C.-L.; Wu, C.-L.; Huang, L.-T. Impaired SynGAP expression and long-term spatial learning and memory in hippocampal CA1 area from rats previously exposed to perinatal hypoxia-induced insults: Beneficial effects of A68930. Neurosci. Lett. 2004, 371, 73–78. [Google Scholar] [CrossRef]
- Lippman-Bell, J.J.; Zhou, C.; Sun, H.; Feske, J.S.; Jensen, F.E. Early-life seizures alter synaptic calcium-permeable AMPA receptor function and plasticity. Mol. Cell. Neurosci. 2016, 76, 11–20. [Google Scholar] [CrossRef] [Green Version]
- Cornejo, B.J.; Mesches, M.H.; Benke, T.A. A single early-life seizure impairs short-term memory but does not alter spatial learning, recognition memory, or anxiety. Epilepsy Behav. 2008, 13, 585–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jensen, F.E.; Wang, C.; Stafstrom, C.E.; Liu, Z.; Geary, C.; Stevens, M.C. Acute and chronic increases in excitability in rat hippocampal slices after perinatal hypoxia In vivo. J. Neurophysiol. 1998, 79, 73–81. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhou, C.; Bell, J.J.L.; Sun, H.; Jensen, F.E. Hypoxia-induced neonatal seizures diminish silent synapses and long-term potentiation in hippocampal CA1 neurons. J. Neurosci. 2011, 31, 18211–18222. [Google Scholar] [CrossRef] [Green Version]
- Tanabe, T.; Hara, K.; Shimakawa, S.; Fukui, M.; Tamai, H. Hippocampal damage after prolonged febrile seizure: One case in a consecutive prospective series. Epilepsia 2011, 52, 837–840. [Google Scholar] [CrossRef]
- Postnikova, T.Y.; Amakhin, D.V.; Trofimova, A.M.; Smolensky, I.V.; Zaitsev, A.V. Changes in Functional Properties of Rat Hippocampal Neurons Following Pentylenetetrazole-induced Status Epilepticus. Neuroscience 2019, 399, 103–116. [Google Scholar] [CrossRef]
- Zucker, R.S.; Regehr, W.G. Short-Term Synaptic Plasticity. Annu. Rev. Physiol. 2002, 64, 355–405. [Google Scholar] [CrossRef] [Green Version]
- Bliss, T.V.P.; Collingridge, G.L. A synaptic model of memory: Long-term potentiation in the hippocampus. Nature 1993, 361, 31–39. [Google Scholar] [CrossRef] [PubMed]
- Citri, A.; Malenka, R.C. Synaptic Plasticity: Multiple Forms, Functions, and Mechanisms. Neuropsychopharmacology 2008, 33, 18–41. [Google Scholar] [CrossRef] [Green Version]
- Lujan, B.; Liu, X.; Wan, Q. Differential roles of GluN2A- and GluN2B-containing NMDA receptors in neuronal survival and death. Int. J. Physiol. Pathophysiol. Pharmacol. 2012, 4, 211–218. [Google Scholar]
- Sibarov, D.A.; Antonov, S.M. Calcium-Dependent Desensitization of NMDA Receptors. Biochemistry 2018, 83, 1173–1183. [Google Scholar] [CrossRef]
- Amakhin, D.V.; Malkin, S.L.; Ergina, J.L.; Kryukov, K.A.; Veniaminova, E.A.; Zubareva, O.E.; Zaitsev, A.V. Alterations in Properties of Glutamatergic Transmission in the Temporal Cortex and Hippocampus Following Pilocarpine-Induced Acute Seizures in Wistar Rats. Front. Cell. Neurosci. 2017, 11, 264. [Google Scholar] [CrossRef]
- Benveniste, M.; Clements, J.; Vyklický, L.; Mayer, M.L. A kinetic analysis of the modulation of N-methyl-D-aspartic acid receptors by glycine in mouse cultured hippocampal neurones. J. Physiol. 1990, 428, 333–357. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.S.; Tu, Y.C.; Lai, Y.C.; Liu, E.; Yang, Y.C.; Kuo, C.C. Desensitization of NMDA channels requires ligand binding to both GluN1 and GluN2 subunits to constrict the pore beside the activation gate. J. Neurochem. 2020, 153, 549–566. [Google Scholar] [CrossRef] [PubMed]
- Henneberger, C.; Papouin, T.; Oliet, S.H.R.; Rusakov, D.A. Long-term potentiation depends on release of d-serine from astrocytes. Nature 2010. [Google Scholar] [CrossRef]
- Clasadonte, J.; Haydon, P.G. Astrocytes and epilepsy. Epilepsia 2010. [Google Scholar] [CrossRef]
- Plata, A.; Lebedeva, A.; Denisov, P.; Nosova, O.; Postnikova, T.Y.; Pimashkin, A.; Brazhe, A.; Zaitsev, A.V.; Rusakov, D.A.; Semyanov, A. Astrocytic Atrophy Following Status Epilepticus Parallels Reduced Ca2+ Activity and Impaired Synaptic Plasticity in the Rat Hippocampus. Front. Mol. Neurosci. 2018, 11, 215. [Google Scholar] [CrossRef] [PubMed]
- Rakhade, S.N.; Klein, P.M.; Huynh, T.; Hilario-Gomez, C.; Kosaras, B.; Rotenberg, A.; Jensen, F.E. Development of later life spontaneous seizures in a rodent model of hypoxia-induced neonatal seizures. Epilepsia 2011, 52, 753–765. [Google Scholar] [CrossRef] [Green Version]
- Rajab, E.; Abdeen, Z.; Hassan, Z.; Alsaffar, Y.; Mandeel, M.; Shawaaf, F.A.; Al-Ansari, S.; Kamal, A. Cognitive performance and convulsion risk after experimentally-induced febrile-seizures in rat. Int. J. Dev. Neurosci. 2014, 34, 19–23. [Google Scholar] [CrossRef]
- Rakhade, S.N.; Zhou, C.; Aujla, P.K.; Fishman, R.; Sucher, N.J.; Jensen, F.E. Early Alterations of AMPA Receptors Mediate Synaptic Potentiation Induced by Neonatal Seizures. J. Neurosci. 2008, 28, 7979–7990. [Google Scholar] [CrossRef] [Green Version]
- Jiang, X.; Mu, D.; Biran, V.; Faustino, J.; Chang, S.; Rincón, C.M.; Ann Sheldon, R.; Ferriero, D.M. Activated Src Kinases Interact with the N-methyl-D-aspartate Receptor after Neonatal Brain Ischemia. Ann Neurol 2008, 63, 632–641. [Google Scholar] [CrossRef] [PubMed]
- Zhou, C.; Sun, H.; Klein, P.M.; Jensen, F.E. Neonatal seizures alter NMDA glutamate receptor GluN2A and 3A subunit expression and function in hippocampal CA1 neurons. Front. Cell. Neurosci. 2015, 9, 362. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanchez, R.M.; Koh, S.; Rio, C.; Wang, C.; Lamperti, E.D.; Sharma, D.; Corfas, G.; Jensen, F.E. Decreased glutamate receptor 2 expression and enhanced epileptogenesis in immature rat hippocampus after perinatal hypoxia-induced seizures. J. Neurosci. 2001, 21, 8154–8163. [Google Scholar] [CrossRef]
- Di Maio, R.; Mastroberardino, P.G.; Hu, X.; Montero, L.; Greenamyre, J.T. Pilocapine alters NMDA receptor expression and function in hippocampal neurons: NADPH oxidase and ERK1/2 mechanisms. Neurobiol. Dis. 2011, 42, 482–495. [Google Scholar] [CrossRef]
- Di Maio, R.; Mastroberardino, P.G.; Hu, X.; Montero, L.M.; Greenamyre, J.T. Thiol oxidation and altered NR2B/NMDA receptor functions in in vitro and in vivo pilocarpine models: Implications for epileptogenesis. Neurobiol. Dis. 2013, 49, 87–98. [Google Scholar] [CrossRef]
- Zhu, X.; Dong, J.; Shen, K.; Bai, Y.; Zhang, Y.; Lv, X.; Chao, J.; Yao, H. NMDA receptor NR2B subunits contribute to PTZ-kindling-induced hippocampal astrocytosis and oxidative stress. Brain Res. Bull. 2015, 114, 70–78. [Google Scholar] [CrossRef]
- Chen, B.; Feng, B.; Tang, Y.; You, Y.; Wang, Y.; Hou, W.; Hu, W.; Chen, Z. Blocking GluN2B subunits reverses the enhanced seizure susceptibility after prolonged febrile seizures with a wide therapeutic time-window. Exp. Neurol. 2016, 283, 29–38. [Google Scholar] [CrossRef]
- Cummings, K.A.; Popescu, G.K. Glycine-dependent activation of NMDA receptors. J. Gen. Physiol. 2015, 145, 513–527. [Google Scholar] [CrossRef] [Green Version]
- Harsing, L.G.; Matyus, P. Mechanisms of glycine release, which build up synaptic and extrasynaptic glycine levels: The role of synaptic and non-synaptic glycine transporters. Brain Res. Bull. 2013, 93, 110–119. [Google Scholar] [CrossRef]
- Zafra, F.; Ibáñez, I.; Bartolomé-Martín, D.; Piniella, D.; Arribas-Blázquez, M.; Giménez, C. Glycine Transporters and Its Coupling with NMDA Receptors. In Advances in Neurobiology; Springer International Publishing AG: New York, NY, USA, 2017; Volume 16, pp. 55–83. [Google Scholar]
- Shen, H.-Y.; van Vliet, E.A.; Bright, K.-A.; Hanthorn, M.; Lytle, N.K.; Gorter, J.; Aronica, E.; Boison, D. Glycine transporter 1 is a target for the treatment of epilepsy. Neuropharmacology 2015, 99, 554–565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eichler, S.A.; Kirischuk, S.; Jüttner, R.; Schafermeier, P.K.; Legendre, P.; Lehmann, T.N.; Gloveli, T.; Grantyn, R.; Meier, J.C. Glycinergic tonic inhibition of hippocampal neurons with depolarizing GABAergic transmission elicits histopathological signs of temporal lobe epilepsy. J. Cell. Mol. Med. 2008, 12, 2848–2866. [Google Scholar] [CrossRef] [Green Version]
- Khan, D.; Dupper, A.; Deshpande, T.; Graan, P.N.E.D.; Steinhäuser, C.; Bedner, P. Experimental febrile seizures impair interastrocytic gap junction coupling in juvenile mice. J. Neurosci. Res. 2016, 94, 804–813. [Google Scholar] [CrossRef]
- Yang, L.; Li, F.; Zhang, H.; Ge, W.; Mi, C.; Sun, R.; Liu, C. Astrocyte activation and memory impairment in the repetitive febrile seizures model. Epilepsy Res. 2009, 86, 209–220. [Google Scholar] [CrossRef]
- Łotowska, J.M.; Sobaniec-Łotowska, M.E.; Sobaniec, W. Ultrastructural features of astrocytes in the cortex of the hippocampal gyrus and in the neocortex of the temporal lobe in an experimental model of febrile seizures and with the use of topiramate. Folia Neuropathol. 2009, 47, 268–277. [Google Scholar]
- Chapman, K.E.; Raol, Y.H.; Brooks-Kayal, A. Neonatal seizures: Controversies and challenges in translating new therapies from the lab to the isolette. Eur. J. Neurosci. 2012, 35, 1857–1865. [Google Scholar] [CrossRef]
- Dubé, C.; Brunson, K.L.; Eghbal-Ahmadi, M.; Gonzalez-Vega, R.; Baram, T.Z. Endogenous neuropeptide Y prevents recurrence of experimental febrile seizures by increasing seizure threshold. J. Mol. Neurosci. 2005, 25, 275–284. [Google Scholar] [CrossRef] [Green Version]
- Postnikova, T.Y.; Trofimova, A.M.; Ergina, J.L.; Zubareva, O.E.; Kalemenev, S.V.; Zaitsev, A.V. Transient Switching of NMDA-Dependent Long-Term Synaptic Potentiation in CA3-CA1 Hippocampal Synapses to mGluR1-Dependent Potentiation After Pentylenetetrazole-Induced Acute Seizures in Young Rats. Cell. Mol. Neurobiol. 2019, 39, 287–300. [Google Scholar] [CrossRef]
- Paxinos, G.; Watson, C. The Rat Brain in Stereotaxic Coordinates; Elsevier: Amsterdam, The Netherlands, 2007. [Google Scholar]
- Livak, K.J.J.; Schmittgen, T.D.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2-ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Swijsen, A.; Nelissen, K.; Janssen, D.; Rigo, J.M.; Hoogland, G. Validation of reference genes for quantitative real-time PCR studies in the dentate gyrus after experimental febrile seizures. BMC Res. Notes 2012, 5, 685. [Google Scholar] [CrossRef] [Green Version]
- Malkin, S.L.; Amakhin, D.V.; Veniaminova, E.A.; Kim, K.K.; Zubareva, O.E.; Magazanik, L.G.; Zaitsev, A. V Changes of ampa receptor properties in the neocortex and hippocampus following pilocarpine-induced status epilepticus in rats. Neuroscience 2016, 327, 146–155. [Google Scholar] [CrossRef]
- Giza, C.C.; Maria, N.S.S.; Hovda, D.A. N-methyl-D-aspartate receptor subunit changes after traumatic injury to the developing brain. J. Neurotrauma 2006, 23, 950–961. [Google Scholar] [CrossRef] [Green Version]
- Floyd, D.W.; Jung, K.-Y.; McCool, B.A. Chronic ethanol ingestion facilitates N-methyl-D-aspartate receptor function and expression in rat lateral/basolateral amygdala neurons. J. Pharmacol. Exp. Ther. 2003, 307, 1020–1029. [Google Scholar] [CrossRef] [PubMed]
- Proudnikov, D.; Yuferov, V.; Zhou, Y.; LaForge, K.S.; Ho, A.; Kreek, M.J. Optimizing primer--probe design for fluorescent PCR. J. Neurosci. Methods 2003, 123, 31–45. [Google Scholar] [CrossRef]
- Popescu, G.; Robert, A.; Howe, J.R.; Auerbach, A. Reaction mechanism determines NMDA receptor response to repetitive stimulation. Nature 2004. [Google Scholar] [CrossRef]
- Iacobucci, G.J.; Popescu, G.K. Kinetic models for activation and modulation of NMDA receptor subtypes. Curr. Opin. Physiol. 2018, 2, 114–122. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene Symbol RefSeq Accession Number | Nucleotide Sequences (Forward, Reverse, TaqMan-Probe) | Reference |
|---|---|---|
| Ppia NM_017101 | AGGATTCATGTGCCAGGGTG CTCAGTCTTGGCAGTGCAGA ROX-CACGCCATAATGGCACTGGTGGCA-BHQ1 | [72] |
| Grin1 NM_017010 | GTTCTTCCGCTCAGGCTTTG AGGGAAACGTTCTGCTTCCA FAM-CGGCATGCGCAAGGACAGCC-BHQ1 | [73] |
| Grin2a NM_012573 | GCTACACACCCTGCACCAATT CACCTGGTAACCTTCCTCAGTGA FAM-TGGTCAATGTGACTTGGGATGGCAA-BHQ1 | [74] |
| Grin2b NM_012574 | CCCAACATGCTCTCTCCCTTAA CAGCTAGTCGGCTCTCTTGGTT FAM-GACGCCAAACCTCTAGGCGGACAG-BHQ1 | [74] |
| Gria1 NM_031608 | TCAGAACGCCTCAACGCC TGTAGTGGTACCCGATGCCA ROX-TCCTGGGCCAGATCGTGAAGCTAGAAAA-BHQ1 | [75] |
| Gria2 NM_017261 | CAGTGCATTTCGGGTAGGGA TGCGAAACTGTTGGCTACCT FAM-TCGGAGTTCAGACTGACACCCCA-BHQ1 | [75] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Postnikova, T.Y.; Griflyuk, A.V.; Amakhin, D.V.; Kovalenko, A.A.; Soboleva, E.B.; Zubareva, O.E.; Zaitsev, A.V. Early Life Febrile Seizures Impair Hippocampal Synaptic Plasticity in Young Rats. Int. J. Mol. Sci. 2021, 22, 8218. https://doi.org/10.3390/ijms22158218
Postnikova TY, Griflyuk AV, Amakhin DV, Kovalenko AA, Soboleva EB, Zubareva OE, Zaitsev AV. Early Life Febrile Seizures Impair Hippocampal Synaptic Plasticity in Young Rats. International Journal of Molecular Sciences. 2021; 22(15):8218. https://doi.org/10.3390/ijms22158218
Chicago/Turabian StylePostnikova, Tatyana Y., Alexandra V. Griflyuk, Dmitry V. Amakhin, Anna A. Kovalenko, Elena B. Soboleva, Olga E. Zubareva, and Aleksey V. Zaitsev. 2021. "Early Life Febrile Seizures Impair Hippocampal Synaptic Plasticity in Young Rats" International Journal of Molecular Sciences 22, no. 15: 8218. https://doi.org/10.3390/ijms22158218
APA StylePostnikova, T. Y., Griflyuk, A. V., Amakhin, D. V., Kovalenko, A. A., Soboleva, E. B., Zubareva, O. E., & Zaitsev, A. V. (2021). Early Life Febrile Seizures Impair Hippocampal Synaptic Plasticity in Young Rats. International Journal of Molecular Sciences, 22(15), 8218. https://doi.org/10.3390/ijms22158218