
Efficient Neuroprotective Rescue of Sacsin-Related Disease Phenotypes in Zebrafish

 , , , ,
, , , ,  ,
,  ,
,  ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
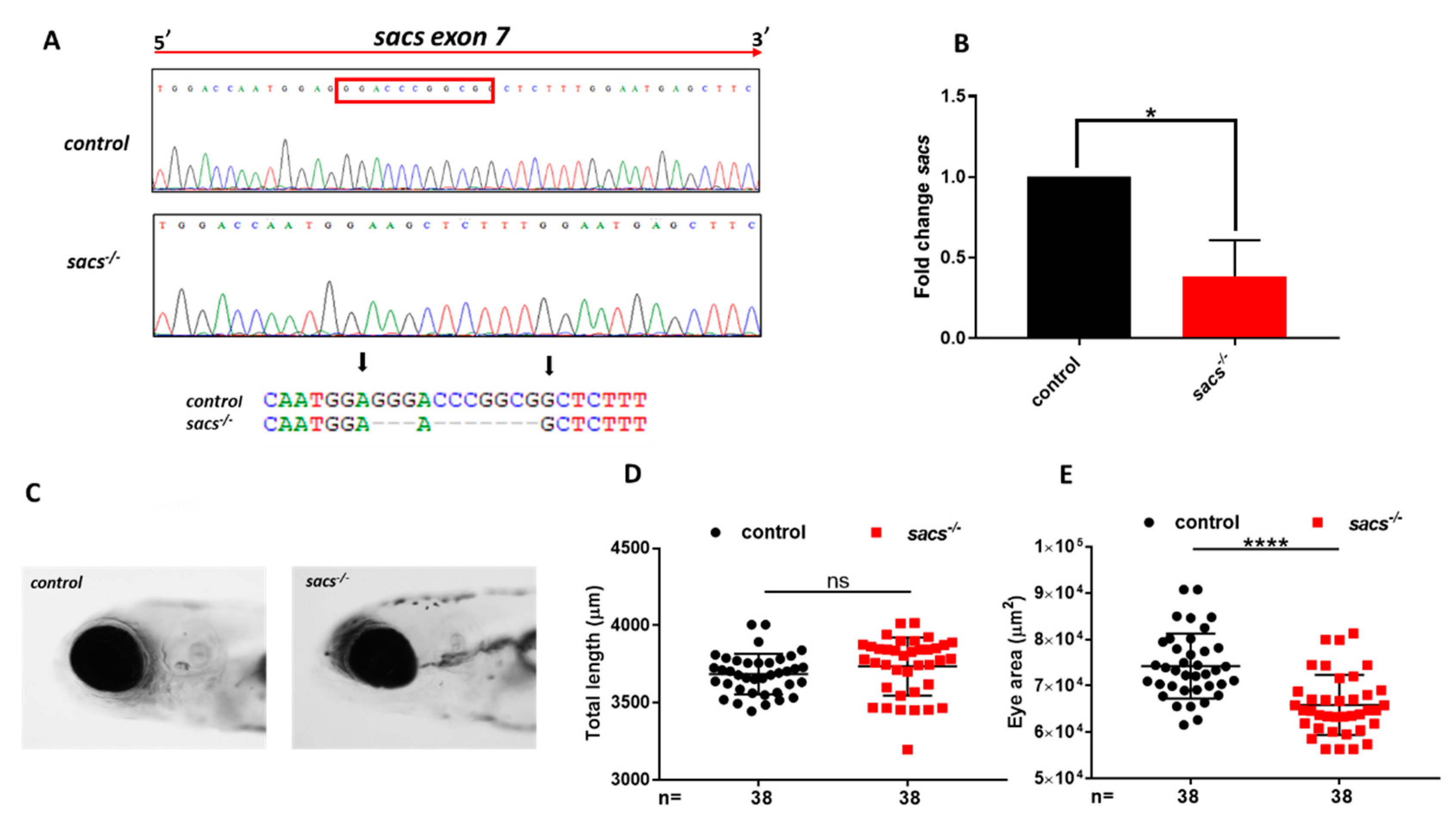
2.1. Generation of Sacs-Null Mutant Zebrafish
2.2. Sacs−/− Mutant Zebrafish Displays Motor Impairment and Reduced “Cerebellar” Area
2.3. Sacs−/− Mutant Zebrafish Manifest Mitochondrial and Autophagic Dysfunction and ROS Accumulation
2.4. Acetyl-DL-leucine and TUDCA Prevent Impairment of Locomotor Activity in Sacs−/− Larvae and Enhance Responses to Light and Dark Transitions
2.5. ADLL and TUDCA Restore the Gene Expression Profile and Prevent Apoptosis in Sacs−/− Larvae
3. Discussion
4. Materials and Methods
4.1. Zebrafish Husbandry
4.2. Multiple Alignments of Sacsin Amino Acid Sequences
4.3. Establishing the Mutant Line
4.4. Genotyping
4.5. Quantitative Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
4.6. Immunohistochemistry Staining of Whole-Mount Zebrafish Embryos
4.7. Analysis of Larval Morphology
4.8. Locomotor Behavior
4.9. Mitochondrial Respiratory Analysis
4.10. Calcium Imaging
4.11. Oxidative Stress Measurement
4.12. Western Blotting
4.13. Detection of Apoptotic Cells
4.14. Pharmacological Treatments
4.15. Birefringence Assay
4.16. Statistics
Supplementary Materials
Author Contributions
Funding
Ethics Statement
Institutional Review Board Statement
Informed Consent Statement
Acknowledgments
Conflicts of Interest
References
- Parfitt, D.A.; Michael, G.J.; Vermeulen, E.G.; Prodromou, N.V.; Webb, T.; Gallo, J.-M.; Cheetham, M.E.; Nicoll, W.S.; Blatch, G.L.; Chapple, J.P. The ataxia protein sacsin is a functional co-chaperone that protects against polyglutamine-expanded ataxin-1. Hum. Mol. Genet. 2009, 18, 1556–1565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vingolo, E.M.; Di Fabio, R.; Salvatore, S.; Grieco, G.; Bertini, E.; Leuzzi, V.; Nesti, C.; Filla, A.; Tessa, A.; Pierelli, F.; et al. Myelinated retinal fibers in autosomal recessive spastic ataxia of Charlevoix-Saguenay. Eur. J. Neurol. 2011, 18, 1187–1190. [Google Scholar] [CrossRef] [PubMed]
- Parkinson, M.H.; Bartmann, A.P.; Clayton, L.M.S.; Nethisinghe, S.; Pfundt, R.; Chapple, J.P.; Reilly, M.M.; Manji, H.; Wood, N.; Bremner, F.; et al. Optical coherence tomography in autosomal recessive spastic ataxia of Charlevoix-Saguenay. Brain 2018, 141, 989–999. [Google Scholar] [CrossRef] [PubMed]
- Engert, J.; Bérubé, P.; Mercier, J.; Doré, C.; Lepage, P.; Ge, B.; Bouchard, J.-P.; Mathieu, J.; Melançon, S.B.; Schalling, M.; et al. ARSACS, a spastic ataxia common in northeastern Québec, is caused by mutations in a new gene encoding an 11.5-kb ORF. Nat. Genet. 2000, 24, 120–125. [Google Scholar] [CrossRef] [PubMed]
- Anderson, J.F.; Siller, E.; Barral, J.M. The Sacsin Repeating Region (SRR): A Novel Hsp90-Related Supra-Domain Associated with Neurodegeneration. J. Mol. Biol. 2010, 400, 665–674. [Google Scholar] [CrossRef]
- Kozlov, G.; Denisov, A.Y.; Girard, M.; Dicaire, M.-J.; Hamlin, J.; McPherson, P.S.; Brais, B.; Gehring, K. Structural Basis of Defects in the Sacsin HEPN Domain Responsible for Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay (ARSACS). J. Biol. Chem. 2011, 286, 20407–20412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Romano, A.; Tessa, A.; Barca, A.; Fattori, F.; De Leva, M.F.; Terracciano, A.; Storelli, C.; Santorelli, F.M.; Verri, T. Comparative Analysis and Functional Mapping of SACS Mutations Reveal Novel Insights into Sacsin Repeated Architecture. Hum. Mutat. 2013, 34, 525–537. [Google Scholar] [CrossRef]
- Girard, M.; Larivière, R.; Parfitt, D.A.; Deane, E.C.; Gaudet, R.; Nossova, N.; Blondeau, F.; Prenosil, G.; Vermeulen, E.G.M.; Duchen, M.; et al. Mitochondrial dysfunction and Purkinje cell loss in autosomal recessive spastic ataxia of Charlevoix-Saguenay (ARSACS). Proc. Natl. Acad. Sci. USA 2012, 109, 1661–1666. [Google Scholar] [CrossRef] [Green Version]
- Duncan, E.J.; Larivière, R.; Bradshaw, T.Y.; Longo, F.; Sgarioto, N.; Hayes, M.J.; Romano, L.E.; Nethisinghe, S.; Giunti, P.; Bruntraeger, M.B.; et al. Altered organization of the intermediate filament cytoskeleton and relocalization of proteostasis modulators in cells lacking the ataxia protein sacsin. Hum. Mol. Genet. 2017, 26, 3130–3143. [Google Scholar] [CrossRef] [Green Version]
- Larivière, R.; Gaudet, R.; Gentil, B.J.; Girard, M.; Conte, T.C.; Minotti, S.; Leclerc-Desaulniers, K.; Gehring, K.; McKinney, R.A.; Shoubridge, E.A.; et al. Sacs knockout mice present pathophysiological defects underlying autosomal recessive spastic ataxia of Charlevoix-Saguenay. Hum. Mol. Genet. 2014, 24, 727–739. [Google Scholar] [CrossRef] [Green Version]
- Larivière, R.; Sgarioto, N.; Márquez, B.T.; Gaudet, R.; Choquet, K.; McKinney, R.A.; Watt, A.J.; Brais, B. Sacs R272C missense homozygous mice develop an ataxia phenotype. Mol. Brain 2019, 12, 19. [Google Scholar] [CrossRef] [PubMed]
- Morani, F.; Doccini, S.; Sirica, R.; Paterno, M.; Pezzini, F.; Ricca, I.; Simonati, A.; Delledonne, M.; Santorelli, F.M. Functional Transcriptome Analysis in ARSACS KO Cell Model Reveals a Role of Sacsin in Autophagy. Sci. Rep. 2019, 9, 11878. [Google Scholar] [CrossRef] [PubMed]
- Morani, F.; Doccini, S.; Chiorino, G.; Fattori, F.; Galatolo, D.; Sciarrillo, E.; Gemignani, F.; Züchner, S.; Bertini, E.S.; Santorelli, F.M. Functional Network Profiles in ARSACS Disclosed by Aptamer-Based Proteomic Technology. Front. Neurol. 2021, 11, 604744. [Google Scholar] [CrossRef]
- Stewart, A.M.; Braubach, O.; Spitsbergen, J.; Gerlai, R.; Kalueff, A.V. Zebrafish models for translational neuroscience research: From tank to bedside. Trends Neurosci. 2014, 37, 264–278. [Google Scholar] [CrossRef] [Green Version]
- Saleem, S.; Kannan, R.R. Zebrafish: An emerging real-time model system to study Alzheimer’s disease and neurospecific drug discovery. Cell Death Discov. 2018, 4, 45. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naef, V.; Mero, S.; Fichi, G.; D’Amore, A.; Ogi, A.; Gemignani, F.; Santorelli, F.M.; Marchese, M. Swimming in Deep Water: Zebrafish Modeling of Complicated Forms of Hereditary Spastic Paraplegia and Spastic Ataxia. Front. Neurosci. 2019, 13, 1311. [Google Scholar] [CrossRef] [PubMed]
- Fichi, G.; Naef, V.; Barca, A.; Longo, G.; Fronte, B.; Verri, T.; Santorelli, F.M.; Marchese, M.; Petruzzella, V. Fishing in the Cell Powerhouse: Zebrafish as A Tool for Exploration of Mitochondrial Defects Affecting the Nervous System. Int. J. Mol. Sci. 2019, 20, 2409. [Google Scholar] [CrossRef] [Green Version]
- Clark, J.M. The 3Rs in research: A contemporary approach to replacement, reduction and refinement. Br. J. Nutr. 2017, 120, S1–S7. [Google Scholar] [CrossRef]
- Pasupuleti, M.K.; Molahally, S.S.; Salwaji, S. Ethical guidelines, animal profile, various animal models used in periodontal research with alternatives and future perspectives. J. Indian Soc. Periodontol. 2016, 20, 360–368. [Google Scholar] [CrossRef]
- Ogi, A.; Licitra, R.; Naef, V.; Marchese, M.; Fronte, B.; Gazzano, A.; Santorelli, F.M. Social Preference Tests in Zebrafish: A Systematic Review. Front. Vet. Sci. 2021, 7. [Google Scholar] [CrossRef]
- Noyes, P.D.; Garcia, G.R.; Tanguay, R.L. Zebrafish as an in vivo model for sustainable chemical design. Green Chem. 2016, 18, 6410–6430. [Google Scholar] [CrossRef] [Green Version]
- Sukardi, H.; Chng, H.T.; Chan, E.C.Y.; Gong, Z.; Lam, S.H. Zebrafish for drug toxicity screening: Bridging thein vitrocell-based models andin vivomammalian models. Expert Opin. Drug Metab. Toxicol. 2011, 7, 579–589. [Google Scholar] [CrossRef]
- Du, X.-F.; Xu, B.; Zhang, Y.; Chen, M.-J.; Du, J.-L. A transgenic zebrafish model for in vivo long-term imaging of retinotectal synaptogenesis. Sci. Rep. 2018, 8, 14077. [Google Scholar] [CrossRef]
- Matsui, H.; Namikawa, K.; Babaryka, A.; Köster, R. Functional regionalization of the teleost cerebellum analyzed in vivo. Proc. Natl. Acad. Sci. USA 2014, 111, 11846–11851. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacRae, C.A.; Peterson, R.T. Zebrafish as tools for drug discovery. Nat. Rev. Drug Discov. 2015, 14, 721–731. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhou, Y.; Qi, X.; Chen, J.; Chen, W.; Qiu, G.; Wu, Z.; Wu, N. CRISPR/Cas9 in zebrafish: An efficient combination for human genetic diseases modeling. Qual. Life Res. 2016, 136, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buglo, E.; Sarmiento, E.; Martuscelli, N.B.; Sant, D.W.; Danzi, M.C.; Abrams, A.J.; Dallman, J.E.; Züchner, S. Genetic compensation in a stable slc25a46 mutant zebrafish: A case for using F0 CRISPR mutagenesis to study phenotypes caused by inherited disease. PLoS ONE 2020, 15, e0230566. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colwill, R.M.; Creton, R. Locomotor behaviors in zebrafish (Danio rerio) larvae. Behav. Process. 2011, 86, 222–229. [Google Scholar] [CrossRef] [Green Version]
- Martin, M.-H.; Bouchard, J.-P.; Sylvain, M.; St-Onge, O.; Truchon, S. Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay: A Report of MR Imaging in 5 Patients. Am. J. Neuroradiol. 2007, 28, 1606–1608. [Google Scholar] [CrossRef] [Green Version]
- Aizenberg, M.; Schuman, E.M. Cerebellar-Dependent Learning in Larval Zebrafish. J. Neurosci. 2011, 31, 8708–8712. [Google Scholar] [CrossRef]
- Harmon, T.C.; Magaram, U.; McLean, D.L.; Raman, I.M. Distinct responses of Purkinje neurons and roles of simple spikes during associative motor learning in larval zebrafish. eLife 2017, 6, e22537. [Google Scholar] [CrossRef]
- Portugues, R.; Feierstein, C.; Engert, F.; Orger, M.B. Whole-Brain Activity Maps Reveal Stereotyped, Distributed Networks for Visuomotor Behavior. Neuron 2014, 81, 1328–1343. [Google Scholar] [CrossRef] [Green Version]
- Namikawa, K.; Dorigo, A.; Zagrebelsky, M.; Russo, G.; Kirmann, T.; Fahr, W.; Dübel, S.; Korte, M.; Köster, R.W. Modeling Neurodegenerative Spinocerebellar Ataxia Type 13 in Zebrafish Using a Purkinje Neuron Specific Tunable Coexpression System. J. Neurosci. 2019, 39, 3948–3969. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bae, Y.-K.; Kani, S.; Shimizu, T.; Tanabe, K.; Nojima, H.; Kimura, Y.; Higashijima, S.-I.; Hibi, M. Anatomy of zebrafish cerebellum and screen for mutations affecting its development. Dev. Biol. 2009, 330, 406–426. [Google Scholar] [CrossRef] [Green Version]
- Kasumu, A.; Bezprozvanny, I. Deranged Calcium Signaling in Purkinje Cells and Pathogenesis in Spinocerebellar Ataxia 2 (SCA2) and Other Ataxias. Cerebellum 2010, 11, 630–639. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calvo-Rodriguez, M.; Hou, S.S.; Snyder, A.C.; Kharitonova, E.K.; Russ, A.N.; Das, S.; Fan, Z.; Muzikansky, A.; Garcia-Alloza, M.; Serrano-Pozo, A.; et al. Increased mitochondrial calcium levels associated with neuronal death in a mouse model of Alzheimer’s disease. Nat. Commun. 2020, 11, 2146. [Google Scholar] [CrossRef] [PubMed]
- Cozzolino, O.; Sicca, F.; Paoli, E.; Trovato, F.; Santorelli, F.M.; Ratto, G.M.; Marchese, M. Evolution of Epileptiform Activity in Zebrafish by Statistical-Based Integration of Electrophysiology and 2-Photon Ca2+ Imaging. Cells 2020, 9, 769. [Google Scholar] [CrossRef] [Green Version]
- Criscuolo, C.; Procaccini, C.; Meschini, M.C.; Cianflone, A.; Carbone, R.; Doccini, S.; Devos, D.; Nesti, C.; Vuillaume, I.; Pellegrino, M.; et al. Powerhouse failure and oxidative damage in autosomal recessive spastic ataxia of Charlevoix-Saguenay. J. Neurol. 2015, 262, 2755–2763. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Gehring, K. Structural studies of parkin and sacsin: Mitochondrial dynamics in neurodegenerative diseases. Mov. Disord. 2015, 30, 1610–1619. [Google Scholar] [CrossRef] [PubMed]
- Ashrafi, G.H.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2012, 20, 31–42. [Google Scholar] [CrossRef] [Green Version]
- Runwal, G.; Stamatakou, E.; Siddiqi, F.H.; Puri, C.; Zhu, Y.; Rubinsztein, D.C. LC3-positive structures are prominent in autophagy-deficient cells. Sci. Rep. 2019, 9, 10147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshii, S.R.; Mizushima, N. Monitoring and Measuring Autophagy. Int. J. Mol. Sci. 2017, 18, 1865. [Google Scholar] [CrossRef] [PubMed]
- Zesiewicz, T.A.; Wilmot, G.; Kuo, S.-H.; Perlman, S.; Greenstein, P.E.; Ying, S.H.; Ashizawa, T.; Subramony, S.; Schmahmann, J.D.; Figueroa, K.; et al. Comprehensive systematic review summary: Treatment of cerebellar motor dysfunction and ataxia. Neurology 2018, 90, 464–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sarva, H.; Shanker, V.L. Treatment Options in Degenerative Cerebellar Ataxia: A Systematic Review. Mov. Disord. Clin. Pract. 2014, 1, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Mori, M.; Adachi, Y.; Mori, N.; Kurihara, S.; Kashiwaya, Y.; Kusumi, M.; Takeshima, T.; Nakashima, K. Double-blind crossover study of branched-chain amino acid therapy in patients with spinocerebellar degeneration. J. Neurol. Sci. 2002, 195, 149–152. [Google Scholar] [CrossRef]
- Strupp, M.; Teufel, J.; Habs, M.; Feuerecker, R.; Muth, C.; Van De Warrenburg, B.P.; Klopstock, T.; Feil, K. Effects of acetyl-dl-leucine in patients with cerebellar ataxia: A case series. J. Neurol. 2013, 260, 2556–2561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kalla, R. Aminopyridines and Acetyl-DL-leucine: New Therapies in Cerebellar Disorders. Curr. Neuropharmacol. 2018, 17, 7–13. [Google Scholar] [CrossRef]
- Kaya, E.; Smith, D.A.; Smith, C.; Boland, B.; Strupp, M.; Platt, F.M. Beneficial Effects of Acetyl-DL-Leucine (ADLL) in a Mouse Model of Sandhoff Disease. J. Clin. Med. 2020, 9, 1050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosa, A.; Duarte-Silva, S.; Silva-Fernandes, A.; Nunes, M.J.; Carvalho, A.N.; Rodrigues, E.; Gama, M.J.; Rodrigues, C.; Maciel, P.; Castro-Caldas, M. Tauroursodeoxycholic Acid Improves Motor Symptoms in a Mouse Model of Parkinson’s Disease. Mol. Neurobiol. 2018, 55, 9139–9155. [Google Scholar] [CrossRef]
- Thams, S.; Lowry, E.R.; Larraufie, M.-H.; Spiller, K.J.; Li, H.; Williams, D.; Hoang, P.; Jiang, E.; Williams, L.A.; Sandoe, J.; et al. A Stem Cell-Based Screening Platform Identifies Compounds that Desensitize Motor Neurons to Endoplasmic Reticulum Stress. Mol. Ther. 2019, 27, 87–101. [Google Scholar] [CrossRef] [Green Version]
- Kashimada, A.; Hasegawa, S.; Isagai, T.; Uchiyama, T.; Matsuo, M.; Kawai, M.; Goto, M.; Morio, T.; Hayashi, Y.K.; Takagi, M. Targeting the enhanced ER stress response in Marinesco-Sjögren syndrome. J. Neurol. Sci. 2018, 385, 49–56. [Google Scholar] [CrossRef] [PubMed]
- Bhargava, P.; Smith, M.D.; Mische, L.; Harrington, E.P.; Fitzgerald, K.C.; Martin, K.A.; Kim, S.; Reyes, A.A.A.; Gonzalez-Cardona, J.; Volsko, C.; et al. Bile acid metabolism is altered in multiple sclerosis and supplementation ameliorates neuroinflammation. J. Clin. Investig. 2020, 130, 3467–3482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Basnet, R.M.; Zizioli, D.; Taweedet, S.; Finazzi, D.; Memo, M. Zebrafish Larvae as a Behavioral Model in Neuropharmacology. Biomedicines 2019, 7, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bassett, D.I.; Bryson-Richardson, R.; Daggett, D.F.; Gautier, P.; Keenan, D.G.; Currie, P. Dystrophin is required for the formation of stable muscle attachments in the zebrafish embryo. Development 2003, 130, 5851–5860. [Google Scholar] [CrossRef] [Green Version]
- Synofzik, M.; Németh, A.H. Recessive ataxias. Handb. Clin. Neurology 2018, 155, 73–89. [Google Scholar] [CrossRef]
- Kani, S.; Bae, Y.-K.; Shimizu, T.; Tanabe, K.; Satou, C.; Parsons, M.J.; Scott, E.; Higashijima, S.-I.; Hibi, M. Proneural gene-linked neurogenesis in zebrafish cerebellum. Dev. Biol. 2010, 343, 1–17. [Google Scholar] [CrossRef] [Green Version]
- Kaslin, J.; Brand, M. The zebrafish cerebellum. In Essentials of Cerebellum and Cerebellar Disorders; Springer International Publishing: Cham, Switzerland, 2016; pp. 411–421. [Google Scholar]
- Namikawa, K.; Dorigo, A.; Köster, R.W. Neurological Disease Modelling for Spinocerebellar Ataxia Using Zebrafish. J. Exp. Neurosci. 2019, 13. [Google Scholar] [CrossRef] [Green Version]
- Pinton, P.; Giorgi, C.; Siviero, R.; Zecchini, E.; Rizzuto, R. Calcium and apoptosis: ER-mitochondria Ca2+ transfer in the control of apoptosis. Oncogene 2008, 27, 6407–6418. [Google Scholar] [CrossRef] [Green Version]
- Ady, V.; Toscano-Márquez, B.; Nath, M.; Chang, P.K.; Hui, J.; Cook, A.; Charron, F.; Larivière, R.; Brais, B.; McKinney, R.A.; et al. Altered synaptic and firing properties of cerebellar Purkinje cells in a mouse model of ARSACS. J. Physiol. 2018, 596, 4253–4267. [Google Scholar] [CrossRef]
- Rodino-Klapac, L.R.; Beattie, C.E. Zebrafish topped is required for ventral motor axon guidance. Dev. Biol. 2004, 273, 308–320. [Google Scholar] [CrossRef] [Green Version]
- Bradshaw, T.Y.; Romano, L.E.; Duncan, E.J.; Nethisinghe, S.; Abeti, R.; Michael, G.J.; Giunti, P.; Vermeer, S.; Chapple, J.P. A reduction in Drp1-mediated fission compromises mitochondrial health in autosomal recessive spastic ataxia of Charlevoix Saguenay. Hum. Mol. Genet. 2016, 25, 3232–3244. [Google Scholar] [CrossRef] [Green Version]
- Michalak, M.; Corbett, E.F.; Mesaeli, N.; Nakamura, K.; Opas, M. Calreticulin: One protein, one gene, many functions. Biochem. J. 1999, 344, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Salati, S.; Genovese, E.; Carretta, C.; Zini, R.; Bartalucci, N.; Prudente, Z.; Pennucci, V.; Ruberti, S.; Rossi, C.; Rontauroli, S.; et al. Calreticulin Ins5 and Del52 mutations impair unfolded protein and oxidative stress responses in K562 cells expressing CALR mutants. Sci. Rep. 2019, 9, 10558. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sinha, K.; Das, J.; Pal, P.B.; Sil, P.C. Oxidative stress: The mitochondria-dependent and mitochondria-independent pathways of apoptosis. Arch. Toxicol. 2013, 87, 1157–1180. [Google Scholar] [CrossRef]
- Cole, L.; Ross, L. Apoptosis in the Developing Zebrafish Embryo. Dev. Biol. 2001, 240, 123–142. [Google Scholar] [CrossRef] [Green Version]
- Saydmohammed, M.; Tsang, M. High-Throughput Automated Chemical Screens in Zebrafish. Methods Mol. Biol. 2017, 1683, 383–393. [Google Scholar] [CrossRef]
- Hull, J.; El Hindy, M.; Kehoe, P.; Chalmers, K.; Love, S.; Conway, M.E. Distribution of the branched chain aminotransferase proteins in the human brain and their role in glutamate regulation. J. Neurochem. 2012, 123, 997–1009. [Google Scholar] [CrossRef] [PubMed]
- Omura, T.; Asari, M.; Yamamoto, J.; Oka, K.; Hoshina, C.; Maseda, C.; Awaya, T.; Tasaki, Y.; Shiono, H.; Yonezawa, A.; et al. Sodium tauroursodeoxycholate prevents paraquat-induced cell death by suppressing endoplasmic reticulum stress responses in human lung epithelial A549 cells. Biochem. Biophys. Res. Commun. 2013, 432, 689–694. [Google Scholar] [CrossRef]
- Amaral, J.; Viana, R.J.; Ramalho, R.; Steer, C.J.; Rodrigues, C.M. Bile acids: Regulation of apoptosis by ursodeoxycholic acid. J. Lipid Res. 2009, 50, 1721–1734. [Google Scholar] [CrossRef] [Green Version]
- Parry, G.J.; Rodrigues, C.; Aranha, M.; Hilbert, S.J.; Davey, C.; Kelkar, P.; Low, W.C.; Steer, C.J. Safety, Tolerability, and Cerebrospinal Fluid Penetration of Ursodeoxycholic Acid in Patients with Amyotrophic Lateral Sclerosis. Clin. Neuropharmacol. 2010, 33, 17–21. [Google Scholar] [CrossRef] [Green Version]
- Martinelli, C.; Battaglini, M.; Pucci, C.; Gioi, S.; Caracci, C.; Macaluso, G.; Doccini, S.; Santorelli, F.M.; Ciofani, G. Development of Nanostructured Lipid Carriers for the Delivery of Idebenone in Autosomal Recessive Spastic Ataxia of Charlevoix-Saguenay. ACS Omega 2020, 5, 12451–12466. [Google Scholar] [CrossRef]
- Kimmel, C.B.; Ballard, W.W.; Kimmel, S.R.; Ullmann, B.; Schilling, T.F. Stages of embryonic development of the zebrafish. Dev. Dyn. 1995, 203, 253–310. [Google Scholar] [CrossRef]
- Varshney, G.K.; Carrington, B.; Pei, W.; Bishop, K.; Chen, Z.; Fan, C.; Xu, L.; Jones, M.; LaFave, M.C.; Ledin, J.; et al. A high-throughput functional genomics workflow based on CRISPR/Cas9-mediated targeted mutagenesis in zebrafish. Nat. Protoc. 2016, 11, 2357–2375. [Google Scholar] [CrossRef]
- Marchese, M.; Pappalardo, A.; Baldacci, J.; Verri, T.; Doccini, S.; Cassandrini, D.; Bruno, C.; Fiorillo, C.; Garcia-Gil, M.; Bertini, E.; et al. Dolichol-phosphate mannose synthase depletion in zebrafish leads to dystrophic muscle with hypoglycosylated α-dystroglycan. Biochem. Biophys. Res. Commun. 2016, 477, 137–143. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of Relative Gene Expression Data Using Real-Time Quantitative PCR and the 2−ΔΔCT Method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Takeuchi, M.; Matsuda, K.; Yamaguchi, S.; Asakawa, K.; Miyasaka, N.; Lal, P.; Yoshihara, Y.; Koga, A.; Kawakami, K.; Shimizu, T.; et al. Establishment of Gal4 transgenic zebrafish lines for analysis of development of cerebellar neural circuitry. Dev. Biol. 2015, 397, 1–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Müller, J.S.; Burns, D.T.; Griffin, H.; Wells, G.; Zendah, R.A.; Munro, B.; Schneider, C.; Horvath, R. RNA exosome mutations in pontocerebellar hypoplasia alter ribosome biogenesis and p53 levels. Life Sci. Alliance 2020, 3, e202000678. [Google Scholar] [CrossRef] [PubMed]
- Mero, S.; Salviati, L.; Leuzzi, V.; Rubegni, A.; Calderan, C.; Nardecchia, F.; Galatolo, D.; Desbats, M.A.; Naef, V.; Gemignani, F.; et al. New pathogenic variants in COQ4 cause ataxia and neurodevelopmental disorder without detectable CoQ10 deficiency in muscle or skin fibroblasts. J. Neurol. 2021. [Google Scholar] [CrossRef]
- Di Nottia, M.; Marchese, M.; Verrigni, D.; Mutti, C.D.; Torraco, A.; Oliva, R.; Fernandez-Vizarra, E.; Morani, F.; Trani, G.; Rizza, T.; et al. A homozygous MRPL24 mutation causes a complex movement disorder and affects the mitoribosome assembly. Neurobiol. Dis. 2020, 141, 104880. [Google Scholar] [CrossRef]
- D’Amore, A.; Tessa, A.; Naef, V.; Bassi, M.T.; Citterio, A.; Romaniello, R.; Fichi, G.; Galatolo, D.; Mero, S.; Battini, R.; et al. Loss of ap4s1 in zebrafish leads to neurodevelopmental defects resembling spastic paraplegia 52. Ann. Clin. Transl. Neurol. 2020, 7, 584–589. [Google Scholar] [CrossRef] [Green Version]
- Xia, Q.; Wei, L.; Zhang, Y.; Kong, H.; Shi, Y.; Wang, X.; Chen, X.; Han, L.; Liu, K. Psoralen Induces Developmental Toxicity in Zebrafish Embryos/Larvae Through Oxidative Stress, Apoptosis, and Energy Metabolism Disorder. Front. Pharmacol. 2018, 9, 1457. [Google Scholar] [CrossRef] [PubMed]
- Colón-Cruz, L.; Kristofco, L.; Crooke-Rosado, J.; Acevedo, A.; Torrado, A.; Brooks, B.W.; Sosa, M.A.; Behra, M. Alterations of larval photo-dependent swimming responses (PDR): New endpoints for rapid and diagnostic screening of aquatic contamination. Ecotoxicol. Environ. Saf. 2018, 147, 670–680. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.L.; Beggs, A.H.; Gupta, V.A. Analysis of skeletal muscle defects in larval zebrafish by birefringence and touch-evoke escape response assays. J. Vis. Exp. 2013, 82, e50925. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brogi, L.; Marchese, M.; Cellerino, A.; Licitra, R.; Naef, V.; Mero, S.; Bibbiani, C.; Fronte, B. β-Glucans as Dietary Supplement to Improve Locomotion and Mitochondrial Respiration in a Model of Duchenne Muscular Dystrophy. Nutrients 2021, 13, 1619. [Google Scholar] [CrossRef] [PubMed]




Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Naef, V.; Marchese, M.; Ogi, A.; Fichi, G.; Galatolo, D.; Licitra, R.; Doccini, S.; Verri, T.; Argenton, F.; Morani, F.; et al. Efficient Neuroprotective Rescue of Sacsin-Related Disease Phenotypes in Zebrafish. Int. J. Mol. Sci. 2021, 22, 8401. https://doi.org/10.3390/ijms22168401
Naef V, Marchese M, Ogi A, Fichi G, Galatolo D, Licitra R, Doccini S, Verri T, Argenton F, Morani F, et al. Efficient Neuroprotective Rescue of Sacsin-Related Disease Phenotypes in Zebrafish. International Journal of Molecular Sciences. 2021; 22(16):8401. https://doi.org/10.3390/ijms22168401
Chicago/Turabian StyleNaef, Valentina, Maria Marchese, Asahi Ogi, Gianluca Fichi, Daniele Galatolo, Rosario Licitra, Stefano Doccini, Tiziano Verri, Francesco Argenton, Federica Morani, and et al. 2021. "Efficient Neuroprotective Rescue of Sacsin-Related Disease Phenotypes in Zebrafish" International Journal of Molecular Sciences 22, no. 16: 8401. https://doi.org/10.3390/ijms22168401
APA StyleNaef, V., Marchese, M., Ogi, A., Fichi, G., Galatolo, D., Licitra, R., Doccini, S., Verri, T., Argenton, F., Morani, F., & Santorelli, F. M. (2021). Efficient Neuroprotective Rescue of Sacsin-Related Disease Phenotypes in Zebrafish. International Journal of Molecular Sciences, 22(16), 8401. https://doi.org/10.3390/ijms22168401