
Hierarchical Structure of Protein Sequence

 ,
,  and
and 
Abstract
:1. Introduction
2. Results
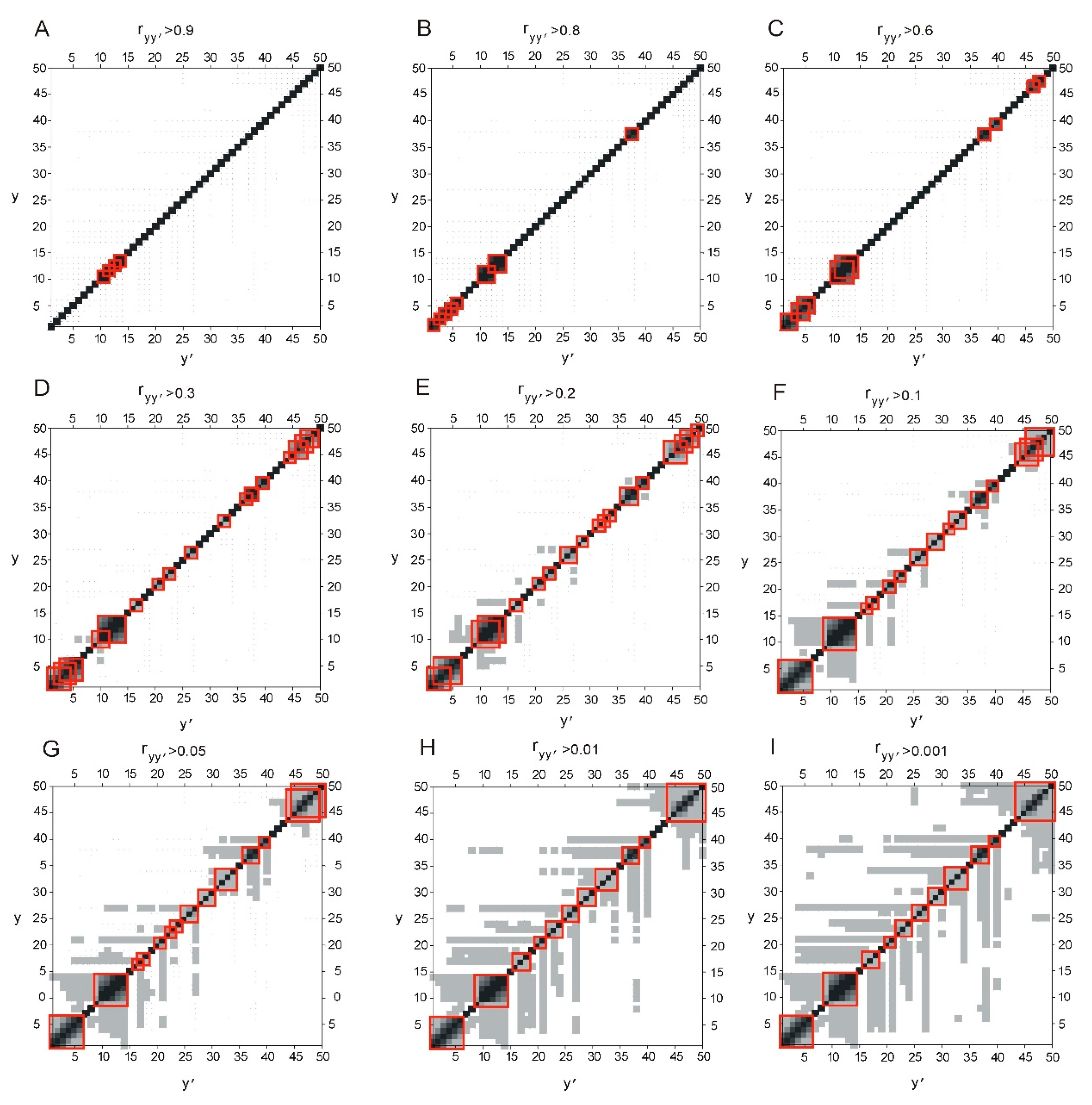
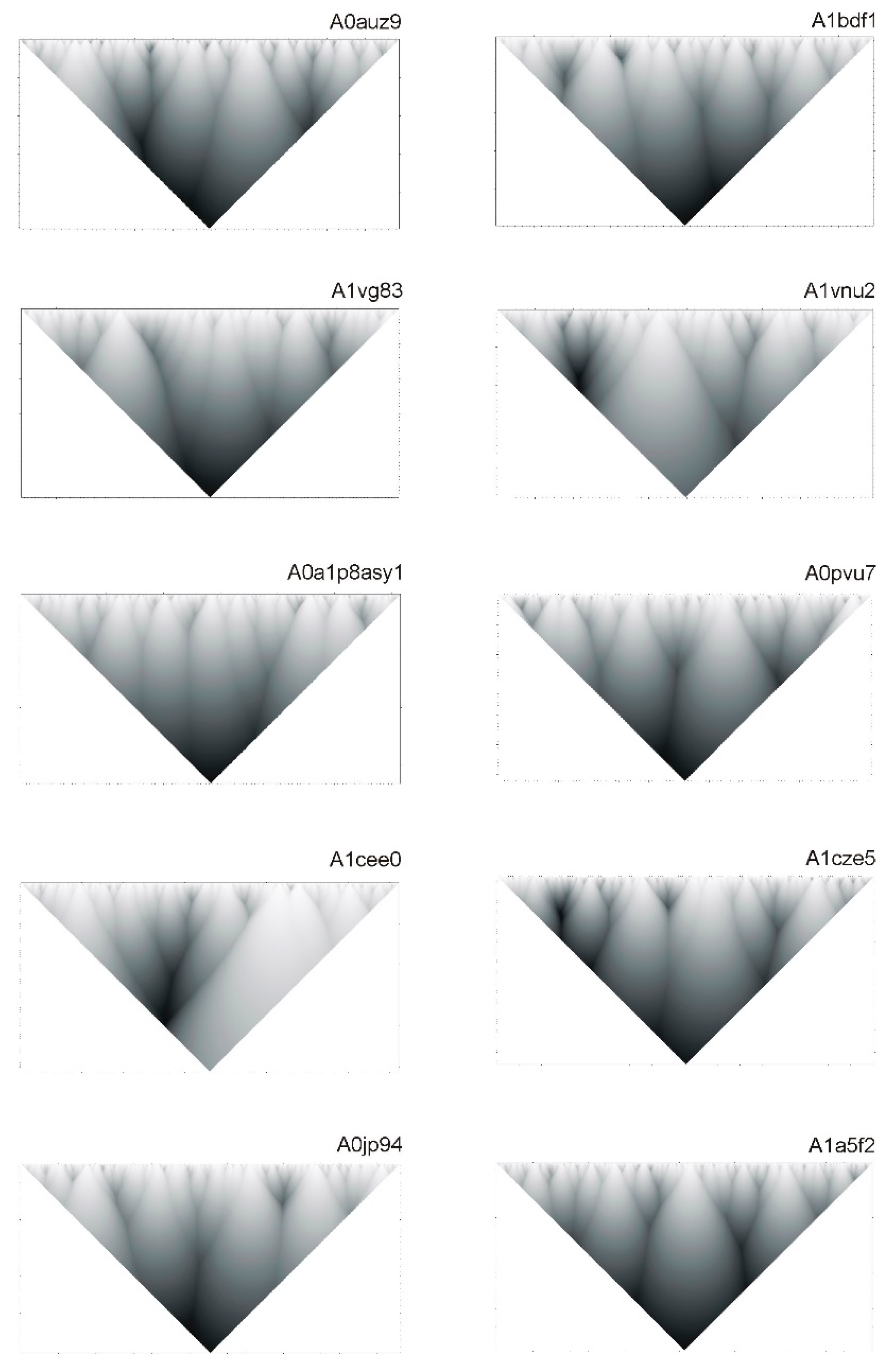
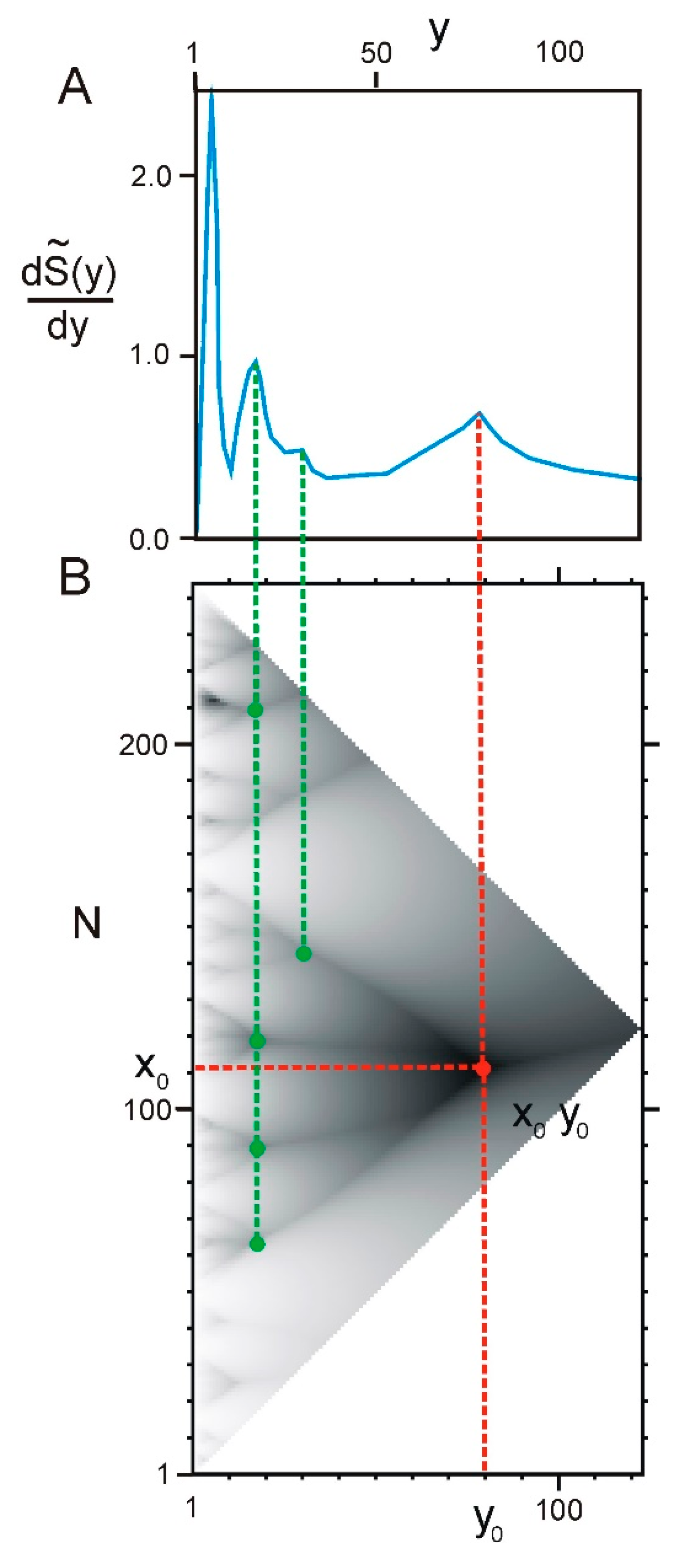
2.1. Correllation between ELements of Informational Structure (ELIS) of Different Hierarchical Levels
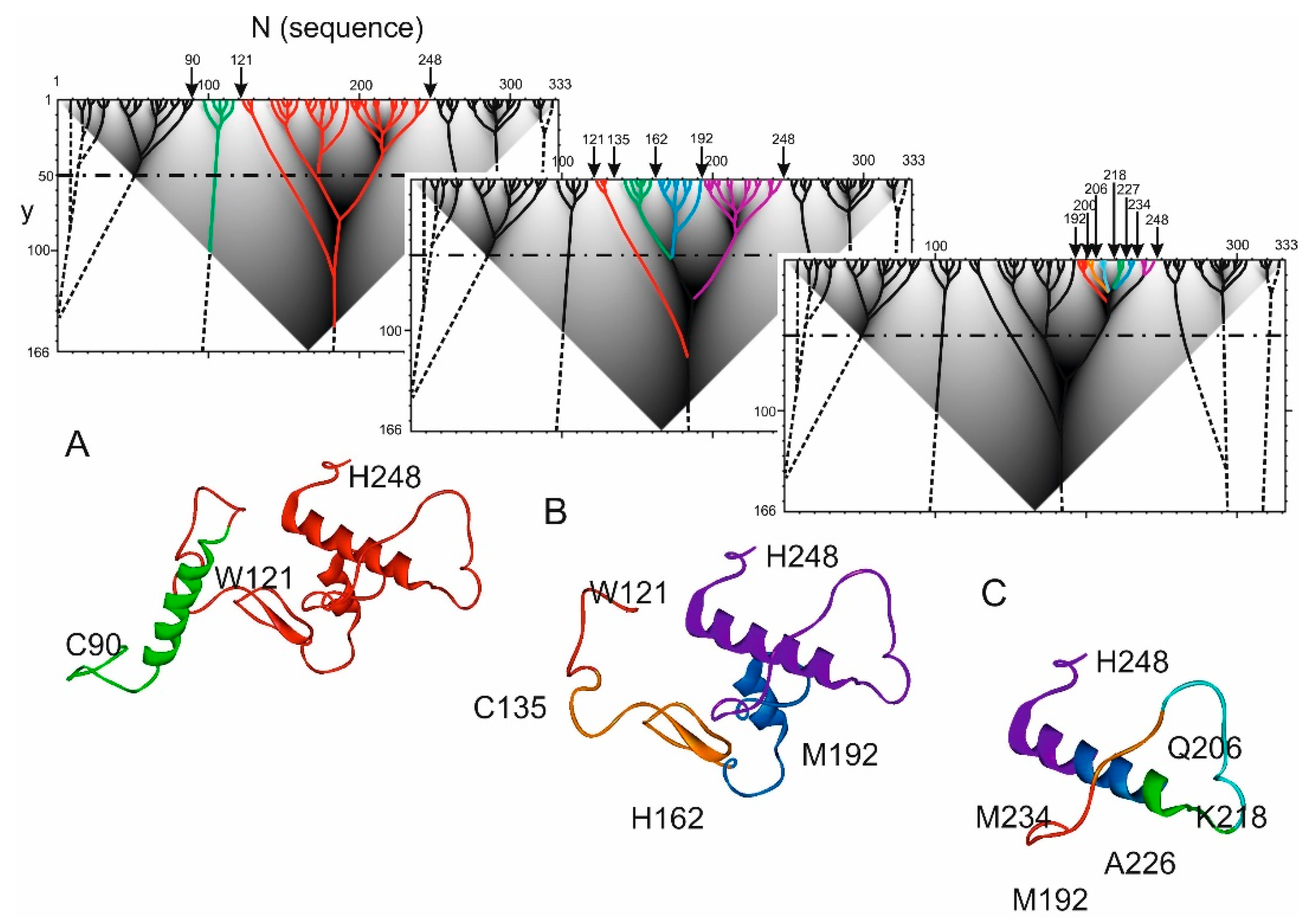
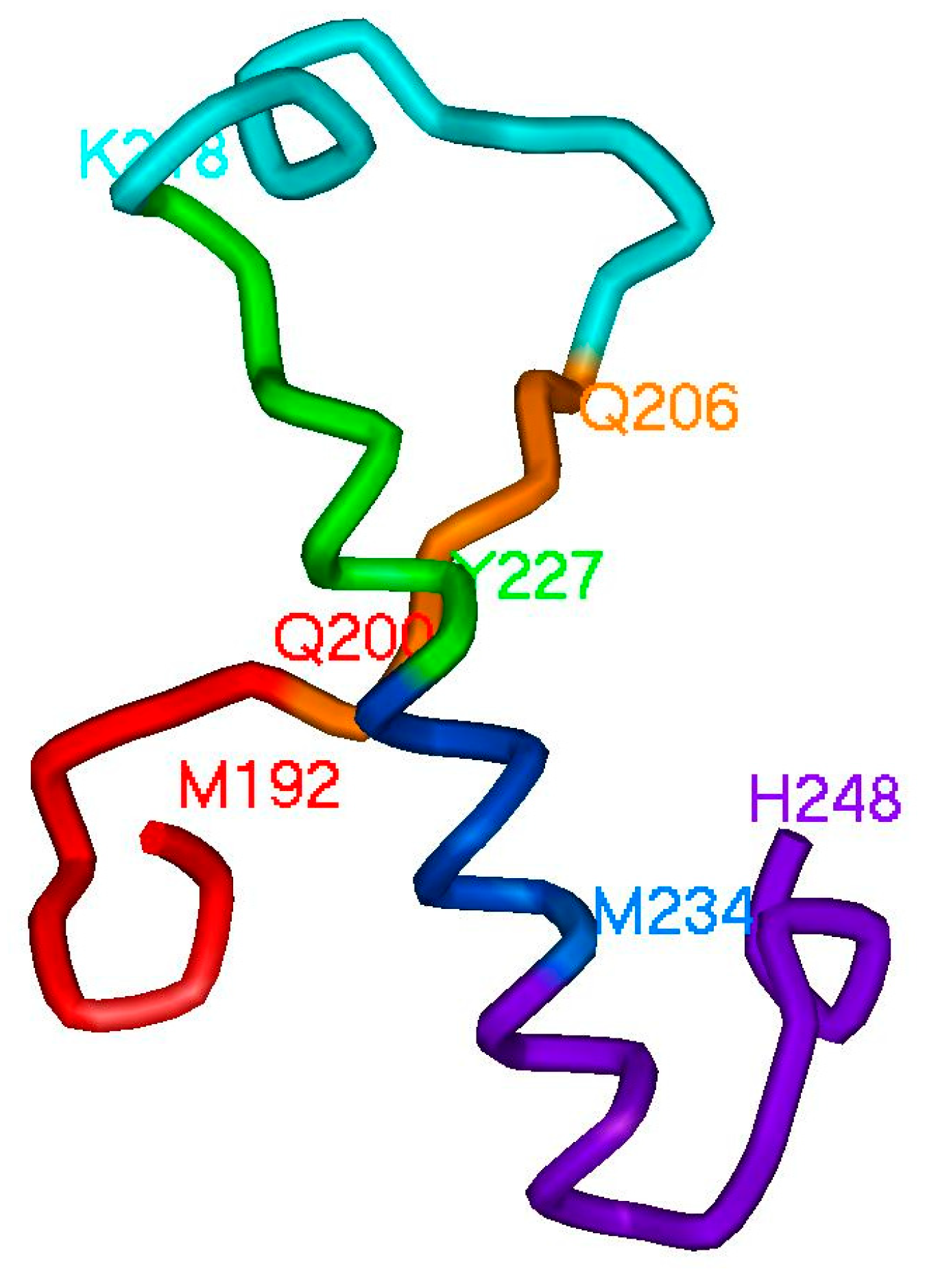
2.2. Hierarchy in the Spatial Structure of Proteins
3. Discussion
4. Materials and Methods
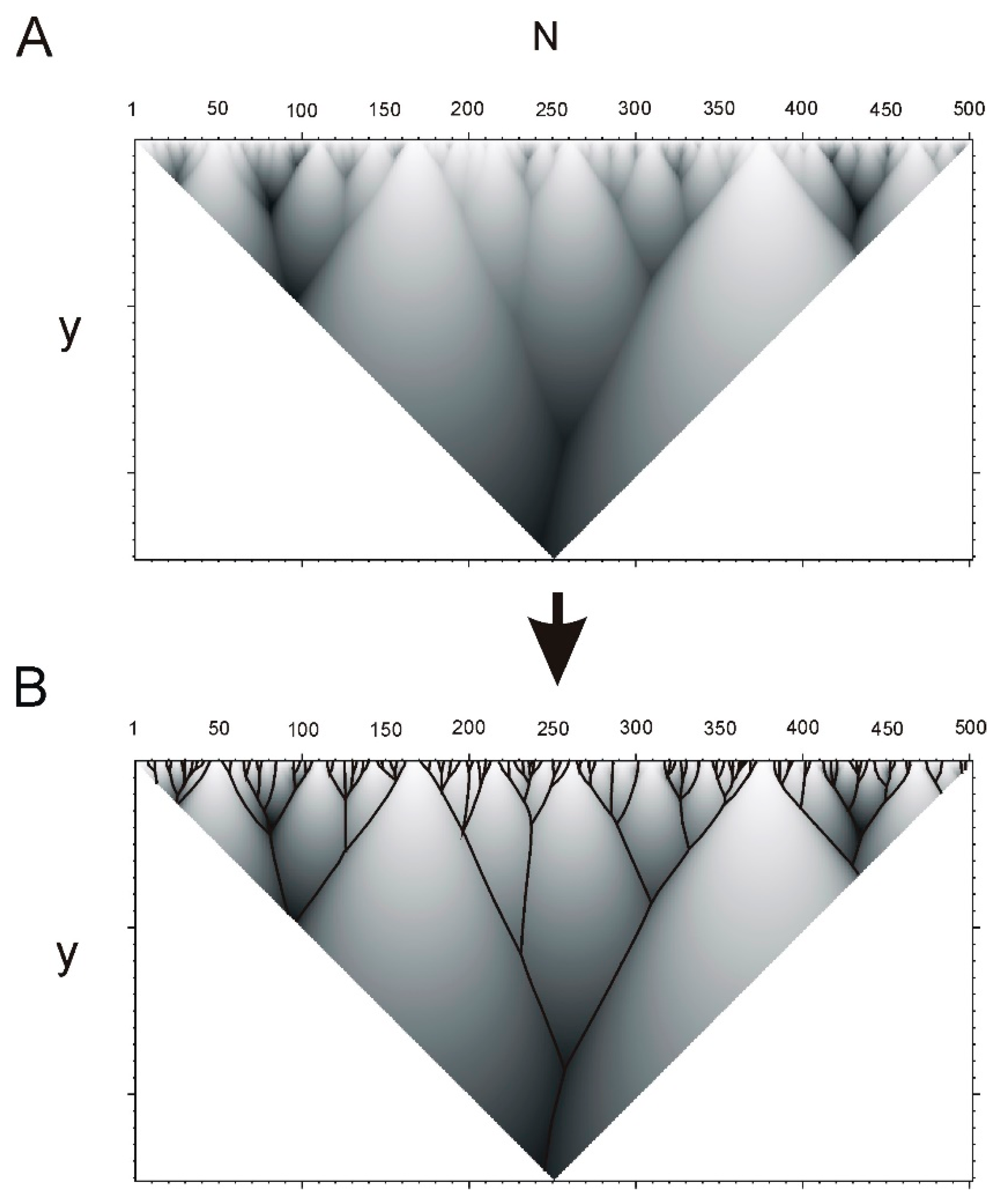
4.1. Fragments and Hierarchical Structure of a Protein
4.2. Hierarchy in Structure of Protein Sequences
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Rose, G.D. Hierarchic Organization of Domains in Globular Proteins. J. Mol. Biol. 1979, 134, 447–470. [Google Scholar] [CrossRef]
- Vriend, G.; Sander, C. Detection of Common Three-Dimensional Substructures in Proteins. Proteins 1991, 11, 52–58. [Google Scholar] [CrossRef] [PubMed]
- Zehfus, M.H. Continuous Compact Protein Domains. Proteins Struct. Funct. Bioinform. 1987, 2, 90–110. [Google Scholar] [CrossRef] [PubMed]
- Gelly, J.-C.; de Brevern, A.G.; Hazout, S. “Protein Peeling”: An Approach for Splitting a 3D Protein Structure into Compact Fragments. Bioinformatics 2006, 22, 129–133. [Google Scholar] [CrossRef] [Green Version]
- Berezovsky, I.N.; Tumanyan, V.G.; Esipova, N.G. Representation of Amino Acid Sequences in Terms of Interaction Energy in Protein Globules. FEBS Lett. 1997, 418, 43–46. [Google Scholar] [CrossRef]
- Dułak, D.; Gadzała, M.; Banach, M.; Ptak, M.; Wiśniowski, Z.; Konieczny, L.; Roterman, I. Filamentous Aggregates of Tau Proteins Fulfil Standard Amyloid Criteria Provided by the Fuzzy Oil Drop (FOD) Model. Int. J. Mol. Sci. 2018, 19, 2910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Galzitskaya, O.V.; Ivankov, D.N.; Finkelstein, A.V. Folding Nuclei in Proteins. Mol. Biol. 2001, 35, 605–613. [Google Scholar] [CrossRef]
- Lesk, A.M.; Rose, G.D. Folding Units in Globular Proteins. Proc. Natl. Acad. Sci. USA 1981, 78, 4304–4308. [Google Scholar] [CrossRef] [Green Version]
- Wetlaufer, D.B. Nucleation, Rapid Folding, and Globular Intrachain Regions in Proteins. Proc. Natl. Acad. Sci. USA 1973, 70, 697–701. [Google Scholar] [CrossRef] [Green Version]
- Zaki, M.J.; Nadimpally, V.; Bardhan, D.; Bystroff, C. Predicting Protein Folding Pathways. Bioinformatics 2004, 20 (Suppl. 1), i386–i393. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brylinski, M.; Konieczny, L.; Kononowicz, A.; Roterman, I. Conservative Secondary Structure Motifs Already Present in Early-Stage Folding (in Silico) as Found in Serpines Family. J. Theor. Biol. 2008, 251, 275–285. [Google Scholar] [CrossRef]
- Weiss, O.; Jiménez-Montaño, M.A.; Herzel, H. Information Content of Protein Sequences. J. Theor. Biol. 2000, 206, 379–386. [Google Scholar] [CrossRef] [PubMed]
- Ptitsyn, O.B.; Volkenstein, M.V. Protein Structure and Neutral Theory of Evolution. J. Biomol. Struct. Dyn. 1986, 4, 137–156. [Google Scholar] [CrossRef]
- Szoniec, G.; Ogorzalek, M.J. Entropy of Never Born Protein Sequences. Springerplus 2013, 2. [Google Scholar] [CrossRef] [Green Version]
- Yoon, B.-J. Hidden Markov Models and Their Applications in Biological Sequence Analysis. Curr. Genom. 2009, 10, 402–415. [Google Scholar] [CrossRef] [Green Version]
- Nekrasov, A.N. Entropy of Protein Sequences: An Integral Approach. J. Biomol. Struct. Dyn. 2002, 20, 87–92. [Google Scholar] [CrossRef] [PubMed]
- Anashkina, A.A.; Nekrasov, A.N. The Method for Identification of Hierarchical Organization of Protein Sequences. Russ. J. Numer. Anal. Math. Model. 2014, 29, 265–273. [Google Scholar] [CrossRef]
- Nekrasov, A.N.; Anashkina, A.A.; Zinchenko, A.A. A New Paradigm of Protein Structural Organization. In Proceedings of the 2nd International Conference “Theoretical Approaches to Bioinformation Systems” (TABIS 2013); Institute of Physics: Belgrade, Serbia, 2014; pp. 1–22. [Google Scholar]
- Shannon, C.E. A Mathematical Theory of Communication. Bell Syst. Tech. J. 1948, 27, 379–423. [Google Scholar] [CrossRef] [Green Version]
- Nekrasov, A.N.; Zinchenko, A.A. Hydrolases: The Correlation between Informational Structure and the Catalytic Sites Organization. J. Biomol. Struct. Dyn. 2008, 25, 553–561. [Google Scholar] [CrossRef]
- Chertkova, R.V.; Brazhe, N.A.; Bryantseva, T.V.; Nekrasov, A.N.; Dolgikh, D.A.; Yusipovich, A.I.; Sosnovtseva, O.; Maksimov, G.V.; Rubin, A.B.; Kirpichnikov, M.P. New Insight into the Mechanism of Mitochondrial Cytochrome c Function. PLoS ONE 2017, 12. [Google Scholar] [CrossRef]
- Shingarova, L.N.; Petrovskaya, L.E.; Nekrasov, A.N.; Kryukova, E.A.; Boldyreva, E.F.; Yakimov, S.A.; Guryanova, S.V.; Dolgih, D.A.; Kirpichnikov, M.P. Expression and Properties of Human TNF Peptide Fragments. Russ. J. Bioorganic Chem. 2010, 36, 301–309. [Google Scholar] [CrossRef] [PubMed]
- Nekrasov, A.N.; Radchenko, V.V.; Shuvaeva, T.M.; Novoselov, V.I.; Fesenko, E.E.; Lipkin, V.M. The Novel Approach to the Protein Design: Active Truncated Forms of Human 1-CYS Peroxiredoxin. J. Biomol. Struct. Dyn. 2007, 24, 455–462. [Google Scholar] [CrossRef]
- Briers, Y.; Miroshnikov, K.; Chertkov, O.; Nekrasov, A.; Mesyanzhinov, V.; Volckaert, G.; Lavigne, R. The Structural Peptidoglycan Hydrolase Gp181 of Bacteriophage PhiKZ. Biochem. Biophys. Res. Commun. 2008, 374, 747–751. [Google Scholar] [CrossRef]
- Nekrasov, A.N.; Petrovskaya, L.E.; Toporova, V.A.; Kryukova, E.A.; Rodina, A.V.; Moskaleva, E.Y.; Kirpichnikov, M.P. Design of a Novel Interleukin-13 Antagonist from Analysis of Informational Structure. Biochemistry 2009, 74, 399–405. [Google Scholar] [CrossRef]
- Mikhailova, A.G.; Nekrasov, A.N.; Zinchenko, A.A.; Rakitina, T.V.; Korzhenevsky, D.A.; Lipkin, A.V.; Razguljaeva, O.A.; Ovchinnikova, M.V.; Gorlenko, V.A.; Rumsh, L.D. Truncated Variants of Serratia Proteamaculans Oligopeptidase B Having Different Activities. Biochemistry 2015, 80, 1331–1343. [Google Scholar] [CrossRef] [PubMed]
- De Brevern, A.G. Protein Local Conformations at the Light of a Structural Alphabet. Biophys. J. 2018, 114, 231a. [Google Scholar] [CrossRef]
- Nekrasov, A.N.; Alekseeva, L.G.; Pogosyan, R.A.; Dolgikh, D.A.; Kirpichnikov, M.P.; de Brevern, A.G.; Anashkina, A.A. A Minimum Set of Stable Blocks for Rational Design of Polypeptide Chains. Biochimie 2019, 160, 88–92. [Google Scholar] [CrossRef] [Green Version]
- Jurkowski, W.; Brylinski, M.; Konieczny, L.; Wiíniowski, Z.; Roterman, I. Conformational Subspace in Simulation of Early-Stage Protein Folding. Proteins 2004, 55, 115–127. [Google Scholar] [CrossRef] [PubMed]
- Jurkowski, W.; Kułaga, T.; Roterman, I. Geometric Parameters Defining the Structure of Proteins--Relation to Early-Stage Folding Step. J. Biomol. Struct. Dyn. 2011, 29, 79–104. [Google Scholar] [CrossRef]
- Kaushik, R.; Zhang, K.Y.J. A Protein Sequence Fitness Function for Identifying Natural and Nonnatural Proteins. Proteins Struct. Funct. Bioinform. 2020, 88, 1271–1284. [Google Scholar] [CrossRef]
- Holm, L.; Sander, C. Removing Near-Neighbour Redundancy from Large Protein Sequence Collections. Bioinformatics 1998, 14, 423–429. [Google Scholar] [CrossRef] [PubMed]
- Trifonov, E.N.; Frenkel, Z.M. Evolution of Protein Modularity. Curr. Opin. Struct. Biol. 2009, 19, 335–340. [Google Scholar] [CrossRef] [PubMed]
- Alva, V.; Söding, J.; Lupas, A.N. A Vocabulary of Ancient Peptides at the Origin of Folded Proteins. eLife 2015, 4, e09410. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Continuous Square Areas, Index Number | L1 | L2 | L3 | L4 | L5 | L6 | L7 | L8 | L9 | L10 | L11 |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Range of smoothing function width value y | 1–6 | 9–14 | 16–18 | 20–21 | 22–24 | 25–27 | 28–30 | 31–34 | 36–38 | 39–40 | 44–50 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nekrasov, A.N.; Kozmin, Y.P.; Kozyrev, S.V.; Ziganshin, R.H.; de Brevern, A.G.; Anashkina, A.A. Hierarchical Structure of Protein Sequence. Int. J. Mol. Sci. 2021, 22, 8339. https://doi.org/10.3390/ijms22158339
Nekrasov AN, Kozmin YP, Kozyrev SV, Ziganshin RH, de Brevern AG, Anashkina AA. Hierarchical Structure of Protein Sequence. International Journal of Molecular Sciences. 2021; 22(15):8339. https://doi.org/10.3390/ijms22158339
Chicago/Turabian StyleNekrasov, Alexei N., Yuri P. Kozmin, Sergey V. Kozyrev, Rustam H. Ziganshin, Alexandre G. de Brevern, and Anastasia A. Anashkina. 2021. "Hierarchical Structure of Protein Sequence" International Journal of Molecular Sciences 22, no. 15: 8339. https://doi.org/10.3390/ijms22158339
APA StyleNekrasov, A. N., Kozmin, Y. P., Kozyrev, S. V., Ziganshin, R. H., de Brevern, A. G., & Anashkina, A. A. (2021). Hierarchical Structure of Protein Sequence. International Journal of Molecular Sciences, 22(15), 8339. https://doi.org/10.3390/ijms22158339