
TRPM2 Oxidation Activates Two Distinct Potassium Channels in Melanoma Cells through Intracellular Calcium Increase

 , , ,
, , ,  ,
,  ,
,
Abstract
:1. Introduction
2. Results
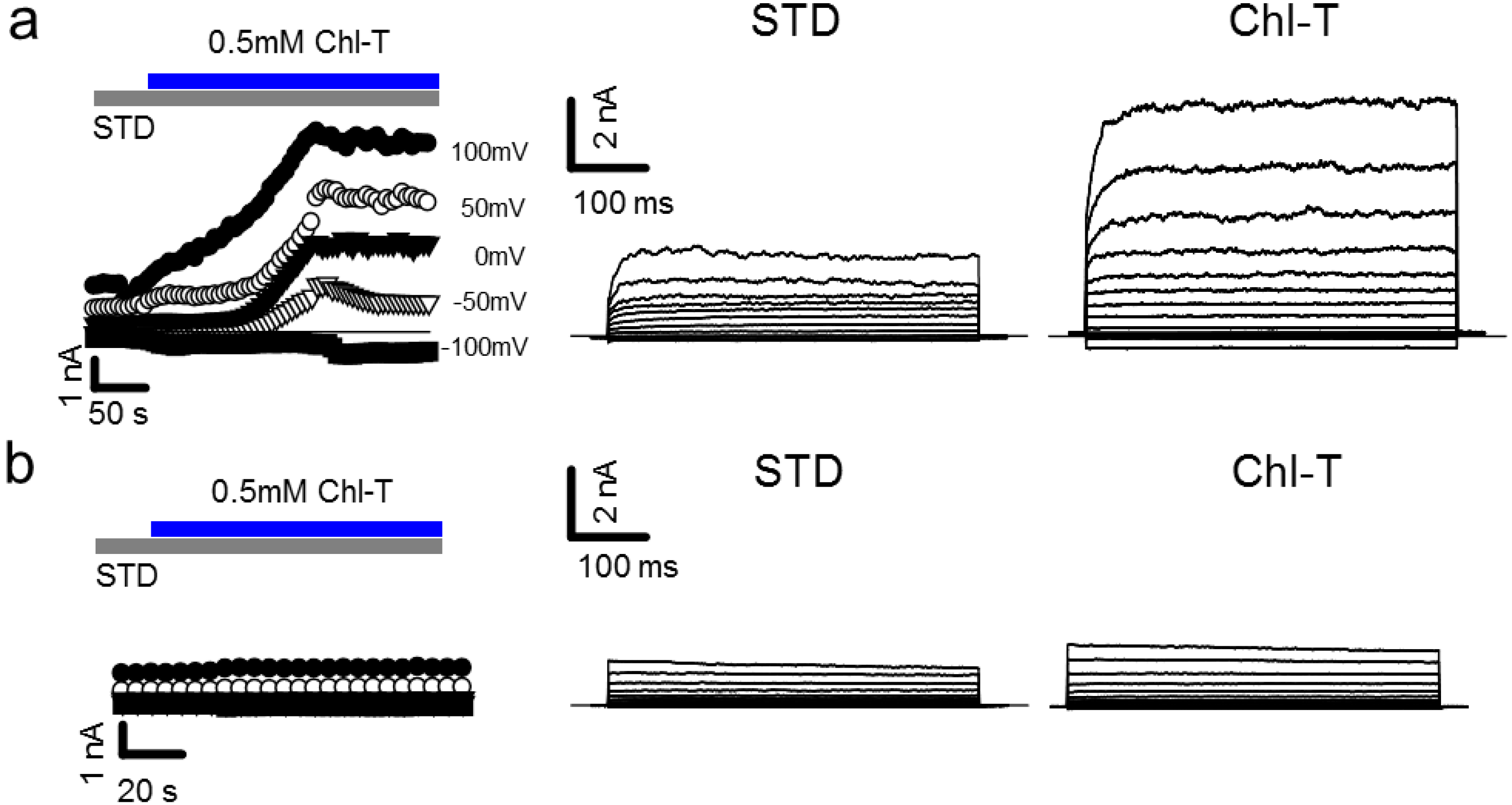
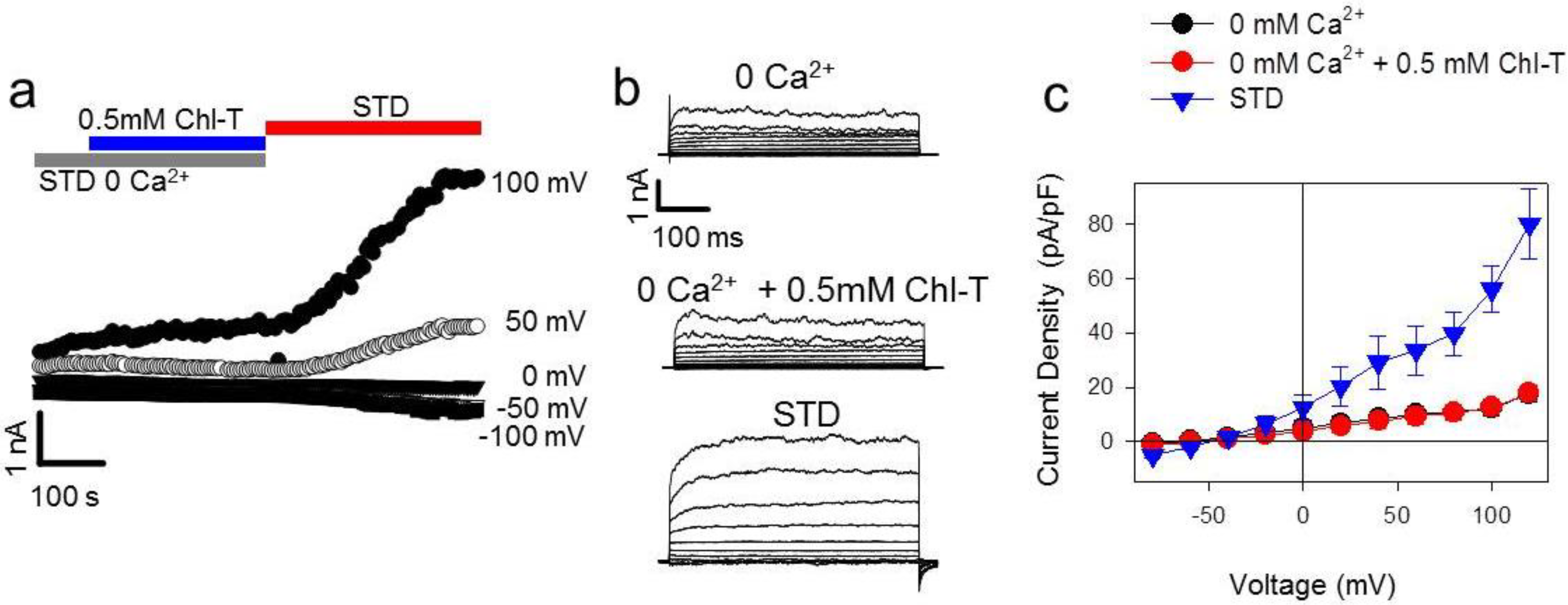
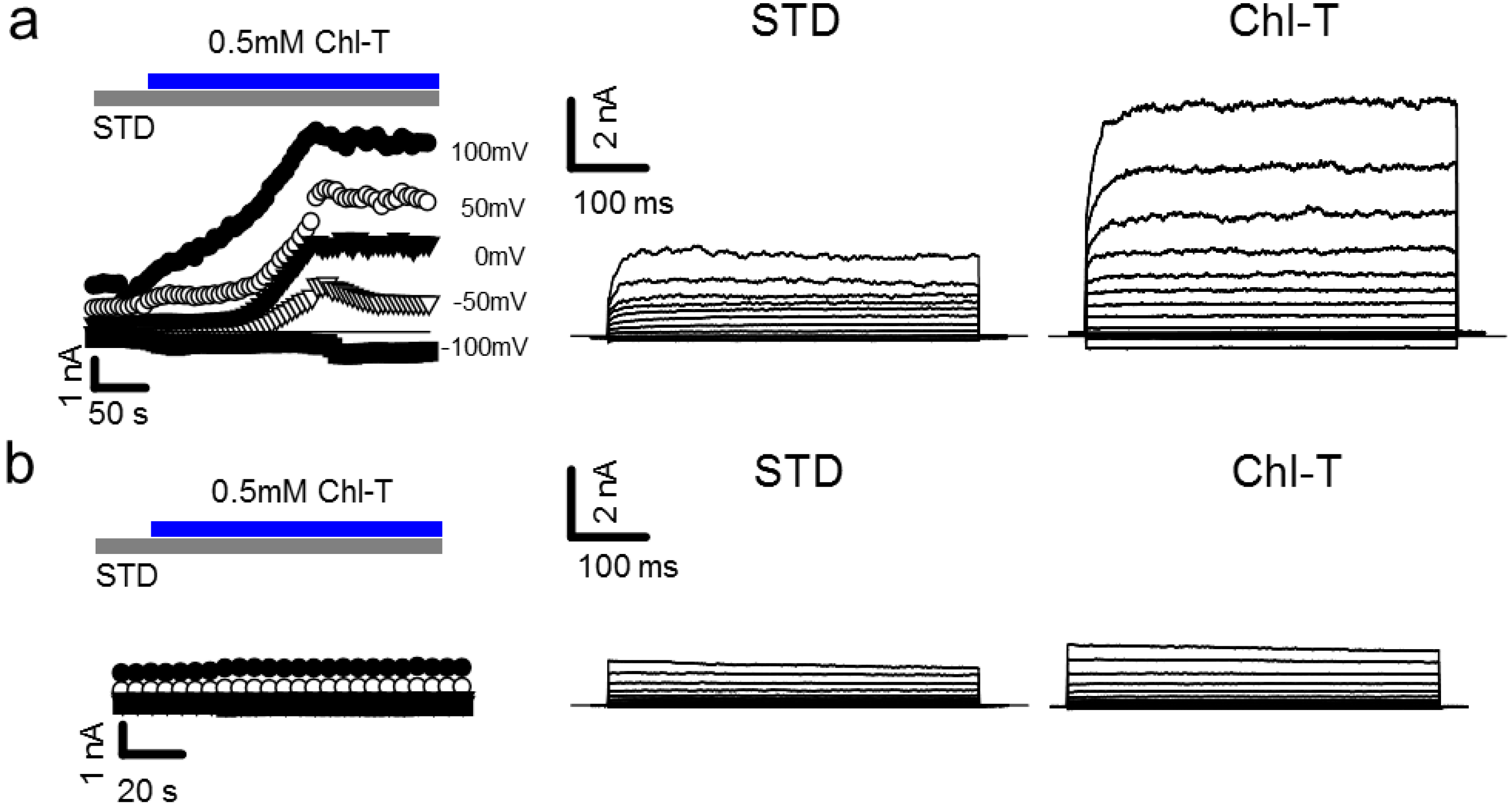
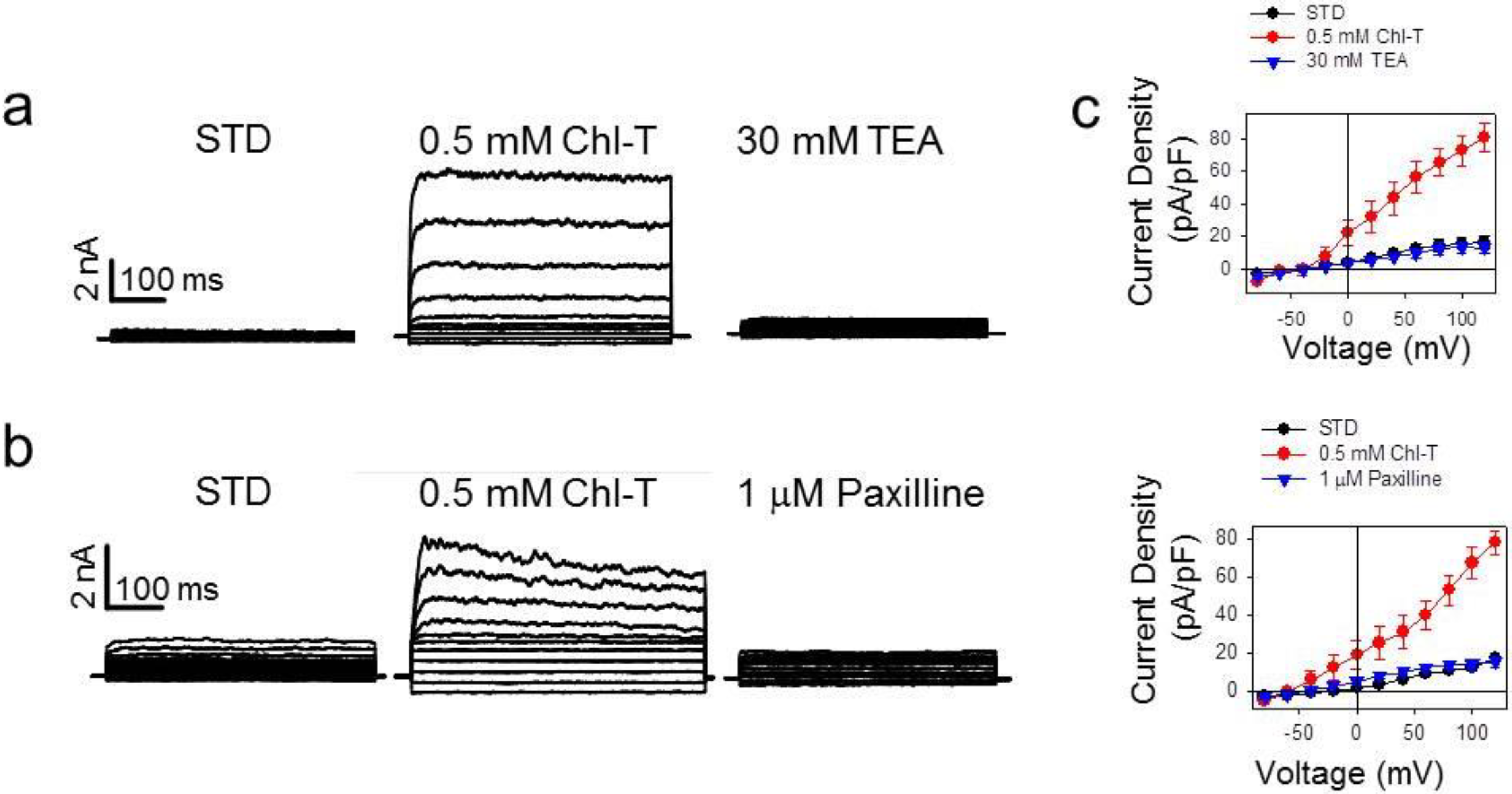
2.1. Activation of Large Currents by Chl-T in IGR39 but Not in IGR37 Cells
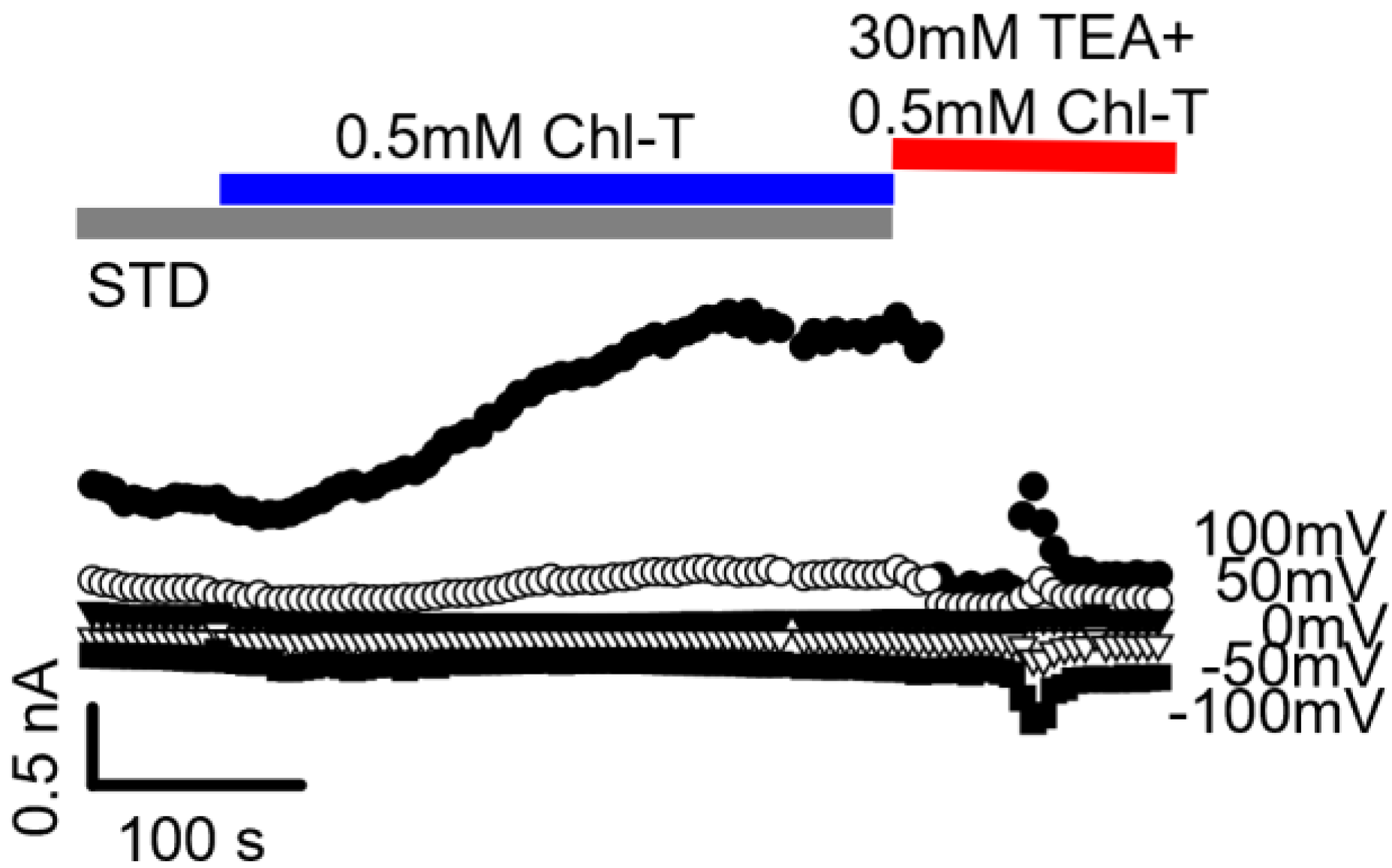
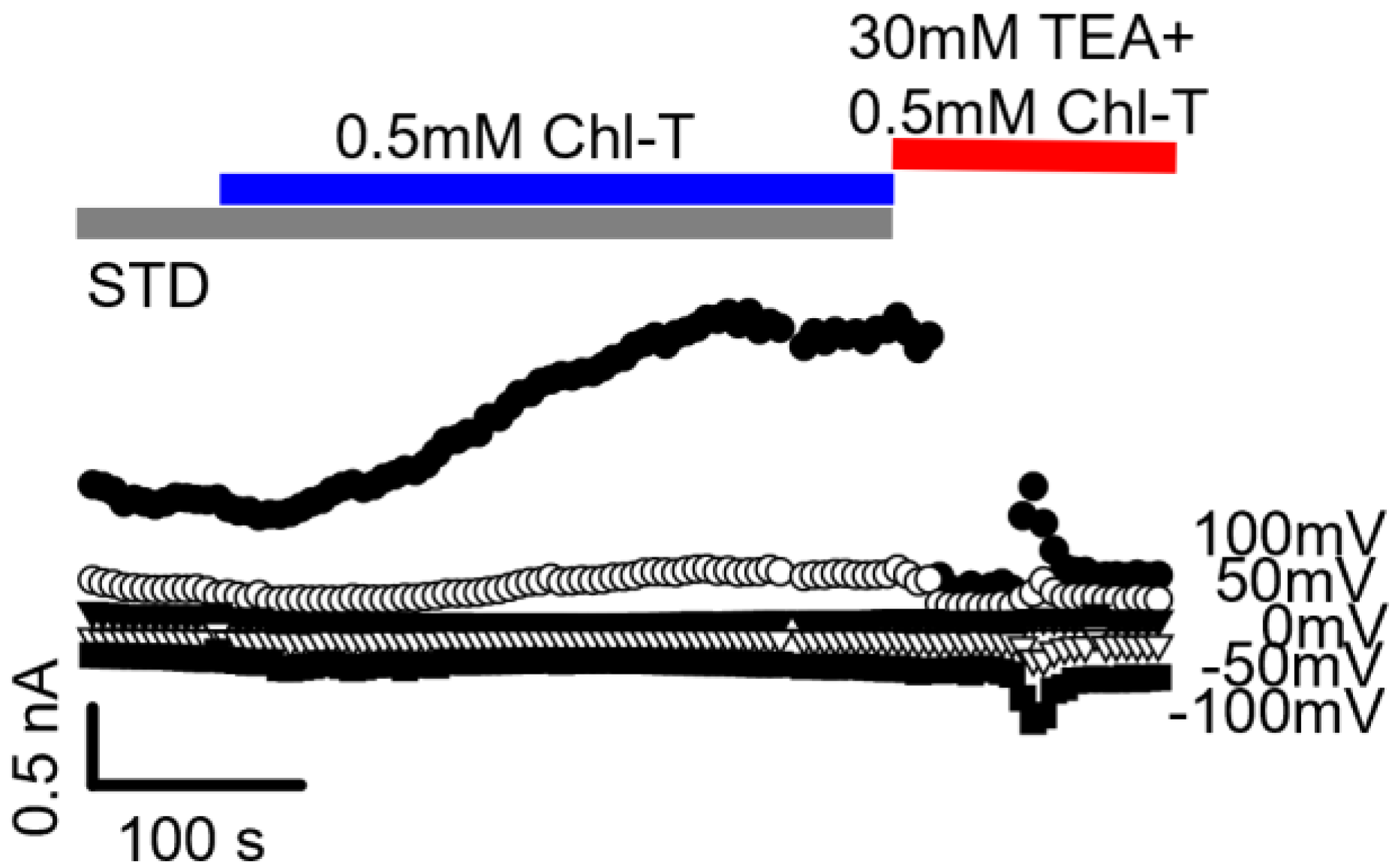
2.2. Identification of the Large-Conductance (BK) K+ Channel as a Major Current Component Activated by Chl-T
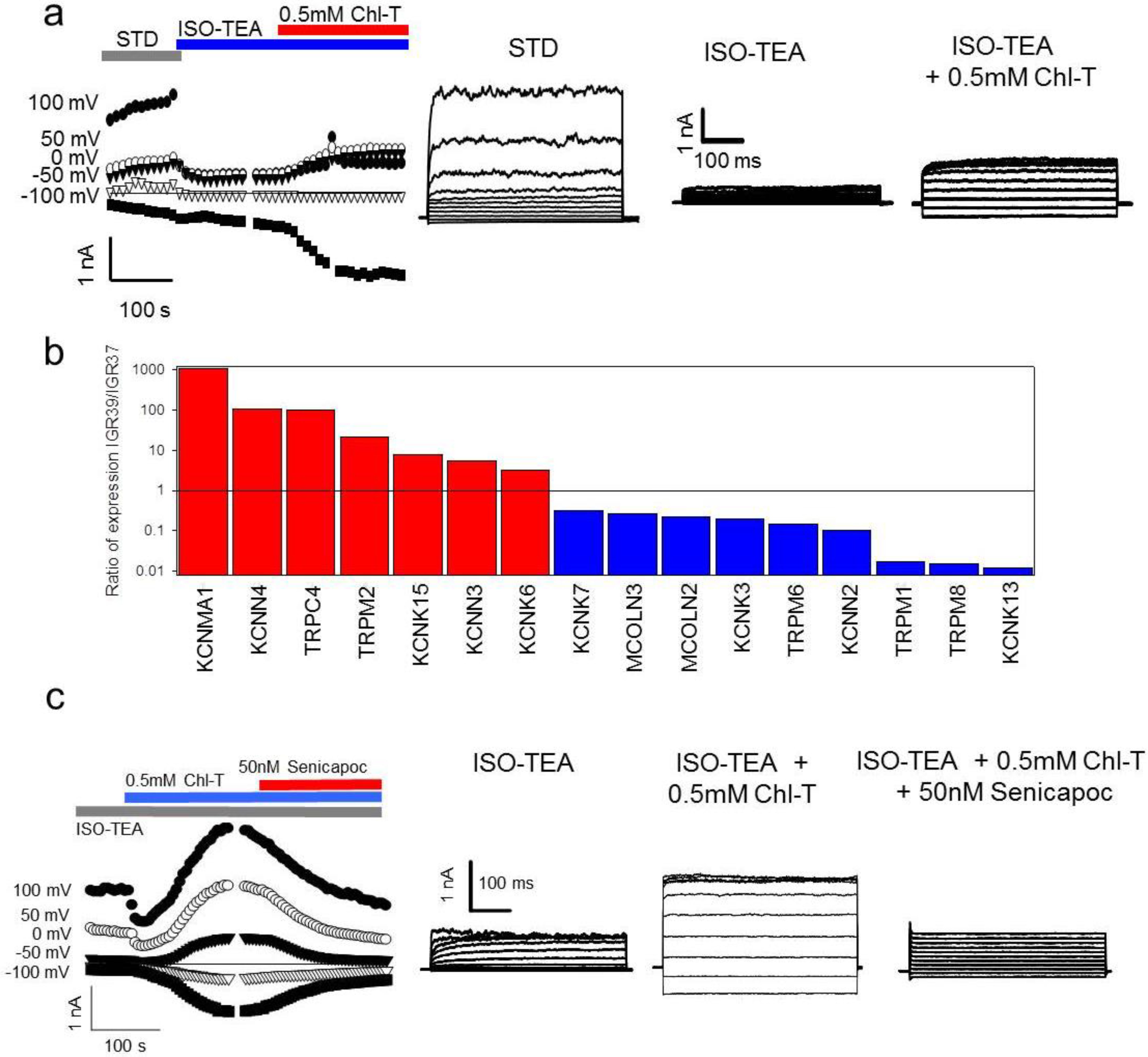
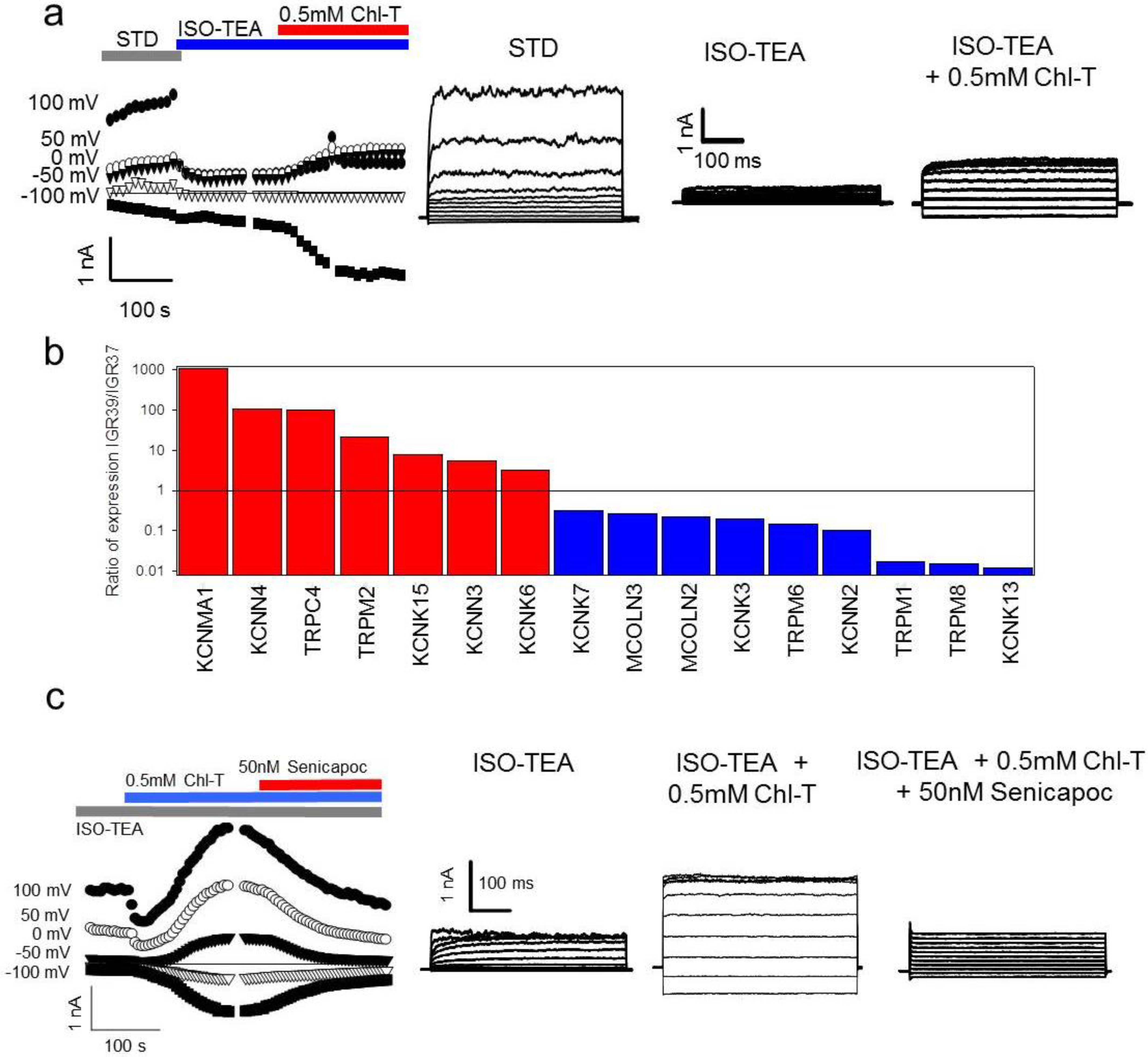
2.3. Additional TEA- and Paxilline-Insensitive K+ Current Activated by Chl-T
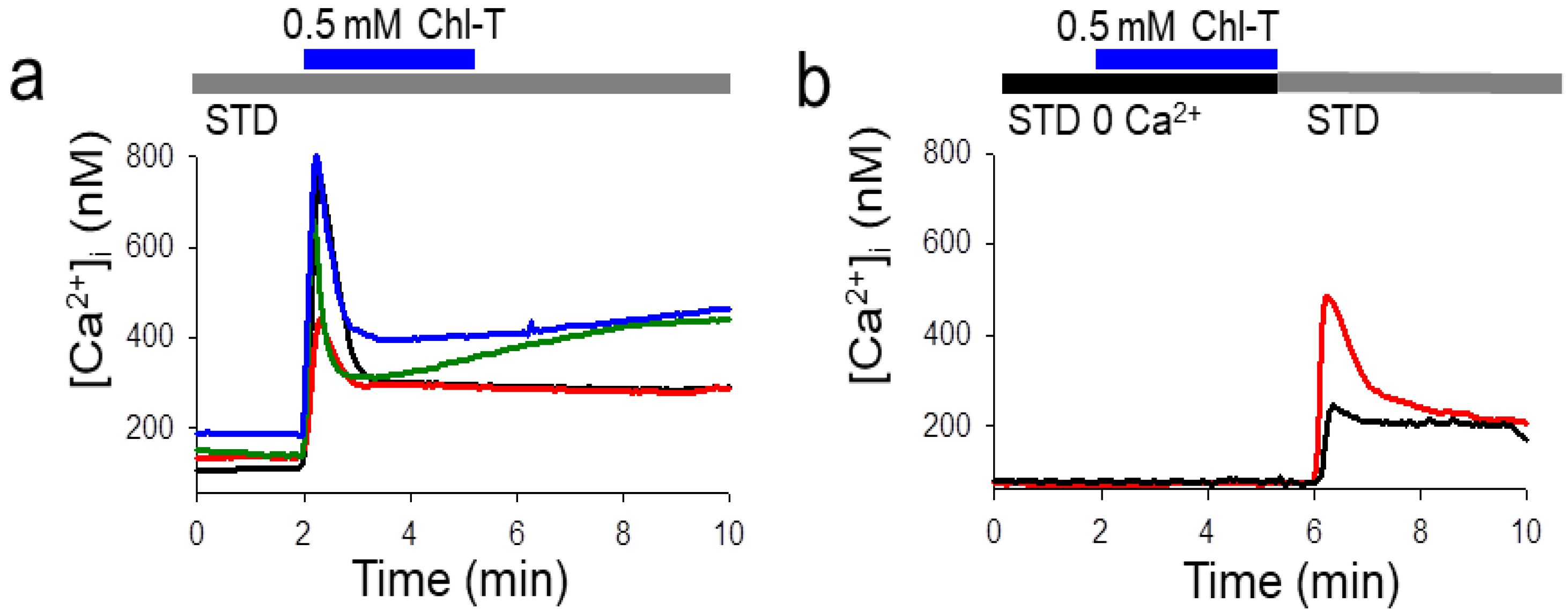
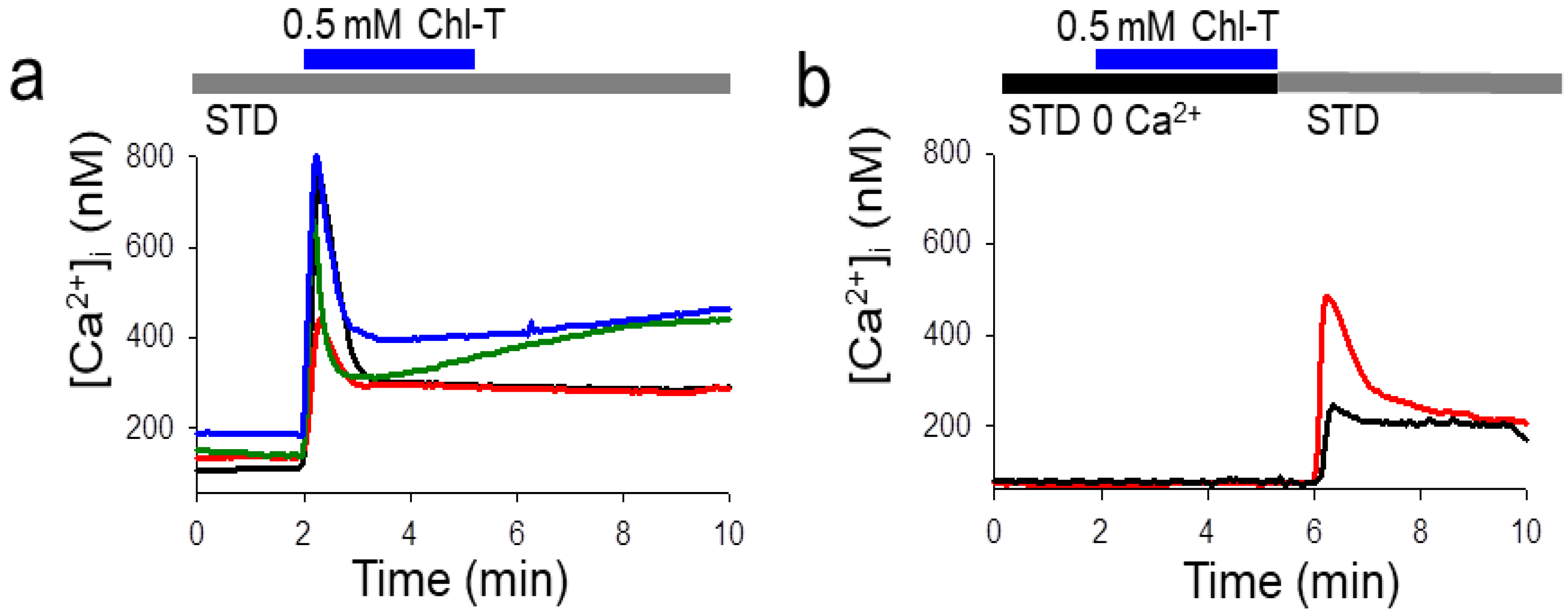
2.4. Intracellular Ca2+ Increase by the Application of Chl-T
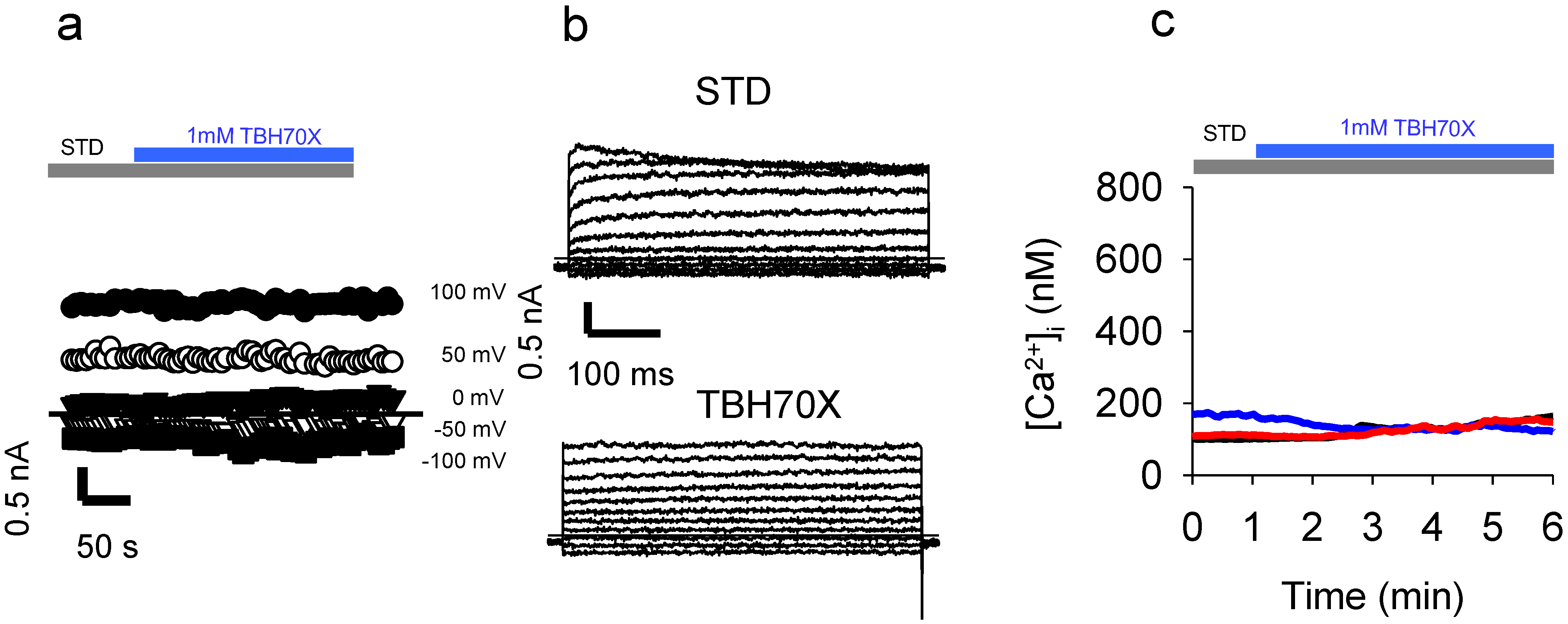
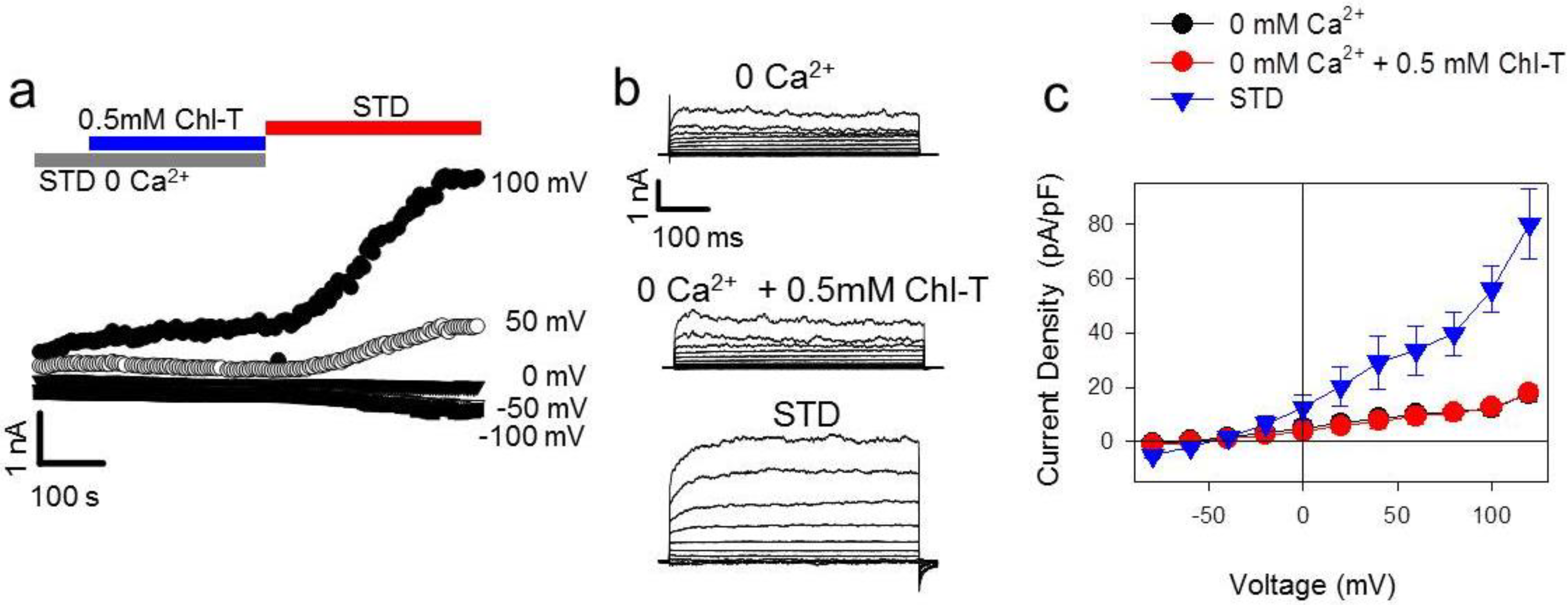
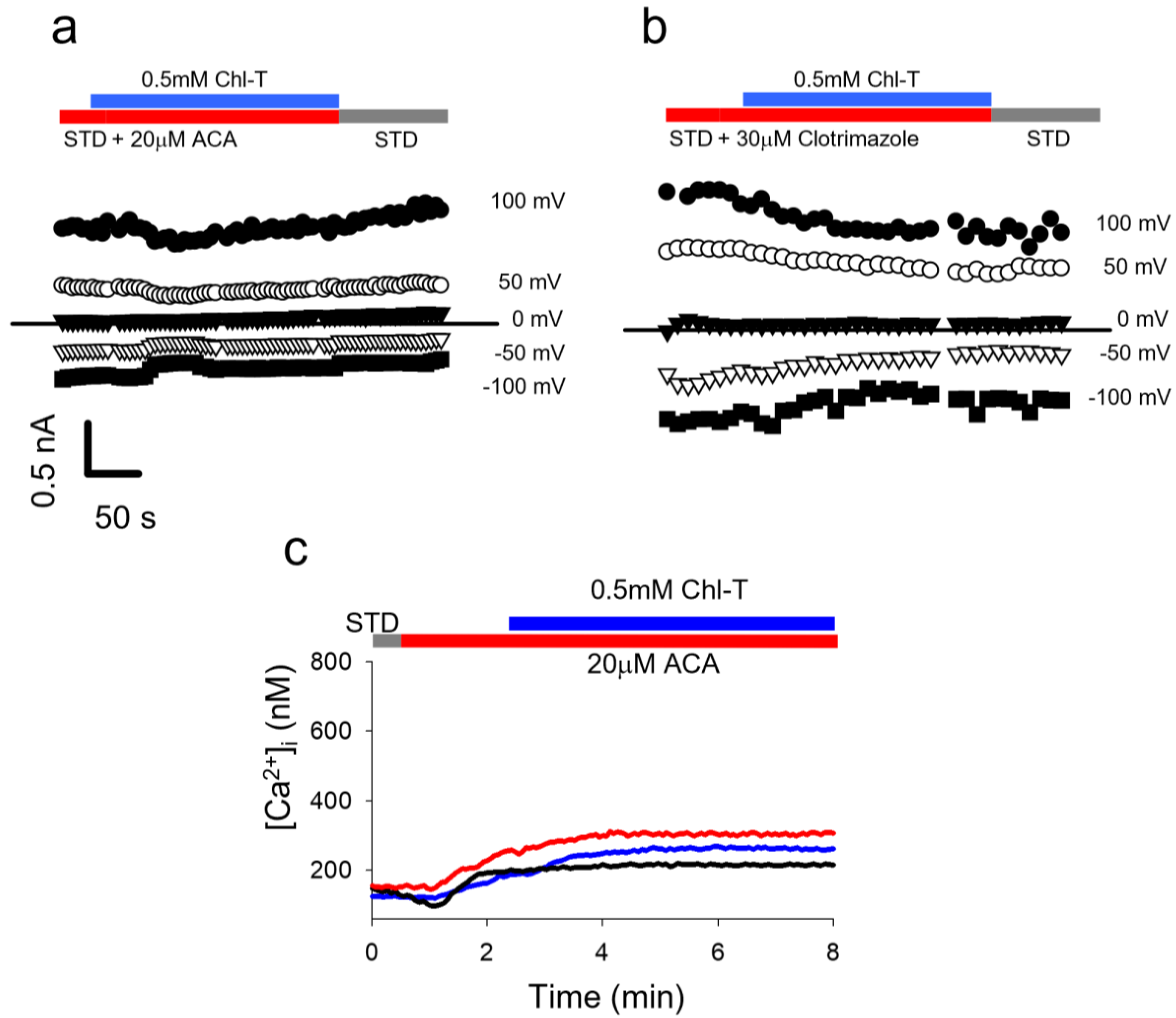
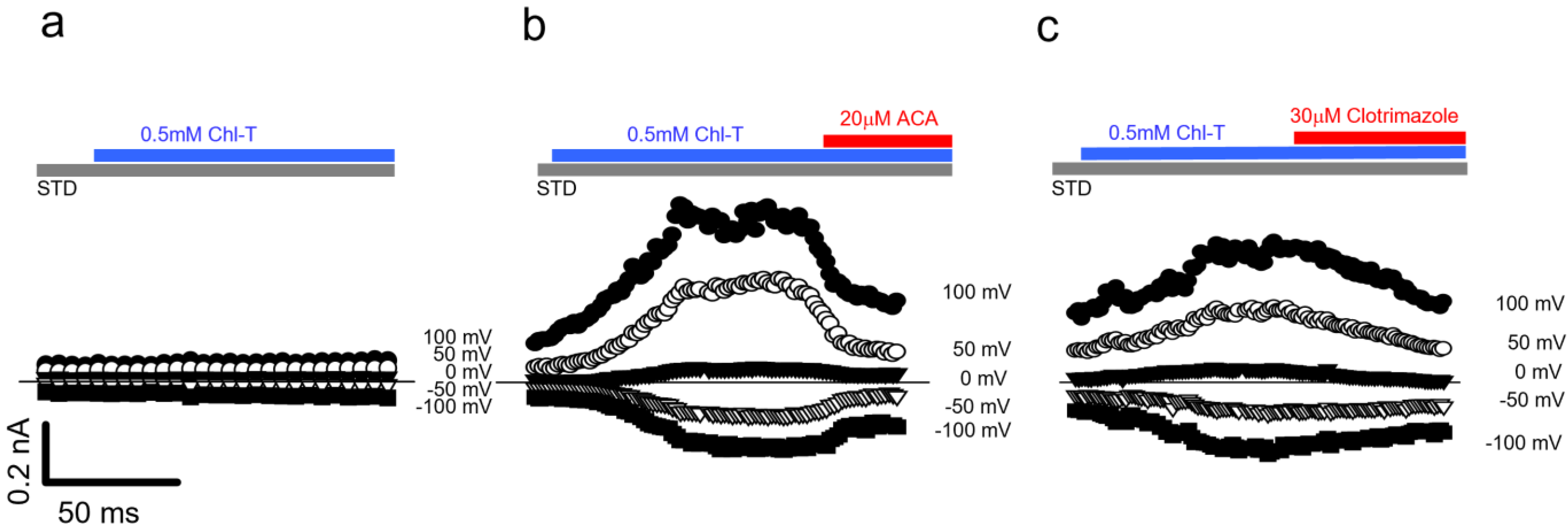
2.5. Identification of a Plasma Membrane Ca2+-Permeable Channel Sensitive to Oxidation
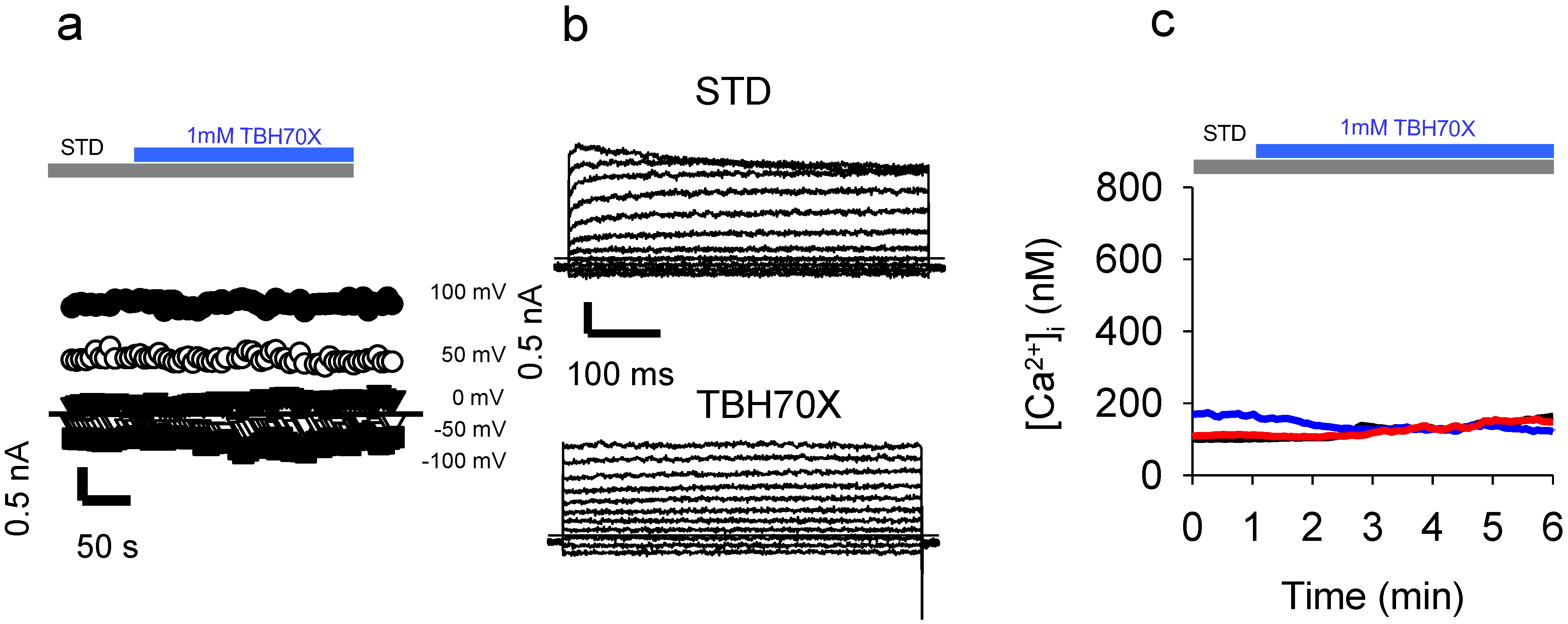
2.6. Application of the Oxidizing Agent TBH70X Does Not Activate KROS
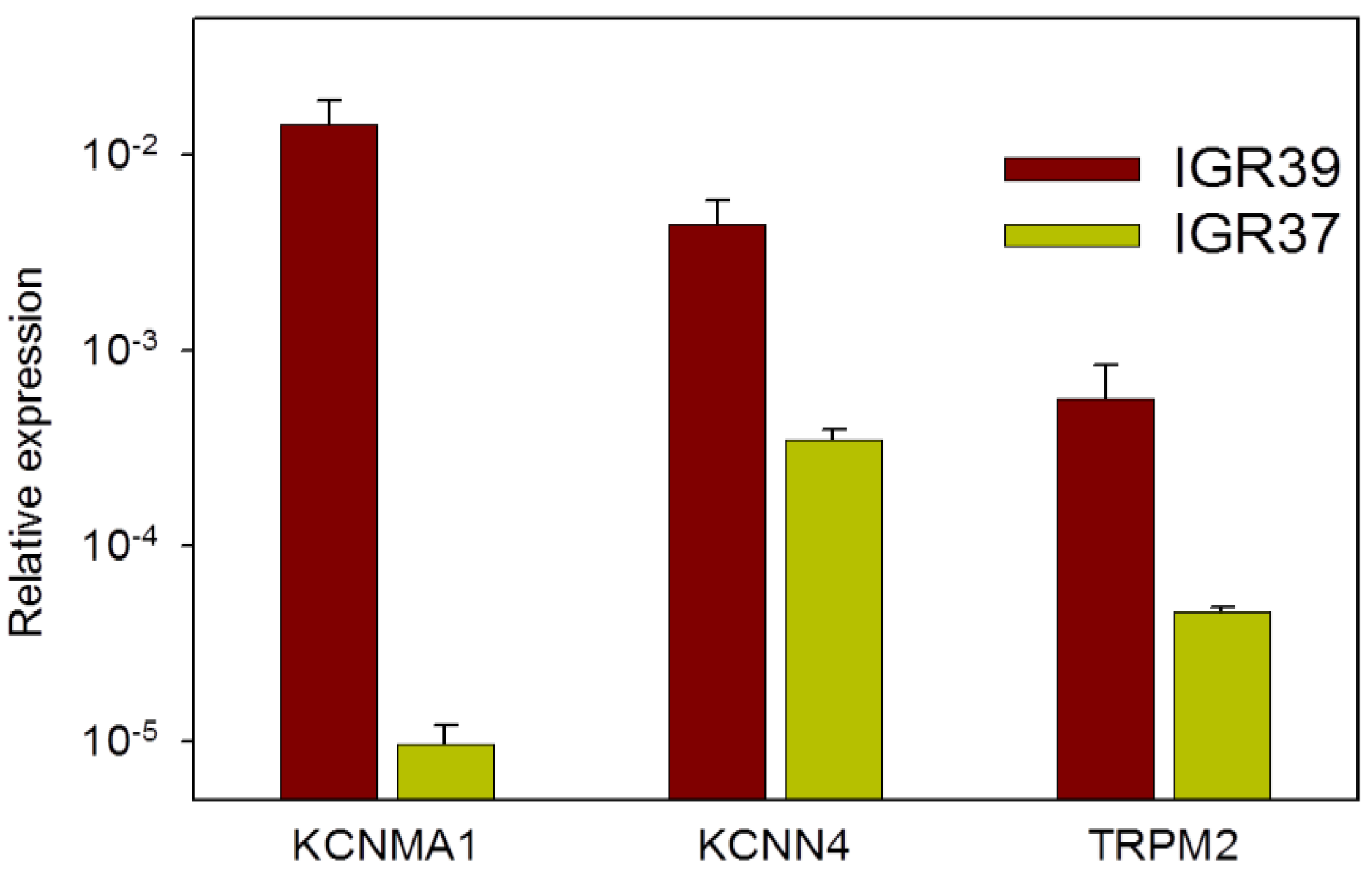
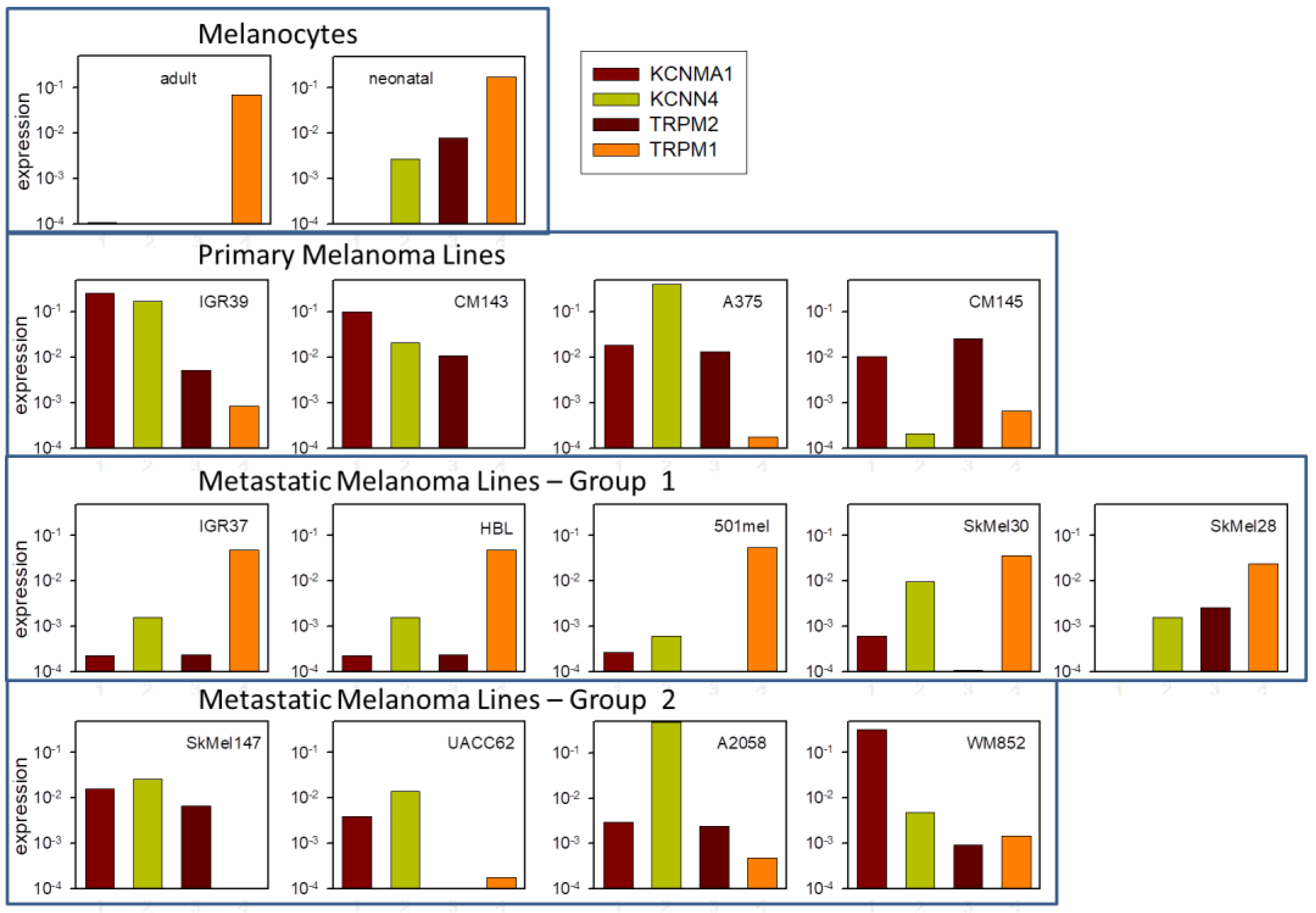
2.7. Channel Gene Expression Analysis
3. Discussion and Conclusions
4. Materials and Methods
4.1. Cell Culture
4.2. Chemicals
4.3. Patch-Clamp Experiments
4.4. Ca2+ Imaging with Fura-2-AM
4.5. Real-Time qPCR
4.6. In Silico Gene Expression Analysis
4.7. Statistics
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Hille, B. Ion Channels of Excitable Membranes, 3rd ed.; Sinauer: Sunderland, UK, 2001. [Google Scholar]
- Zheng, J.; Trudeau, M.C. Handbook of Ion Channels; CRC Press: Boca Raton, FL, USA, 2016; p. 691. [Google Scholar]
- Marino, A.; Dossena, S.; Tamma, G.; Donnini, S. Oxidative stress and membrane transport systems. Oxid. Med. Cell Longev. 2018, 2018, 9625213. [Google Scholar] [CrossRef] [Green Version]
- Singh, A.; Kukreti, R.; Saso, L.; Kukreti, S. Oxidative stress: A key modulator in neurodegenerative diseases. Molecules 2019, 24, 1583. [Google Scholar] [CrossRef] [Green Version]
- Xu, T.; Ding, W.; Ji, X.; Ao, X.; Liu, Y.; Yu, W.; Wang, J. Oxidative stress in cell death and cardiovascular diseases. Oxid. Med. Cell Longev. 2019, 2019, 9030563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rehman, K.; Akash, M.S.H. Mechanism of generation of oxidative stress and pathophysiology of type 2 diabetes mellitus: How are they interlinked? J. Cell. Biochem. 2017, 118, 3577–3585. [Google Scholar] [CrossRef] [PubMed]
- Davalli, P.; Mitic, T.; Caporali, A.; Lauriola, A.; D’Arca, D. ROS, Cell Senescence, and Novel Molecular Mechanisms in Aging and Age-Related Diseases. Oxid. Med. Cell. Longev. 2016, 2016, 3565127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorrini, C.; Harris, I.S.; Mak, T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013, 12, 931–947. [Google Scholar] [CrossRef]
- Gupta, R.K.; Patel, A.K.; Shah, N.; Chaudhary, A.K.; Jha, U.K.; Yadav, U.C.; Gupta, P.K.; Pakuwal, U. Oxidative stress and antioxidants in disease and cancer: A review. Asian Pac. J. Cancer Prev. 2014, 15, 4405–4409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sosa, V.; Moline, T.; Somoza, R.; Paciucci, R.; Kondoh, H.; ME, L.L. Oxidative stress and cancer: An overview. Ageing Res. Rev. 2013, 12, 376–390. [Google Scholar] [CrossRef]
- Sies, H. Oxidative stress: A concept in redox biology and medicine. Redox Biol. 2015, 4, 180–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Remigante, A.; Morabito, R.; Marino, A. Band 3 protein function and oxidative stress in erythrocytes. J. Cell Physiol. 2021, 236, 6225–6234. [Google Scholar] [CrossRef]
- Remigante, A.; Morabito, R.; Marino, A. Natural Antioxidants Beneficial Effects on Anion Exchange through Band 3 Protein in Human Erythrocytes. Antioxidants 2019, 9, 25. [Google Scholar] [CrossRef] [Green Version]
- Zuniga-Garcia, V.; Chavez-Lopez Mde, G.; Quintanar-Jurado, V.; Gabino-Lopez, N.B.; Hernandez-Gallegos, E.; Soriano-Rosas, J.; Perez-Carreon, J.I.; Camacho, J. Differential Expression of Ion Channels and Transporters During Hepatocellular Carcinoma Development. Dig. Dis. Sci. 2015, 60, 2373–2383. [Google Scholar] [CrossRef]
- Vogt, W. Oxidation of methionyl residues in proteins: Tools, targets, and reversal. Free Radic. Biol. Med. 1995, 18, 93–105. [Google Scholar] [CrossRef]
- Santarelli, L.C.; Wassef, R.; Heinemann, S.H.; Hoshi, T. Three methionine residues located within the regulator of conductance for K+ (RCK) domains confer oxidative sensitivity to large-conductance Ca2+-activated K+ channels. J. Physiol. 2006, 571, 329–348. [Google Scholar] [CrossRef] [PubMed]
- Fricke, T.C.; Echtermeyer, F.; Zielke, J.; de la Roche, J.; Filipovic, M.R.; Claverol, S.; Herzog, C.; Tominaga, M.; Pumroy, R.A.; Moiseenkova-Bell, V.Y.; et al. Oxidation of methionine residues activates the high-threshold heat-sensitive ion channel TRPV2. Proc. Natl. Acad. Sci. USA 2019, 116, 24359–24365. [Google Scholar] [CrossRef] [PubMed]
- Anderson, K.J.; Cormier, R.T.; Scott, P.M. Role of ion channels in gastrointestinal cancer. World J. Gastroenterol. 2019, 25, 5732–5772. [Google Scholar] [CrossRef]
- Ahmad, S.; Khan, M.Y.; Rafi, Z.; Khan, H.; Siddiqui, Z.; Rehman, S.; Shahab, U.; Khan, M.S.; Saeed, M.; Alouffi, S. Oxidation, glycation and glycoxidation-The vicious cycle and lung cancer. Semin. Cancer Biol. 2018, 49, 29–36. [Google Scholar] [CrossRef]
- Venza, M.; Visalli, M.; Beninati, C.; De Gaetano, G.V.; Teti, D.; Venza, I. Cellular Mechanisms of Oxidative Stress and Action in Melanoma. Oxid. Med. Cell. Longev. 2015, 2015, 481782. [Google Scholar] [CrossRef] [Green Version]
- Pardo, L.A.; Stühmer, W. The roles of K(+) channels in cancer. Nat. Rev. Cancer 2014, 14, 39–48. [Google Scholar] [CrossRef]
- Böhme, I.; Schönherr, R.; Eberle, J.; Bosserhoff, A.K. Membrane transporters and channels in melanoma. Rev. Physiol. Biochem. Pharmacol. 2020. [Google Scholar] [CrossRef]
- Yde, S.S.; Sjoegren, P.; Heje, M.; Stolle, L.B. Mucosal Melanoma: A Literature Review. Curr. Oncol. Rep. 2018, 20, 28. [Google Scholar] [CrossRef] [PubMed]
- Abbas, O.; Miller, D.D.; Bhawan, J. Cutaneous malignant melanoma: Update on diagnostic and prognostic biomarkers. Am. J. Dermatopathol. 2014, 36, 363–379. [Google Scholar] [CrossRef] [PubMed]
- Rosa, P.; Catacuzzeno, L.; Sforna, L.; Mangino, G.; Carlomagno, S.; Mincione, G.; Petrozza, V.; Ragona, G.; Franciolini, F.; Calogero, A. BK channels blockage inhibits hypoxia-induced migration and chemoresistance to cisplatin in human glioblastoma cells. J. Cell. Physiol. 2018, 233, 6866–6877. [Google Scholar] [CrossRef]
- Sizemore, G.; McLaughlin, S.; Newman, M.; Brundage, K.; Ammer, A.; Martin, K.; Pugacheva, E.; Coad, J.; Mattes, M.D.; Yu, H.G. Opening large-conductance potassium channels selectively induced cell death of triple-negative breast cancer. BMC Cancer 2020, 20, 595. [Google Scholar] [CrossRef] [PubMed]
- Goda, A.A.; Siddique, A.B.; Mohyeldin, M.; Ayoub, N.M.; El Sayed, K.A. The Maxi-K (BK) Channel Antagonist Penitrem A as a Novel Breast Cancer-Targeted Therapeutic. Mar. Drugs 2018, 16, 0157. [Google Scholar] [CrossRef] [Green Version]
- Tajima, N.; Itokazu, Y.; Korpi, E.R.; Somerharju, P.; Kakela, R. Activity of BK(Ca) channel is modulated by membrane cholesterol content and association with Na+/K+-ATPase in human melanoma IGR39 cells. J. Biol. Chem. 2011, 286, 5624–5638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cambien, B.; Rezzonico, R.; Vitale, S.; Rouzaire-Dubois, B.; Dubois, J.M.; Barthel, R.; Soilihi, B.K.; Mograbi, B.; Schmid-Alliana, A.; Schmid-Antomarchi, H. Silencing of hSlo potassium channels in human osteosarcoma cells promotes tumorigenesis. Int. J. Cancer 2008, 123, 365–371. [Google Scholar] [CrossRef]
- Latorre, R.; Castillo, K.; Carrasquel-Ursulaez, W.; Sepulveda, R.V.; Gonzalez-Nilo, F.; Gonzalez, C.; Alvarez, O. Molecular Determinants of BK Channel Functional Diversity and Functioning. Physiol. Rev. 2017, 97, 39–87. [Google Scholar] [CrossRef] [PubMed]
- Feng, B.; Ye, W.L.; Ma, L.J.; Fang, Y.; Mei, Y.A.; Wei, S.M. Hydrogen peroxide enhanced Ca(2+)-activated BK currents and promoted cell injury in human dermal fibroblasts. Life Sci. 2012, 90, 424–431. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Sun, X.; Zhu, Y.; Gan, L.; Xu, H.; Yang, X. Biphasic effects of H(2)O(2) on BK(Ca) channels. Free Radic. Res. 2010, 44, 1004–1012. [Google Scholar] [CrossRef] [PubMed]
- Hermann, A.; Sitdikova, G.F.; Weiger, T.M. Oxidative Stress and Maxi Calcium-Activated Potassium (BK) Channels. Biomolecules 2015, 5, 1870–1911. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klumpp, L.; Sezgin, E.C.; Skardelly, M.; Eckert, F.; Huber, S.M. KCa3.1 Channels and Glioblastoma: In Vitro Studies. Curr. Neuropharmacol. 2018, 16, 627–635. [Google Scholar] [CrossRef]
- De Marchi, U.; Sassi, N.; Fioretti, B.; Catacuzzeno, L.; Cereghetti, G.M.; Szabo, I.; Zoratti, M. Intermediate conductance Ca2+-activated potassium channel (KCa3.1) in the inner mitochondrial membrane of human colon cancer cells. Cell Calcium 2009, 45, 509–516. [Google Scholar] [CrossRef] [PubMed]
- Sassi, N.; De Marchi, U.; Fioretti, B.; Biasutto, L.; Gulbins, E.; Franciolini, F.; Szabo, I.; Zoratti, M. An investigation of the occurrence and properties of the mitochondrial intermediate-conductance Ca2+-activated K+ channel mtKCa3.1. Biochim. Biophys. Acta 2010, 1797, 1260–1267. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovalenko, I.; Glasauer, A.; Schockel, L.; Sauter, D.R.; Ehrmann, A.; Sohler, F.; Hagebarth, A.; Novak, I.; Christian, S. Identification of KCa3.1 channel as a novel regulator of oxidative phosphorylation in a subset of pancreatic carcinoma cell lines. PLoS ONE 2016, 11, e0160658. [Google Scholar] [CrossRef]
- Tajada, S.; Villalobos, C. Calcium permeable channels in cancer hallmarks. Front. Pharmacol. 2020, 11, 968. [Google Scholar] [CrossRef]
- Girault, A.; Ahidouch, A.; Ouadid-Ahidouch, H. Roles for Ca(2+) and K(+) channels in cancer cells exposed to the hypoxic tumour microenvironment. Biochim. Biophys. Acta Mol. Cell. Res. 2020, 1867, 118644. [Google Scholar] [CrossRef]
- Moloney, J.N.; Cotter, T.G. ROS signalling in the biology of cancer. Semin. Cell Dev. Biol. 2018, 80, 50–64. [Google Scholar] [CrossRef]
- Stueber, T.; Eberhardt, M.J.; Caspi, Y.; Lev, S.; Binshtok, A.; Leffler, A. Differential cytotoxicity and intracellular calcium-signalling following activation of the calcium-permeable ion channels TRPV1 and TRPA1. Cell Calcium 2017, 68, 34–44. [Google Scholar] [CrossRef]
- Kraft, R.; Grimm, C.; Grosse, K.; Hoffmann, A.; Sauerbruch, S.; Kettenmann, H.; Schultz, G.; Harteneck, C. Hydrogen peroxide and ADP-ribose induce TRPM2-mediated calcium influx and cation currents in microglia. Am. J. Physiol. Cell Physiol. 2004, 286, C129–C137. [Google Scholar] [CrossRef]
- Miller, B.A. TRPM2 in cancer. Cell Calcium 2019, 80, 8–17. [Google Scholar] [CrossRef]
- Ji, D.; Luo, Z.W.; Ovcjak, A.; Alanazi, R.; Bao, M.H.; Feng, Z.P.; Sun, H.S. Role of TRPM2 in brain tumours and potential as a drug target. Acta Pharmacol. Sin. 2021, 1–12. [Google Scholar] [CrossRef]
- Gokce Kutuk, S.; Gokce, G.; Kutuk, M.; Gurses Cila, H.E.; Naziroglu, M. Curcumin enhances cisplatin-induced human laryngeal squamous cancer cell death through activation of TRPM2 channel and mitochondrial oxidative stress. Sci. Rep. 2019, 9, 17784. [Google Scholar] [CrossRef] [PubMed]
- Wang, Q.; Huang, L.; Yue, J. Oxidative stress activates the TRPM2-Ca(2+)-CaMKII-ROS signaling loop to induce cell death in cancer cells. Biochim. Biophys. Acta. Mol. Cell Res. 2017, 1864, 957–967. [Google Scholar] [CrossRef]
- Miller, B.A.; Wang, J.; Song, J.; Zhang, X.Q.; Hirschler-Laszkiewicz, I.; Shanmughapriya, S.; Tomar, D.; Rajan, S.; Feldman, A.M.; Madesh, M.; et al. Trpm2 enhances physiological bioenergetics and protects against pathological oxidative cardiac injury: Role of Pyk2 phosphorylation. J. Cell Physiol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Miller, B.A.; Cheung, J.Y. TRPM2 protects against tissue damage following oxidative stress and ischaemia-reperfusion. J. Physiol. 2016, 594, 4181–4191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ge, L.; Hoa, N.T.; Wilson, Z.; Arismendi-Morillo, G.; Kong, X.T.; Tajhya, R.B.; Beeton, C.; Jadus, M.R. Big Potassium (BK) ion channels in biology, disease and possible targets for cancer immunotherapy. Int. Immunopharmacol. 2014, 22, 427–443. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bentzen, B.H.; Olesen, S.P.; Ronn, L.C.; Grunnet, M. BK channel activators and their therapeutic perspectives. Front. Physiol. 2014, 5, 389. [Google Scholar] [CrossRef] [Green Version]
- Xia, J.; Wang, H.; Li, S.; Wu, Q.; Sun, L.; Huang, H.; Zeng, M. Ion channels or aquaporins as novel molecular targets in gastric cancer. Mol. Cancer 2017, 16, 54. [Google Scholar] [CrossRef] [Green Version]
- Sterea, A.M.; Egom, E.E.; El Hiani, Y. TRP channels in gastric cancer: New hopes and clinical perspectives. Cell Calcium 2019, 82, 102053. [Google Scholar] [CrossRef]
- Meyer, R.; Schönherr, R.; Gavrilova-Ruch, O.; Wohlrab, W.; Heinemann, S.H. Identification of ether a go-go and calcium-activated potassium channels in human melanoma cells. J. Membr. Biol. 1999, 171, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Tajima, N.; Schonherr, K.; Niedling, S.; Kaatz, M.; Kanno, H.; Schonherr, R.; Heinemann, S.H. Ca2+-activated K+ channels in human melanoma cells are up-regulated by hypoxia involving hypoxia-inducible factor-1alpha and the von Hippel-Lindau protein. J. Physiol. 2006, 571, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Langton, P.D.; Nelson, M.T.; Huang, Y.; Standen, N.B. Block of calcium-activated potassium channels in mammalian arterial myocytes by tetraethylammonium ions. Am. J. Physiol. 1991, 260, H927–H934. [Google Scholar] [CrossRef]
- Zhou, Y.; Lingle, C.J. Paxilline inhibits BK channels by an almost exclusively closed-channel block mechanism. J. Gen. Physiol. 2014, 144, 415–440. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duprat, F.; Girard, C.; Jarretou, G.; Lazdunski, M. Pancreatic two P domain K+ channels TALK-1 and TALK-2 are activated by nitric oxide and reactive oxygen species. J. Physiol. 2005, 562, 235–244. [Google Scholar] [CrossRef] [PubMed]
- Tarasov, M.V.; Kotova, P.D.; Bystrova, M.F.; Kabanova, N.V.; Sysoeva, V.Y.; Kolesnikov, S.S. Arachidonic acid hyperpolarizes mesenchymal stromal cells from the human adipose tissue by stimulating TREK1 K(+) channels. Channels 2019, 13, 36–47. [Google Scholar] [CrossRef] [Green Version]
- Papreck, J.R.; Martin, E.A.; Lazzarini, P.; Kang, D.; Kim, D. Modulation of K2P3.1 (TASK-1), K2P9.1 (TASK-3), and TASK-1/3 heteromer by reactive oxygen species. Pflugers Arch. 2012, 464, 471–480. [Google Scholar] [CrossRef] [Green Version]
- Wei, A.D.; Gutman, G.A.; Aldrich, R.; Chandy, K.G.; Grissmer, S.; Wulff, H. International Union of Pharmacology. LII. Nomenclature and molecular relationships of calcium-activated potassium channels. Pharmacol. Rev. 2005, 57, 463–472. [Google Scholar] [CrossRef] [Green Version]
- McNaughton-Smith, G.A.; Burns, J.F.; Stocker, J.W.; Rigdon, G.C.; Creech, C.; Arrington, S.; Shelton, T.; de Franceschi, L. Novel inhibitors of the Gardos channel for the treatment of sickle cell disease. J. Med. Chem. 2008, 51, 976–982. [Google Scholar] [CrossRef]
- Nguyen, H.M.; Singh, V.; Pressly, B.; Jenkins, D.P.; Wulff, H.; Yarov-Yarovoy, V. Structural Insights into the Atomistic Mechanisms of Action of Small Molecule Inhibitors Targeting the KCa3.1 Channel Pore. Mol. Pharmacol. 2017, 91, 392–402. [Google Scholar] [CrossRef] [Green Version]
- Ataga, K.I.; Smith, W.R.; De Castro, L.M.; Swerdlow, P.; Saunthararajah, Y.; Castro, O.; Vichinsky, E.; Kutlar, A.; Orringer, E.P.; Rigdon, G.C.; et al. Efficacy and safety of the Gardos channel blocker, senicapoc (ICA-17043), in patients with sickle cell anemia. Blood 2008, 111, 3991–3997. [Google Scholar] [CrossRef]
- Bessac, B.F.; Sivula, M.; von Hehn, C.A.; Escalera, J.; Cohn, L.; Jordt, S.E. TRPA1 is a major oxidant sensor in murine airway sensory neurons. J. Clin. Investig. 2008, 118, 1899–1910. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hack, C.T.; Buck, T.; Bagnjuk, K.; Eubler, K.; Kunz, L.; Mayr, D.; Mayerhofer, A. A Role for H2O2 and TRPM2 in the Induction of Cell Death: Studies in KGN Cells. Antioxidants 2019, 8, 518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hill, K.; McNulty, S.; Randall, A.D. Inhibition of TRPM2 channels by the antifungal agents clotrimazole and econazole. Naunyn Schmiedebergs Arch. Pharmacol. 2004, 370, 227–237. [Google Scholar] [CrossRef]
- Kraft, R.; Grimm, C.; Frenzel, H.; Harteneck, C. Inhibition of TRPM2 cation channels by N-(p-amylcinnamoyl)anthranilic acid. Br. J. Pharmacol. 2006, 148, 264–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ito, C.; Tecchio, C.; Coustan-Smith, E.; Suzuki, T.; Behm, F.G.; Raimondi, S.C.; Pui, C.H.; Campana, D. The antifungal antibiotic clotrimazole alters calcium homeostasis of leukemic lymphoblasts and induces apoptosis. Leukemia 2002, 16, 1344–1352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- D’Arcangelo, D.; Scatozza, F.; Giampietri, C.; Marchetti, P.; Facchiano, F.; Facchiano, A. Ion Channel Expression in Human Melanoma Samples: In Silico Identification and Experimental Validation of Molecular Targets. Cancers 2019, 11, 446. [Google Scholar] [CrossRef] [Green Version]
- Salinas, M.; Reyes, R.; Lesage, F.; Fosset, M.; Heurteaux, C.; Romey, G.; Lazdunski, M. Cloning of a new mouse two-P domain channel subunit and a human homologue with a unique pore structure. J. Biol Chem. 1999, 274, 11751–11760. [Google Scholar] [CrossRef] [Green Version]
- Daenen, K.; Andries, A.; Mekahli, D.; Van Schepdael, A.; Jouret, F.; Bammens, B. Oxidative stress in chronic kidney disease. Pediatr. Nephrol. 2019, 34, 975–991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eisfeld, J.; Luckhoff, A. Trpm2. Handbook of Experimental Pharmacology; Springer: Berlin/Heidelberg, Germany, 2007; pp. 237–252. [Google Scholar] [CrossRef]
- Meseguer, V.; Karashima, Y.; Talavera, K.; D’Hoedt, D.; Donovan-Rodriguez, T.; Viana, F.; Nilius, B.; Voets, T. Transient receptor potential channels in sensory neurons are targets of the antimycotic agent clotrimazole. J. Neurosci. 2008, 28, 576–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harteneck, C.; Frenzel, H.; Kraft, R. N-(p-amylcinnamoyl)anthranilic acid (ACA): A phospholipase A(2) inhibitor and TRP channel blocker. Cardiovasc. Drug Rev. 2007, 25, 61–75. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhao, S.; Yu, J.; Yang, W.; Liu, Z.; Zhang, L. Medicinal chemistry perspective of TRPM2 channel inhibitors: Where we are and where we might be heading? Drug Discov. Today 2020, 25, 2326–2334. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Zeng, B.; Chen, G.L.; Bot, D.; Eastmond, S.; Elsenussi, S.E.; Atkin, S.L.; Boa, A.N.; Xu, S.Z. Effect of non-steroidal anti-inflammatory drugs and new fenamate analogues on TRPC4 and TRPC5 channels. Biochem. Pharmacol. 2012, 83, 923–931. [Google Scholar] [CrossRef]
- Kashio, M.; Sokabe, T.; Shintaku, K.; Uematsu, T.; Fukuta, N.; Kobayashi, N.; Mori, Y.; Tominaga, M. Redox signal-mediated sensitization of transient receptor potential melastatin 2 (TRPM2) to temperature affects macrophage functions. Proc. Natl. Acad. Sci. USA 2012, 109, 6745–6750. [Google Scholar] [CrossRef] [Green Version]
- Huang, C.; Gan, D.; Fan, C.; Wen, C.; Li, A.; Li, Q.; Zhao, J.; Wang, Z.; Zhu, L.; Lu, D. The Secretion from Neural Stem Cells Pretreated with Lycopene Protects against tert-Butyl Hydroperoxide-Induced Neuron Oxidative Damage. Oxid. Med. Cell Longev. 2018, 2018, 5490218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gradogna, A.; Gavazzo, P.; Boccaccio, A.; Pusch, M. Subunit-dependent oxidative stress sensitivity of LRRC8 volume-regulated anion channels. J. Physiol. 2017, 595, 6719–6733. [Google Scholar] [CrossRef]
- Tang, X.D.; Daggett, H.; Hanner, M.; Garcia, M.L.; McManus, O.B.; Brot, N.; Weissbach, H.; Heinemann, S.H.; Hoshi, T. Oxidative regulation of large conductance calcium-activated potassium channels. J. Gen. Physiol. 2001, 117, 253–274. [Google Scholar] [CrossRef] [Green Version]
- Mazar, J.; DeYoung, K.; Khaitan, D.; Meister, E.; Almodovar, A.; Goydos, J.; Ray, A.; Perera, R.J. The regulation of miRNA-211 expression and its role in melanoma cell invasiveness. PLoS ONE 2010, 5, e13779. [Google Scholar] [CrossRef] [Green Version]
- Agaimy, A.; Specht, K.; Stoehr, R.; Lorey, T.; Markl, B.; Niedobitek, G.; Straub, M.; Hager, T.; Reis, A.C.; Schilling, B.; et al. Metastatic Malignant Melanoma with Complete Loss of Differentiation Markers (Undifferentiated/Dedifferentiated Melanoma): Analysis of 14 Patients Emphasizing Phenotypic Plasticity and the Value of Molecular Testing as Surrogate Diagnostic Marker. Am. J. Surg. Pathol. 2016, 40, 181–191. [Google Scholar] [CrossRef]
- Lin, R.; Wang, Y.; Chen, Q.; Liu, Z.; Xiao, S.; Wang, B.; Shi, B. TRPM2 promotes the proliferation and invasion of pancreatic ductal adenocarcinoma. Mol. Med. Rep. 2018, 17, 7537–7544. [Google Scholar] [CrossRef]
- Du, C.; Zheng, Z.; Li, D.; Chen, L.; Li, N.; Yi, X.; Yang, Y.; Guo, F.; Liu, W.; Xie, X.; et al. BKCa promotes growth and metastasis of prostate cancer through facilitating the coupling between alphavbeta3 integrin and FAK. Oncotarget 2016, 7, 40174–40188. [Google Scholar] [CrossRef]
- Hoa, N.T.; Ge, L.; Tajhya, R.B.; Beeton, C.; Cornforth, A.N.; Abolhoda, A.; Lambrecht, N.; DaCosta-Iyer, M.; Ouyang, Y.; Mai, A.P.; et al. Small cell lung cancer cells express the late stage gBK tumor antigen: A possible immunotarget for the terminal disease. Am. J. Transl. Res. 2014, 6, 188–205. [Google Scholar]
- Ma, Y.G.; Liu, W.C.; Dong, S.; Du, C.; Wang, X.J.; Li, J.S.; Xie, X.P.; Wu, L.; Ma, D.C.; Yu, Z.B.; et al. Activation of BK(Ca) channels in zoledronic acid-induced apoptosis of MDA-MB-231 breast cancer cells. PLoS ONE 2012, 7, e37451. [Google Scholar] [CrossRef] [Green Version]
- Mohr, C.J.; Schroth, W.; Murdter, T.E.; Gross, D.; Maier, S.; Stegen, B.; Dragoi, A.; Steudel, F.A.; Stehling, S.; Hoppe, R.; et al. Subunits of BK channels promote breast cancer development and modulate responses to endocrine treatment in preclinical models. Br. J. Pharmacol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Khaitan, D.; Sankpal, U.T.; Weksler, B.; Meister, E.A.; Romero, I.A.; Couraud, P.O.; Ningaraj, N.S. Role of KCNMA1 gene in breast cancer invasion and metastasis to brain. BMC Cancer 2009, 9, 258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmidt, J.; Friebel, K.; Schönherr, R.; Coppolino, M.G.; Bosserhoff, A.K. Migration-associated secretion of melanoma inhibitory activity at the cell rear is supported by KCa3.1 potassium channels. Cell Res. 2010, 20, 1224–1238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, B.M.; Pressley, B.; Wulff, H. KCa3.1 Channel Modulators as Potential Therapeutic Compounds for Glioblastoma. Curr. Neuropharmacol. 2018, 16, 618–626. [Google Scholar] [CrossRef] [PubMed]
- Catacuzzeno, L.; Sforna, L.; Esposito, V.; Limatola, C.; Franciolini, F. Ion Channels in Glioma Malignancy. Rev. Physiol. Biochem. Pharmacol. 2020. [Google Scholar] [CrossRef]
- Jurman, M.E.; Boland, L.M.; Liu, Y.; Yellen, G. Visual identification of individual transfected cells for electrophysiology using antibody-coated beads. Biotechniques 1994, 17, 876–881. [Google Scholar]
- Pusch, M.; Zifarelli, G. Large transient capacitive currents in wild-type lysosomal Cl-/H+ antiporter ClC-7 and residual transport activity in the proton glutamate mutant E312A. J. Gen. Physiol. 2021, 153. [Google Scholar] [CrossRef] [PubMed]
- Bertelli, S.; Barbieri, R.; Pusch, M.; Gavazzo, P. Gain of function of sporadic/familial hemiplegic migraine-causing SCN1A mutations: Use of an optimized cDNA. Cephalalgia Int. J. Headache 2018, 39, 477–488. [Google Scholar] [CrossRef] [PubMed]
- Barbieri, R.; Bertelli, S.; Pusch, M.; Gavazzo, P. Late sodium current blocker GS967 inhibits persistent currents induced by familial hemiplegic migraine type 3 mutations of the SCN1A gene. J. Headache Pain 2019, 20, 107. [Google Scholar] [CrossRef]
- Grynkiewicz, G.; Poenie, M.; Tsien, R.Y. A new generation of Ca2+ indicators with greatly improved fluorescence properties. J. Biol. Chem. 1985, 260, 3440–3450. [Google Scholar] [CrossRef]
- Langmead, B.; Trapnell, C.; Pop, M.; Salzberg, S.L. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009, 10, R25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dobin, A.; Davis, C.A.; Schlesinger, F.; Drenkow, J.; Zaleski, C.; Jha, S.; Batut, P.; Chaisson, M.; Gingeras, T.R. STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 2013, 29, 15–21. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| NaCl | KCl | CaCl2 | MgCl2 | HEPES | TEACl | Mannitol | EGTA | |
|---|---|---|---|---|---|---|---|---|
| STD | 145 | 5 | 2 | 1 | 10 | - | - | - |
| STD 0 Ca2+ | 145 | 5 | - | 1 | 10 | - | - | 3 |
| STD TEA | 115 | 5 | 2 | 1 | 10 | 30 | - | - |
| ISO-TEA | 60 | 5 | 2 | 1 | 10 | 30 | 100 | - |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ferrera, L.; Barbieri, R.; Picco, C.; Zuccolini, P.; Remigante, A.; Bertelli, S.; Fumagalli, M.R.; Zifarelli, G.; La Porta, C.A.M.; Gavazzo, P.; et al. TRPM2 Oxidation Activates Two Distinct Potassium Channels in Melanoma Cells through Intracellular Calcium Increase. Int. J. Mol. Sci. 2021, 22, 8359. https://doi.org/10.3390/ijms22168359
Ferrera L, Barbieri R, Picco C, Zuccolini P, Remigante A, Bertelli S, Fumagalli MR, Zifarelli G, La Porta CAM, Gavazzo P, et al. TRPM2 Oxidation Activates Two Distinct Potassium Channels in Melanoma Cells through Intracellular Calcium Increase. International Journal of Molecular Sciences. 2021; 22(16):8359. https://doi.org/10.3390/ijms22168359
Chicago/Turabian StyleFerrera, Loretta, Raffaella Barbieri, Cristiana Picco, Paolo Zuccolini, Alessia Remigante, Sara Bertelli, Maria Rita Fumagalli, Giovanni Zifarelli, Caterina A. M. La Porta, Paola Gavazzo, and et al. 2021. "TRPM2 Oxidation Activates Two Distinct Potassium Channels in Melanoma Cells through Intracellular Calcium Increase" International Journal of Molecular Sciences 22, no. 16: 8359. https://doi.org/10.3390/ijms22168359
APA StyleFerrera, L., Barbieri, R., Picco, C., Zuccolini, P., Remigante, A., Bertelli, S., Fumagalli, M. R., Zifarelli, G., La Porta, C. A. M., Gavazzo, P., & Pusch, M. (2021). TRPM2 Oxidation Activates Two Distinct Potassium Channels in Melanoma Cells through Intracellular Calcium Increase. International Journal of Molecular Sciences, 22(16), 8359. https://doi.org/10.3390/ijms22168359