Abstract
Atopic dermatitis (AD) is a common inflammatory dermatosis affecting up to 30% of children and 10% of adults worldwide. AD is primarily driven by an epidermal barrier defect which triggers immune dysregulation within the skin. According to recent research such phenomena are closely related to the microbial dysbiosis of the skin. There is growing evidence that cutaneous microbiota and bacterial biofilms negatively affect skin barrier function, contributing to the onset and exacerbation of AD. This review summarizes the latest data on the mechanisms leading to microbiome dysbiosis and biofilm formation in AD, and the influence of these phenomena on skin barrier function.
1. Introduction
Atopic dermatitis (AD) is a chronic inflammatory skin disorder characterized by a highly heterogenous clinical picture [1,2]. No specific tests ensure the diagnosis of AD. Therefore, it is based on clinical criteria (i.e., UK Working Party criteria and the Hanifin and Rajka criteria applicable to the pediatric and adult populations, respectively) [3,4]. The essential features of AD include the presence of eczematous lesions, itch, and a chronic or relapsing disease course [2,5]. The distribution and morphology of skin lesions reveals age-dependent variations. Children under 2 years of age typically present with poorly defined, exudative erythematous lesions located on the trunk, face, and cheeks. The majority of pediatric patients >2 years of age exhibit a more localized eczema affecting flexor surfaces and the lichenification of chronic lesions. As regards adults, AD most commonly manifests as chronic hand eczema, flexural dermatitis, and/or head and neck dermatitis. A recently published paper describing the prevalence of AD in adults revealed that the classic adult type, with lichenified/exudative flexural dermatitis frequently associated with head and neck eczema or hand eczema was the most common AD phenotype [6]. Furthermore, the predominant manifestations of adult-onset AD included nummular eczema-like and prurigo-like phenotypes, whereas childhood-onset AD was more often characterized by lichenified/exudative flexural dermatitis alone and/or in association with portrait dermatitis.
The lack of causative treatment, significant psychosocial impact, and high prevalence reaching 10% of adults and 30% of children make AD a considerable challenge for public health worldwide [7,8].
The current view on AD pathogenesis encompasses the following factors [2,9,10]:
- An epidermal barrier defect;
- Immune dysregulation;
- Skin microbial dysbiosis;
- The itch/scratch cycle.
These pathogenic events are strictly related and trigger the occurrence and exacerbations of AD, making it a model disease for the study of interactions between the skin barrier and cutaneous microbiome (Figure 1).
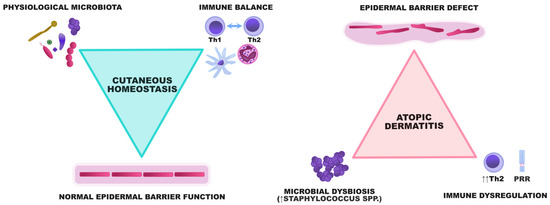
Figure 1.
A graphical representation of the interactions between the epidermal barrier, microbiota, and the immune system. In healthy individuals, the epidermis constitutes a barrier preventing the uncontrolled proliferation of potentially harmful microorganisms. A diverse array of microbes participates in the crosstalk with the immune system and ensures its maturation to maintain immune balance within the skin. In AD, the epidermal barrier defect and dysregulation of innate and acquired immunity result in the enhanced adhesion and proliferation of some bacterial species, especially staphylococci. Microbial dysbiosis subsequently aggravates the skin defect by degrading the components of the epidermal barrier and potentiating Th2-skewing.
Literature data suggest that an epidermal barrier defect is the primary factor driving AD symptoms [11,12]. Individuals with AD exhibit highly variable congenital abnormalities that translate into epidermal barrier impairment. The most frequent polymorphisms affect the expression of filaggrin, tight junctions, epidermal lipids, and endothelial protease activity [13,14,15,16,17,18]. The epidermal barrier may also be secondarily damaged by epigenetic changes and environmental factors [2,13,19].
Recent research in the field of the microbiome has highlighted the crucial role of skin microbes in preserving the intact skin barrier [20,21]. Commensal microorganisms are involved in constant crosstalk with the keratinocytes and the immune system, mediate the formation of tight junctions, maintain immune homeostasis, and “teach” the host to recognize and combat pathogens capable of causing a potential skin infection. Furthermore, the physiological microbiome constitutes an additional physiological barrier against pathogenic microbes by competing for the niches favoring their growth [22].
Microbes may function in a sessile form, but in most cases they form complex multi-species communities within the biofilms [23]. A growing body of evidence points to bacterial biofilms as the primary pathogenic factor in a number of dermatological conditions, e.g., in acne vulgaris [24] and chronic wounds [25]. The importance of bacterial biofilms for the skin barrier in AD has also been recently suggested.
The aim of this narrative review is to summarize the recent insights into the interactions between cutaneous microbes, their biofilms, and skin barrier function in AD.
2. Materials and Methods
The literature search was performed with the use of the following databases: PubMed, Scopus and Web of Science. The keywords used to perform the search were “atopic dermatitis”, “biofilm”, “microbiome”, “S. aureus”, “epidermal differentiation complex”, “tight junctions”, “epidermal proteases”, and “epidermal lipids” in different combinations. There were no language limitations.
3. The Function and Structure of the Physiological Epidermal Barrier
The epidermis is a specialized structure that warrants selective permeability for a wide range of exogenous substances and protects the host from dehydration, infection, loss of endogenous molecules, and negative influence of physical factors (e.g., UV radiation, temperature changes, and mechanical stress) [26,27]. The epidermis is formed by keratinocytes which constantly divide in the basal layer and undergo a gradual differentiation while migrating through the more superficial stratum spinosum and stratum granulosum, and eventually slough once they reach the stratum corneum [27]. The physiological turnover time, i.e., the period during which the keratinocyte reaches the stratum corneum, is approximately 28 days. Epidermal differentiation and desquamation are complex processes that may be altered in various dermatoses, e.g., in AD and psoriasis [28].
The integrity of the epidermal barrier is largely ensured by the stratum granulosum and stratum corneum [18,29,30]. The expression of tight junctions (TJs) is one of the most important functional features of the stratum granulosum [26]. Keratinocytes in this layer are also characterized by the presence of keratohyalin granules containing filaggrin and loricrin precursors which, once modified, ensure the appropriate structure of keratinocytes in the stratum corneum.
The keratinocytes in the stratum corneum are referred to as corneocytes. Corneocytes are flat anucleate cells surrounded by a cornified envelope, a structure composed of highly crosslinked insoluble proteins that provide adequate cell structure and adhesion [29]. Intracellular space is filled with lipid lamellae that prevent transepidermal water loss (TEWL) and ensure adequate skin hydration [31].
The crucial elements providing the integrity of the epidermal barrier which might be affected by microbial dysbiosis will be briefly discussed below.
3.1. Tight Junctions
The physiological barrier function of the epidermis is largely due to the presence of tight junctions (TJs) that connect the epidermal cells within the stratum granulosum. Their complex structure involves strands between the adjacent keratinocytes formed by claudins, transmembrane proteins primarily composed of occludin (Ocln), JAM-A, and angulin, and proteins forming a “scaffold” within the intracellular zone (zonula occludens (ZO)-1, -2 and -3) [26,32]. Initially, TJs were solely regarded as structures regulating the paracellular diffusion of substances based on their electric charge and molecular weight. Therefore, TJs used to be perceived mainly as a barrier controlling the penetration of potentially dangerous exogenous molecules into the dermis. However, recent advances suggest the key importance of TJs in other aspects of skin physiology, such as the maintenance of the basoapical polarity of the epidermis and the influence on gene expression in the epidermal cells [32,33]. TJs are associated with several cellular signaling networks that regulate cell proliferation and junction assembly. Importantly, different components of the tight junctions are targeted by a wide range of pathogens, which degrade them to cross tissue barriers [34].
Polymorphisms in genes encoding TJ components have been linked to an increased risk and severity of atopic dermatitis [35]. Such a predisposition might result in the decreased expression of claudin-1 and -23, causing the defect of the bioelectric barrier within the epidermis of patients with AD [17].
3.2. Filaggrin
As mentioned above, the stratum granulosum is also distinguished by the presence of keratohyalin granules which contain keratin, loricrin, trichohyalin, and profilaggrin [36]. Profilaggrin is a highly phosphorylated, functionally inactive precursor of filament aggregating protein (filaggrin), a crucial component of the physiological skin barrier [37,38]. Moving from the stratum granulosum to the stratum corneum, profilaggrin is gradually dephosphorylated and cleaved into 10–12 filaggrin monomers by endogenous proteases such as CAP1/Prss8 and SASPase/ASPRV1 [39,40,41]. Subsequently, filaggrin binds to keratin filaments and forms them into flattened squames that support the cytoskeleton and provide structure to the corneocytes to ensure adequate cell-to-cell adhesion [37,42]. Keratin filaments in the corneocytes are bound by corneodesmosomes formed by cytoplasmic anchoring proteins (desmoplakin, plakoglobin) and adhesion molecules (desmoglein and desmocollin) [43]. Finally, filaggrin monomers detach from keratin and are cleaved by proteases which degrade them into amino acids and their derivatives. These final products of filaggrin formation are collectively referred to as natural moisturizing factor (NMF) due to their crucial role in maintaining skin hydration [44].
3.3. Endogenous Proteases
The physiological differentiation of the epidermis and adequate cell turnover is largely due to the subtle balance between the activity of epidermal proteases and their inhibitors [45]. Proteases degrade intracellular connections, which is essential for the desquamation of the terminally differentiated corneocytes [38]. The increased activity of proteases or decreased expression of protease inhibitors results in the excessive degradation of the epidermal barrier [18].
According to the literature, gene polymorphisms resulting in the overexpression of certain types of kallikrein-related proteases were linked to AD [46]. Kallikreins (KLKs) coordinate the process of physical desquamation by degrading corneodesmosome components (desmoglein1, desmocollin 1, and corneodesmosin) [47]. Some of them (e.g., KLK5 and KLK7) are also involved in the modification of filaggrin and lipid processing enzymes such as ß-glucocerebrosidase and acidic sphingomyelinase [15,48]. The function of KLKs is regulated by endogenous protease inhibitors, e.g., lymphoepithelial Kazal-type-related inhibitor (LEKTI) encoded by serine protease inhibitor of the Kazal-type 5 (SPINK5) gene [15]. Loss-of-function mutations in the latter may lead to the uncontrolled activation of KLKs which degrade intraepithelial connections and downregulate epidermal lipids. Homozygous mutations in the SPINK5 result in the development of Netherton syndrome, a disease characterized by the presence of atopic diathesis, severe ichthyosiform erythroderma, and hair abnormalities [49].
Importantly, the activity of endogenous proteases is also regulated by physicochemical factors, such as skin pH [50]. The latter is typically higher than physiological values in AD being partly due to microbiome disorders, which causes the excessive activity of proteases [51,52].
3.4. Epidermal Lipids
A frequent analogy used to describe the structure of the stratum corneum is that of “brick” and “mortar” referring to corneocytes and lipid-rich extracellular matrix, respectively [53,54]. The latter is composed of ceramides, free fatty acids, cholesterol, and triacylglycerol species. Along with corneocytes, epidermal lipids form a barrier that prevents TEWL and the penetration of irritants and allergens [54]. AD skin exhibits an altered expression of COUP-TF-interacting protein 1 and 2 (CTIP1 and CTIP2), the transcription factors regulating the activity of lipid forming enzymes [55,56]. A study conducted in CTIP1-deficient mice showed several abnormalities, e.g., significant downregulation of ketodihydrosphingosine reductase (Kdsr), UDP-glucose ceramide glucosyltransferase (Ugcg), N-acylsphingosine amidohydrolase 1 (Asah1), and ceramide synthase 6 (Cers6) [55].
AD was shown to be particularly associated with ceramide abnormalities. A report by Berdyshev et al. revealed that the overall paucity of ultra-long-chain ceramides (i.e., more than 26 carbons in length) and a relative increase in short-chain ceramides was associated with the upregulation of Th2 cytokines and epidermal barrier impairment [57].
Damage to the epidermal barrier triggers the compensatory production of free fatty acids (FFAs) [58]. Physiologically, FFAs show cytotoxic activity and antimicrobial properties [59]. However, as regards AD, FFAs are characterized by shortened fatty acyl chain length and an increased level of unsaturated fatty acids, which compromises their function as factors maintaining epidermal barrier integrity and the homeostasis of the cutaneous microbiota [60].
4. Immune Dysregulation in Atopic Dermatitis
AD is characterized by defects in both innate and acquired immune responses. A detailed description of immune abnormalities in AD exceeds the scope of this review. However, due to a close relationship between the epidermal barrier function, immune dysregulation, and microbiome dysbiosis, we present the essential concepts below.
The innate immune system is responsible for the rapid identification and neutralization of pathogenic microbes and for the recognition of host tissue damage to trigger its repair [61]. These processes are mediated by pattern recognition receptors (PRRs) that are stimulated by danger-associated molecular patterns (DAMPs) and pathogen-associated molecular patterns (PAMPs) [62]. Once activated, PRRs may activate a wide range of immunocompetent cells. AD is associated with abnormalities in the signaling via the following PRRs: (1) Toll-like receptors (TLR) [63]; (2) nucleotide-binding oligomerization domain (NOD) [64]; and some soluble PRRs [65]. These abnormalities result in the reduced reactivity of dendritic cells, polymorphonuclears, and NK lymphocytes, as well as a decreased synthesis of antimicrobial peptides (AMPs) in the skin [65]. Collectively, all these disorders hamper the control of pathogens on the skin.
With respect to acquired immune response, AD has been associated with variable dysregulation of Th lymphocytes dependent on the patient’s phenotype and disease phase [66]. In general, the overexpression of Th2 cytokines (thymic stromal lymphopoietin (TSLP), IL-4, IL-13, IL-31) is a common finding in the acute phase of the disease [9]. Th22 is another axis that is typically upregulated in AD, primarily resulting in the overexpression of IL-22 [67,68]. Both Th2 and Th22 cytokines impair the terminal differentiation of keratinocytes by interfering with filaggrin, loricrin, and involucrin modification and the production of tight junction components, particularly claudins [69]. Chronic AD lesions, apart from a high expression of Th2 and Th22 cytokines, are also characterized by the activation of the Th1 axis and upregulation of markers such as IFN-γ, CXCL9, and CXCL10 [68]. Lastly, in Asians, children, and in patients with intrinsic AD, a marked expression of Th17 cytokines is seen [66].
5. The Skin Microbiome in Atopic Dermatitis
Despite being a dynamic and challenging habitat, the skin is home to a highly diverse array of bacteria, viruses, and fungi collectively referred to as the microbiome [22]. With the advent of next-generation sequencing, it became possible to appreciate the diversity of bacterial species that had previously been impossible to identify with classic culturing methods. The initial studies on the skin microbiome were conducted by Grice et al. who analyzed the bacterial composition in twenty locations on the human body by targeted amplicon sequencing of the 16S rRNA subunit [70]. Four out of nineteen detected bacterial phyla were the most dominant: Actinobacteria (59% of total bacterial species belonging to the Micrococcus, Propionibacterium, Corynebacterium genera), Firmicutes (24%, Lactobacillus, Streptococcus, Staphylococcus), Proteobacteria (17%, Paracoccus, Haematobacter), and Bacteroidetes (7%, Prevotella, Porphyromonas). The composition of the skin microbiome depends on numerous intrinsic (e.g., sex, age, occupation) and extrinsic (moisture, pH, sebum content, UV exposure) factors [71]. These observations were the basis for distinguishing three primary types of microniches: (1) sebaceous (mainly inhabited by Cutibacterium spp.), (2) moist (characterized by the domination of Staphylococcus spp. and Corynebacterium spp.), and (3) dry (harboring diverse microbiota) [22].
Initial reports on the physiological skin microbiome were shortly followed by studies based on the genome sequencing of skin samples in various dermatoses. An investigation in AD confirmed the observations from studies implementing classic culturing methods and identified Staphylococcus aureus as the pathogen dominating the lesional skin of AD patients [72]. A longitudinal analysis of the skin microbiome in the pediatric population with AD demonstrated that disease flares were to a certain extent preceded by the domination of the physiological skin microbiome by S. aureus [73], which contributed to the hypothesis on the fundamental role of microbial dysbiosis in AD. In the mentioned report, a concomitant proliferation of S. epidermidis and the correlation of this phenomenon with disease severity were also demonstrated. Habitually, S. epidermidis was only perceived as an important skin commensal orchestrating the maturation of the immune system and combating pathogens while spontaneously causing opportunistic infections [74]. Based on the results of in vitro studies showing that S. epidermidis inhibited the growth of S. aureus, its proliferation in AD lesions was attributed to the ability to thrive in S. aureus-dominated habitat [75,76]. However, the latest reports suggested that in certain conditions, S. epidermidis might contribute to the inflammatory reaction in AD [77,78]. Additionally, a recent study implementing a 3D human skin model revealed that S. epidermidis elicited a strong bias toward the downregulation of genes in comparison with other bacterial species [79]. With respect to the genes of the epidermal differentiation complex, despite the upregulation of FLG and LOR encoding filaggrin and loricrin, respectively, a significant downregulation was shown for IVL, FLG2, TCHH, SPRR-3, SPRR-4, and S100A7 encoding involucrin, filaggrin-2, trichohyalin, small proline rich protein-3 and -4, and S100 calcium binding protein A7, respectively. The authors hypothesized that due to its high prevalence on the skin, S. epidermidis might have more unique interactions with the host tissue than other microbes. The discrepancies in the presented studies may suggest that the impact of S. epidermidis on AD is more nuanced, and probably dependent on many variables such as strain specificity [80].
Other niches have also been investigated in AD. Similarly to skin lesions, the nonlesional skin was revealed to harbor an increased load of S. aureus [81,82,83]. Furthermore, a report comparing the microbial composition of the nonlesional skin of AD patients and the skin of controls demonstrated the abundance of Streptococcus spp. and Gemella spp. with the simultaneous depletion of Dermacoccus spp. in the former group [51]. The researchers concluded that such changes might result in the excessive production of ammonia and a subsequent increase in skin pH. As discussed above, the latter is a typical feature of AD causing disturbances in the functioning of the skin barrier.
6. Biofilms
Although the microbes multiply most efficiently in the planktonic form, in most cases they form multi-species communities producing extracellular polymeric substance (EPS), which enables the attachment to biotic or abiotic surfaces [84,85]. This form of bacterial life is referred to as biofilm and renders bacteria more resistant to antibiotics, unfavorable environmental conditions, and recognition by the host immune system [84,86,87]. These functions are largely related to the properties of EPS, a complex structure composed of various extracellular polysaccharides, DNA, and proteins, penetrated by channels ensuring the supply of water, air, and nutrients [85,87].
Importantly, the cycle of biofilm formation is strictly associated with the production of virulence factors by the bacteria. These phenomena are regulated by the so-called quorum sensing systems which control the biofilm formation cycle depending on cell density within the bacterial community, environmental factors, the accessibility of nutrients, and other variables [88].
As regards Staphylococcus spp., the ability to sense bacterial quorum is controlled by the accessory gene regulator (Agr) [89]. An Agr product, the autoinducing peptide (AIP), is a molecule acting as an extracellular quorum signal [90]. Once a sufficiently high density of cells is achieved within a bacterial community, Agr triggers the expression of toxins and degradative exoenzymes and simultaneously causes the downregulation of colonization factors. Therefore, high Agr activity contributes to: (1) the upregulation of staphylococcal virulence factors and (2) the degradation of biofilm with the dispersal of microcolonies to new niches [91]. High Agr activity results in acute staphylococcal diseases (e.g., pneumonia), whereas low Agr activity is a characteristic feature of chronic infections mediated by biofilm (e.g., infections of indwelling medical devices) [92,93]. The cycle of biofilm formation by S. aureus and its impact on skin barrier in AD is presented in Figure 2.
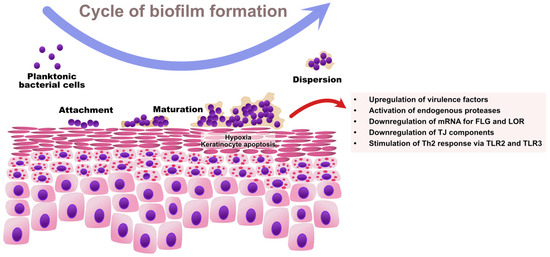
Figure 2.
The cycle of S. aureus biofilm formation and its impact on skin barrier function in AD. The increased expression of adhesion molecules, epidermal lipid aberrations, and paucity of AMPs predispose AD patients to S. aureus colonization. Initially, planktonic S. aureus cells adhere to the skin surface via microbial surface components recognizing adhesive matrix molecules (MSCRAMMs) and start producing extracellular polymeric substances (EPS), which results in the establishment of a bacterial community associated in the biofilm. The growing biofilm causes hypoxia and the apoptosis of keratinocytes. Once the bacterial quorum is achieved, the Agr system activates the upregulation of virulence factors including proteases and toxins, and the downregulation of colonization and adhesion molecules with the dispersal of biofilm to new locations. Collectively, S. aureus biofilm contributes to the destruction of the epidermal barrier by downregulating EDC proteins and TJ components, and by stimulating Th2-skewing as a result of aberrant innate immune response. FLG—filaggrin; LOR—loricrin.
The highly unstable environment of the skin subjected to frequent temperature and humidity changes, variable UV exposure, and bactericidal substances (e.g., in personal hygiene products) likely results in the functioning of most cutaneous microbes within biofilms. In AD, this phenomenon may also be partly responsible for the persistent microbial dysbiosis of the skin [94]. However, during the flares, S. aureus probably tends to degrade the biofilm and upregulate the virulence factors which triggers an even more pronounced epithelial barrier degradation and immune dysregulation.
7. The Influence of Barrier Disruption on Skin Microbiome Dysbiosis
Before addressing the influence of microbiota on skin barrier function, it is essential to describe the molecular mechanisms predisposing AD patients to colonization by pathogenic bacteria.
The skin barrier in AD is characterized by the abnormal expression of various surface molecules which may be used by pathogens to attach to the skin surface. For example, fibrinogen, fibronectin, and collagen may be bound by microbial surface components recognizing adhesive matrix molecules (MSCRAMMs), specialized structures developed by S. aureus [95]. Interestingly, it was reported that the efficient binding of fibronectin and fibrinogen by S. aureus was possible on AD skin and not on healthy skin or in other inflammatory dermatoses [96]. Binding via MSCRAMMs is followed by the secretion of EPS and biofilm production with the resultant establishment of a stable bacterial community on the skin surface.
Although various studies included contradictory results, it seems that microbial dysbiosis might also be associated with filaggrin deficiency. A trial by Clausen et al. revealed that S. aureus colonization of the lesional skin and the nose correlated with the presence of filaggrin loss-of-function mutations [97]. Furthermore, Cai et al. demonstrated that filaggrin mutations were a risk factor for skin infections in children with AD [98]. Nakatsuji et al. showed that filaggrin-deficient mice were prone to the increased penetration of the epidermis by S. aureus [99]. Conversely, Simpson et al. showed that despite the fact that S. aureus carriage correlated with the parameters reflecting epidermal barrier damage, there was no association between this phenomenon and filaggrin mutations [100].
Filaggrin deficiency is associated with the paucity of NMF. One study demonstrated that Staphylococcus aureus implemented clumping factor B to bind to corneocytes and that the strength of bacterium-corneocyte adhesion correlated with NMF levels [101]. The concept was further investigated by Towell et al., who showed that a low concentration of NMF resulted in the aberrant expression of corneodesmosin, a target structure bound by fibronectin-binding protein B (FnBPB) and clumping factor B (ClfB) [102]. The authors demonstrated that blocking the N-terminal region of corneodesmosin inhibited the adhesion of S. aureus. Furthermore, FnBPB- and ClfB-deficient S. aureus mutants were incapable of binding to human keratinocytes.
As mentioned above, alterations in the stratum corneum lipids are another factor predisposing to microbiome disorders in AD. A recent study by Igawa et al. showed that sphingosine-deficient mice were characterized by the increased penetration of S. aureus through the skin [103]. Similar observations were made in an experiment by Lipsky et al., who concluded that the stratum corneum was more easily penetrated by S. aureus when the lipid levels were depleted to simulate AD skin [104]. Baurecht et al. showed that apart from filaggrin deficiency, S. aureus abundance correlated with ceramide α-Hydroxy fatty acid/sphingosine base activity [105], while Cleary et al. demonstrated that ceramide distribution might structurally inhibit the formation of bacterial biofilms, and proposed that the characteristic pattern of their depletion typical for AD probably served as a conduit for S. aureus growth [106]. Lastly, a decrease in monounsaturated fatty acid species (MUFAs) seems to be an additional factor predisposing to dysbacteriosis in AD [107].
8. The Influence of Skin Microbiome Dysbiosis on the Skin Barrier
Microbial dysbiosis impairs skin barrier function through diverse mechanisms. Recent studies on the role of the microbiome in skin physiology revealed its influence on the expression of numerous factors implemented in skin immunity and barrier function. For example, Meisel et al. showed that immune response genes encoding TLRs, antimicrobial peptides, the complement cascade, and IL-1 family cytokine signaling were modulated by the microbiota [108]. Furthermore, the skin microbiome was revealed to influence keratinocyte differentiation and development by modifying the expression of genes in the epidermal differentiation complex (EDC).
The influence of the abnormal microbial composition of the skin is discussed with respect to the previously described elements of the skin barrier.
8.1. Effect on Tight Junctions
Tight junctions are targeted by microorganisms that penetrate the stratum corneum. The degradation of their components and the resultant increased skin permeability contribute to AD exacerbations.
Research in impetigo resulted in the publication of one of the first reports describing the influence of S. aureus on tight junction function. The authors concluded that ZO-1 and Ocln were downregulated in the infected zones and upregulated in the neighboring colonized areas of the skin [109]. More recent studies demonstrated that challenging normal human epidermal keratinocytes (NHEKs) with live S. aureus affected the TJ function in a biphasic way [110]. At early time points S. aureus was shown to strengthen the TJ function which was supported by the increased transepithelial resistance and reduced FITC–dextran permeability. The authors attributed this process to the relocation of Ocln and Cldn-4 to the cell-cell surface. However, long-term exposure caused a decrease in TJ function, probably due to the downregulation of Cldn-1 and Cldn-4. Similar albeit less pronounced effects on tight junction function were demonstrated for S. epidermidis.
Some studies pointed out selected staphylococcal virulence factors as the culprits of decreased TJ function. Wang et al. demonstrated that SspA/V8 protease was the primary secreted factor of S. aureus correlating with skin barrier permeability and tight junction damage [111]. The effect of SspA/V8 was limited by IL-1β-induced production of human β-defensin 2. However, as mentioned in the introduction, Th2 skewing typical of AD likely results in the downregulation of human β-defensin 2 and insufficient protection of TJs from staphylococcal proteases.
TJ function is largely regulated by the innate immune system. In line with these observations, Kuo et al. showed that TLR2 enhanced the production of TJ proteins, such as claudin-1, claudin-23, occludin, and ZO-1 upon the exposure to S. aureus-derived peptidoglycan and synthetic TLR2 agonists [112]. However, patients with AD often exhibit PRR polymorphisms that impaired those processes. Furthermore, some staphylococcal superantigens, such as SEB, were shown to diminish Ocln and ZO-1 in a model of chronic sinusitis [113] which is likely translatable to AD.
8.2. The Influence on Filaggrin and Epidermal Differentiation
It was shown that the microbiota might influence the differentiation and maturation of keratinocytes. According to the literature, the submission of 3D skin tissue cultures to mixed microbiota treatment resulted in the upregulation of filaggrin and epidermal cell proliferation [79]. Such an effect was not observed after skin exposure to single microorganisms. As the microbiota of AD patients is dramatically dominated by Staphylococcus spp., it is very likely that filaggrin deficiency in AD may be aggravated by reduced microbial diversity.
Furthermore, various studies showed that filaggrin and epidermal differentiation were impaired by Th2-cytokines, such as IL-4, IL-13, IL-31, and IL-33. At the molecular level, the downregulation of EDC components such as FLG and LOR by IL-4 and IL-13 is mediated by Janus kinases (JAK) 1 and 2, and tyrosine kinase 2 (TYK2) which induce the phosphorylation of STAT6 and STAT3 resulting in the downregulation of EDC molecules. The accessory mechanism of filaggrin downregulation involves the IL-13/periostin pathway inducing IL-24 production in keratinocytes [114]. Furthermore, IL-31-treated HaCaT cells showed differentiation defects associated with the underexpression of filaggrin, reduced epidermal thickness, and poor development of the stratum granulosum, while IL-33 was shown to downregulate filaggrin and TJ components such as claudin-1 through STAT3 and ERK phosphorylation in human keratinocytes [115,116,117,118]. Therefore, as microbiome disorders in AD are capable of triggering Th2 skewing, they also indirectly exert a negative effect on the skin barrier. Th2 polarization caused by AD-specific pathogens was proven by Nakatsuji et al., who demonstrated that S. aureus was capable of inducing the upregulation of IL-4, IL-13, IL-22, and thymic stromal lymphopoietin after skin penetration [99]. Moreover, Kim et al. showed that mice exposed to S. aureus and recombinant staphylococcal enterotoxin A showed the increased levels of proinflammatory cytokines, such as IL-4, IL-13, INF-γ, IL-17, and IL-18 [119]. Additionally, Kindi et al. discovered that S. aureus second immunoglobulin-binding protein (Sbi) was a type-2 promoting virulence factor that aggravated AD by triggering IL-33 expression and the downregulation of corneodesmosin [120]. Similar observations were also made in a study concerning allergic nasal mucosa, wherein S. aureus was shown to stimulate IL-33 and TSLP [121]. Furthermore, Brauweiler et al. showed that S. aureus lipoteichoic acid worked in combination with IL-4 and IL-13 through p63 to aggravate the suppression of early keratinocyte differentiation [122]. The same group revealed that the lipoteichoic acid of S. aureus aggravated epidermal barrier damage by reducing the expression of filaggrin and loricrin via an IL-1 mediated pathway [123]. Conversely, another study showed that the exposure to staphylococcal enterotoxin B was associated with a decreased expression of filaggrin degradation products [124]. Late keratinocyte differentiation resulting from the exposure to S. aureus antigens was also demonstrated in one report, in which the authors identified IL-6 as the offending molecular mediator [125].
8.3. Skin Barrier Disruption Mediated by Proteases
Pathogens residing on the skin may aggravate epidermal barrier defects by the secreted proteases and by modifying the activity of the endogenous proteases of the host.
According to Williams et al., S. aureus was shown to stimulate the activity of selected kallikreins (KLK6, 13, and 14). It coincided with the degradation of desmoglein-1 and filaggrin and an aberrant barrier function [126]. Another study revealed an increased proteolytic activity within the epidermis following the exposure to phenol-soluble modulin α derived from S. aureus [127].
With respect to the secreted proteases of S. aureus and S. epidermidis, apart from inducing epithelial barrier damage, they were also shown to degrade galectin-3, which orchestrates antimicrobial response in the skin, suggesting a new mechanism for staphylococcal skin colonization and immune evasion in AD [128]. S. epidermidis was also shown to secrete EcpA cysteine protease that has the ability to degrade desmoglein-1 and LL-37 (an antimicrobial peptide) in vitro with the resultant disruption of skin barrier function [129]. The process was dependent on quorum sensing, implying that it might be excessively activated because of the increased S. epidermidis density on the lesional skin of AD patients. Additionally, the observations of the murine model of S. aureus skin infections revealed that proteases were crucial for producing more severe lesions in the fatty acid kinase A (FakA)-deficient strain of S. aureus, underlying their role in orchestrating the cutaneous colonization and infection by this pathogen [130]. Lastly, staphylococcal exfoliative toxins and α-toxin were shown to degrade desmoglein-1 and E-cadherin, respectively [131].
9. The Influence of Skin Microbiome Dysbiosis on Itch
As discussed in the introduction, the itch–scratch cycle plays a detrimental role in aggravating epidermal barrier defect, immune dysregulation, and microbial dysbiosis in AD. However, the latest literature reports suggested that the dysbiotic skin microbiome itself was an important factor stimulating itch [132].
As regards AD, damaged keratinocytes and skin microbes upregulate pro-inflammatory cytokines, proteases, and neuropeptides that stimulate peripheral nerve fibers, thus causing the sensation of itch [132]. Seemingly, in the case of AD this process is largely triggered by S. aureus. Firstly, S. aureus antigens may upregulate IL-31, a cytokine known to mediate itch by operating directly on the sensory neurons via TRPV1 and TRPA1 [133]. Furthermore, the upregulation of serine protease activity results in cleaving protease-activated receptors (PARs) which are present on many cell types including sensory neurons [134,135]. As highlighted above, serine proteases additionally trigger Th2 inflammation, which is associated with the sensation of pruritus [136]. S. aureus also seems to induce histaminergic itch by causing the degranulation of mast cells in a process mediated by δ-toxin [137]. Lastly, it was shown that N-formylated peptides and α-hemolysin from S. aureus directly stimulated the sensory neurons and elicited the sensation of pain [138], which could imply their role in generating itch in a similar manner.
The sensation of pruritus is an important factor causing stress in AD [139]. In this context, it is also important to emphasize that the release of substance P resulting from stress modulates the virulence of Gram-positive bacteria, such as S. aureus [140,141].
10. The Effect of Bacterial Biofilms on the Skin Barrier in Atopic Dermatitis
As outlined in the introduction, biofilms constitute the most prevalent form of bacterial skin colonization. The epidermis of AD patients is a relatively beneficial habitat for the rapid development of biofilms, especially those of Staphylococcus spp. In the case of S. aureus, the endogenous propensity to form biofilm depends on the bacterial genotype. S. aureus strains may be grouped into clonal complexes (CCs) based on multilocus sequence typing of staphylococcal housekeeping genes or Staphylococcus protein A (spa) typing [142]. The dominant CCs colonizing patients with AD and the healthy population tend to differ, which translates into the propensity to form biofilm on the skin surface [143,144].
The pathogenic role of staphylococcal biofilms for the skin barrier is diverse. Firstly, the developing biofilm forms an impermeable barrier that causes hypoxia and the subsequent apoptosis of keratinocytes [145,146]. An in vitro study by Tankersley et al. revealed the cytotoxic effect after only 3 h of keratinocyte exposure to staphylococcal biofilm and was not present upon incubation with a sessile form of S. aureus [147]. Furthermore, in vivo assays showed that biofilms developing in AD skin lesions infiltrated the sweat ducts, which aggravated pruritus with the subsequent destruction of the skin barrier due to scratching [148,149]. Longstanding biofilm infections caused by S. aureus may also prompt the self-destruction of host tissues by the activated immune system, which perpetuates the biofilm-related disease and possibly contributes to the defect of the skin barrier [150].
A study by Gonzalez et al. revealed that the presence of S. aureus showing higher relative biofilm propensity compared with S. epidermidis was associated with increased TEWL on both the lesional and nonlesional skin of pediatric AD patients, pointing to epidermal barrier impairment in this cohort [151]. Conversely, Sonesson et al. revealed that proteases secreted late in the process of biofilm development might inactivate antimicrobial peptides, such as LL-37, and hypothesized that they might also contribute to activating kallikreins and cleaving structural proteins important for maintaining skin barrier [152]. Furthermore, according to the literature, bacterial extracts of biofilm-producing S. aureus reduced the expression of mRNA for loricrin and filaggrin by 70–90%, and upregulated mRNA for filaggrin-degrading enzymes caspase-14 and calpain-1 in NHKs, suggesting abnormalities in the epidermal differentiation of the upper stratum corneum [125]. Biofilms may also impair the restoration of tight junctions. Mixed bacterial biofilms were associated with the downregulation of ZO-1 and ZO-2 with a resultant increase in TEWL in a chronic wound model [25] (Figure 2).
Biofilms are additionally known to cause significant aberrations in the immune response. As outlined above (see the section about filaggrin), immune dysregulation typical of AD subsequently causes the disruption of the epidermal barrier. Biofilm-dependent keratinocyte apoptosis and S. aureus antigens, such as lipoteichoic acid, result in the release of numerous DAMPs which trigger innate immunity via TLRs. TLRs are then responsible for eliciting Th2-immune response with the subsequent downregulation of EDC components. In a murine model, triggering TLR2 was shown to induce chronic AD via IL-4-mediated IL-10 suppression [63]. Conversely, TLR3 activation was associated with the upregulation of TSLP, a potent factor responsible for immune polarization towards Th2 response [153].
Indirect insights into the role of biofilms for the epidermal permeability barrier come from the research on disorders affecting the mucous membranes. For example, biofilm exoproteins from S. aureus strains isolated from chronic rhinosinusitis patients were analyzed for their influence on primary human nasal epithelial cells grown at the air–liquid interface [154]. The study revealed the dose- and time-dependent cytotoxicity of epithelial cells, increased permeability, and the reduction of transepithelial resistance after exposure to the biofilm culture of S. aureus in comparison with planktonic cells. Furthermore, the disruption of tight junctions was demonstrated on electron microscopy and a decrease in zonula occludens-1 and claudin-1 was shown with immunofluorescence imaging.
11. Therapeutic Methods with the Potential of Restoring the Normal Skin Microbiota
The critical role of microbiome dysbiosis in AD prompts the search for treatment methods that could restore the physiological composition of the cutaneous microflora and thus alleviate the disease symptoms. It must be emphasized that antibiotics are not routinely recommended in AD because of the insufficient specificity and the possibility of generating resistant pathogens [155,156].
According to current AD guidelines, the treatment with sodium hypochlorite is recommended due to its beneficial effect in alleviating itch and disease severity with varying impact on skin microbiome in different trials [1,157,158]. A recent study by Eriksson et al. showed that sodium hypochlorite used at high concentrations (i.e., 0.02% vs the recommended 0.005%) exerted a significant anti-staphylococcal and anti-biofilm effect [159]. Therefore, it seems that high-concentration sodium hypochlorite baths could be successful in preventing microbial dysbiosis in AD, provided the safety of such treatment was established.
Various novel microbiome-oriented treatments are being elaborated on in AD. One of the concepts involves topical treatment with probiotic strains hampering the growth of S. aureus and supporting the physiological microbiota. For example, Nakatsuji et al. demonstrated that the antimicrobials produced by S. epidermidis and S. hominis showed selective bactericidal activity towards S. aureus and potentiated the antibacterial effect of LL-37, a human AMP [160]. Furthermore, Myles et al. showed that the topical application of Gram-negative Roseomonas mucosa bacterium resulted in a significant decrease in AD severity, itch, and a steroid-sparing effect in adults and children with AD without significant side effects [161].
S. aureus-specific phages and phage lysins constitute another group of medicines with a potential for use in AD. A recent study showed that a treatment with S. epidermidis and SaGU1, a S. aureus phage, showed a sustained inhibition of S. aureus growth [162]. The ongoing research on Staphefekt, an engineered bacteriophage endolysin showing a specific bactericidal activity towards S. aureus, might confirm the clinical efficacy of this approach in AD treatment [163].
Additionally, it is possible that synthetic AMPs prove successful in combating S. aureus in AD. Dawgul et al. showed a significant anti-staphylococcal and anti-biofilm effect of two amphibian AMPs (citropin 1.1 and temporin A) [164]. Furthermore, a phase II trial of omiganan, an indolicidin analog, revealed its potential for improving microbial dysbiosis on AD skin and a small, but significant effect on clinical scores encouraging further research on the implementation of AMPs [165].
Lastly, it was shown that systemic treatment with dupilumab reduced the proportion of S. aureus on both the lesional and nonlesional skin and caused the increase in the population of coagulase-negative staphylococci, such as S. epidermidis, S. hominis, and S. saprophyticus on the nonlesional skin [166]. A study by Lossius et al. revealed that bacterial diversity was also increased on the lesional skin of patients treated with UVB therapy [167]. Such observations reflect the close relationship between the inflammatory cascade and the microbial dysbiosis, underlying the need to diversify the therapeutic process in AD.
12. Conclusions
The epidermal barrier defect and microbiome disorders are closely related and may contribute to the immune dysregulation in AD with the resultant aggravation of the disease course. The latest literature data revealed a nuanced influence of the skin microbiota on various components of the skin barrier such as tight junctions, proteins belonging to the epidermal differentiation complex, and endogenous proteases either directly, or by aggravating the immune dysregulation typical of AD. Furthermore, there is increasing evidence demonstrating that such pathogenic events are largely caused by bacterial biofilms. Further studies are needed to characterize the spectrum of microbiome–host interactions with respect to the skin barrier and to lay the foundation for elaborating on novel treatment methods in atopic dermatitis.
Author Contributions
All authors made substantial contributions to drafting the paper (conceptualization, L.B., Z.S. and J.C.; writing—original draft preparation, L.B., L.R., J.C., A.W.-B., M.G., M.O. and Z.S.; review and editing, L.B., L.R., J.C., A.W.-B., M.G., M.O. and Z.S.). All authors have read and agreed to the published version of the manuscript.
Funding
This research received no external funding.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Wollenberg, A.; Barbarot, S.; Bieber, T.; Christen-Zaech, S.; Deleuran, M.; Fink-Wagner, A.; Gieler, U.; Girolomoni, G.; Lau, S.; Muraro, A.; et al. Consensus-Based European Guidelines for Treatment of Atopic Eczema (Atopic Dermatitis) in Adults and Children: Part I. J. Eur. Acad. Dermatol. Venereol. 2018, 32, 657–682. [Google Scholar] [CrossRef] [PubMed]
- Langan, S.M.; Irvine, A.D.; Weidinger, S. Atopic Dermatitis. Lancet 2020, 396, 345–360. [Google Scholar] [CrossRef]
- Williams, H.C.; Burney, P.G.; Pembroke, A.C.; Hay, R.J. Validation of the U.K. Diagnostic Criteria for Atopic Dermatitis in a Population Setting. U.K. Diagnostic Criteria for Atopic Dermatitis Working Party. Br. J. Dermatol. 1996, 135, 12–17. [Google Scholar] [CrossRef]
- Rajka Georg, H.; Jon, M. Diagnostic Features of Atopic Dermatitis. Acta Derm. Venereol. 1980, 60, 44–47. [Google Scholar]
- Weidinger, S.; Beck, L.A.; Bieber, T.; Kabashima, K.; Irvine, A.D. Atopic Dermatitis. Nat. Rev. Dis. Primers 2018, 4, 1. [Google Scholar] [CrossRef] [PubMed]
- Nettis, E.; Ortoncelli, M.; Pellacani, G.; Foti, C.; Di Leo, E.; Patruno, C.; Rongioletti, F.; Argenziano, G.; Ferrucci, S.; Macchia, L.; et al. A Multicenter Study on the Prevalence of Clinical Patterns and Clinical Phenotypes in Adult Atopic Dermatitis. J. Investig. Allergol. Clin. Immunol. 2020, 30, 448–450. [Google Scholar] [CrossRef]
- Sacotte, R.; Silverberg, J.I. Epidemiology of Adult Atopic Dermatitis. Clin. Dermatol. 2018, 36, 595–605. [Google Scholar] [CrossRef]
- Odhiambo, J.A.; Williams, H.C.; Clayton, T.O.; Robertson, C.F.; Asher, M.I.; ISAAC Phase Three Study Group. Global Variations in Prevalence of Eczema Symptoms in Children from ISAAC Phase Three. J. Allergy Clin. Immunol. 2009, 124, 1251–1258. [Google Scholar] [CrossRef] [PubMed]
- David Boothe, W.; Tarbox, J.A.; Tarbox, M.B. Atopic Dermatitis: Pathophysiology. Adv. Exp. Med. Biol. 2017, 1027, 21–37. [Google Scholar] [CrossRef] [PubMed]
- Pavlis, J.; Yosipovitch, G. Management of Itch in Atopic Dermatitis. Am. J. Clin. Dermatol. 2018, 19, 319–332. [Google Scholar] [CrossRef] [PubMed]
- Leung, D.Y.M.; Berdyshev, E.; Goleva, E. Cutaneous Barrier Dysfunction in Allergic Diseases. J. Allergy Clin. Immunol. 2020, 145, 1485–1497. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Seok, J.K.; Kang, H.C.; Cho, Y.-Y.; Lee, H.S.; Lee, J.Y. Skin Barrier Abnormalities and Immune Dysfunction in Atopic Dermatitis. Int. J. Mol. Sci. 2020, 21, 2867. [Google Scholar] [CrossRef]
- Liang, Y.; Chang, C.; Lu, Q. The Genetics and Epigenetics of Atopic Dermatitis-Filaggrin and Other Polymorphisms. Clin. Rev. Allergy Immunol. 2016, 51, 315–328. [Google Scholar] [CrossRef] [PubMed]
- Egawa, G.; Kabashima, K. Multifactorial Skin Barrier Deficiency and Atopic Dermatitis: Essential Topics to Prevent the Atopic March. J. Allergy Clin. Immunol. 2016, 138, 350–358. [Google Scholar] [CrossRef] [PubMed]
- Kishibe, M. Physiological and Pathological Roles of Kallikrein-Related Peptidases in the Epidermis. J. Dermatol. Sci. 2019, 95, 50–55. [Google Scholar] [CrossRef]
- Tokumasu, R.; Yamaga, K.; Yamazaki, Y.; Murota, H.; Suzuki, K.; Tamura, A.; Bando, K.; Furuta, Y.; Katayama, I.; Tsukita, S. Dose-Dependent Role of Claudin-1 in Vivo in Orchestrating Features of Atopic Dermatitis. Proc. Natl. Acad. Sci. USA 2016, 113, E4061–E4068. [Google Scholar] [CrossRef] [PubMed]
- De Benedetto, A.; Rafaels, N.M.; McGirt, L.Y.; Ivanov, A.I.; Georas, S.N.; Cheadle, C.; Berger, A.E.; Zhang, K.; Vidyasagar, S.; Yoshida, T.; et al. Tight Junction Defects in Patients with Atopic Dermatitis. J. Allergy Clin. Immunol. 2011, 127, 773–786. [Google Scholar] [CrossRef]
- Egawa, G.; Kabashima, K. Barrier Dysfunction in the Skin Allergy. Allergol. Int. 2018, 67, 3–11. [Google Scholar] [CrossRef]
- Lee, J.; Jang, A.; Seo, S.J.; Myung, S.C. Epigenetic Regulation of Filaggrin Gene Expression in Human Epidermal Keratinocytes. Ann. Dermatol. 2020, 32, 122. [Google Scholar] [CrossRef] [PubMed]
- Dréno, B.; Araviiskaia, E.; Berardesca, E.; Gontijo, G.; Sanchez Viera, M.; Xiang, L.F.; Martin, R.; Bieber, T. Microbiome in Healthy Skin, Update for Dermatologists. J. Eur. Acad. Dermatol. Venereol. 2016, 30, 2038–2047. [Google Scholar] [CrossRef]
- Yamazaki, Y.; Nakamura, Y.; Núñez, G. Role of the Microbiota in Skin Immunity and Atopic Dermatitis. Allergol. Int. 2017, 66, 539–544. [Google Scholar] [CrossRef]
- Byrd, A.L.; Belkaid, Y.; Segre, J.A. The Human Skin Microbiome. Nat. Rev. Microbiol. 2018, 16, 143–155. [Google Scholar] [CrossRef]
- Kumar, A.; Alam, A.; Rani, M.; Ehtesham, N.Z.; Hasnain, S.E. Biofilms: Survival and Defense Strategy for Pathogens. Int. J. Med. Microbiol. 2017, 307, 481–489. [Google Scholar] [CrossRef]
- Dréno, B.; Pécastaings, S.; Corvec, S.; Veraldi, S.; Khammari, A.; Roques, C. Cutibacterium Acnes (Propionibacterium Acnes) and Acne Vulgaris: A Brief Look at the Latest Updates. J. Eur. Acad. Dermatol. Venereol. 2018, 32, 5–14. [Google Scholar] [CrossRef]
- Roy, S.; Elgharably, H.; Sinha, M.; Ganesh, K.; Chaney, S.; Mann, E.; Miller, C.; Khanna, S.; Bergdall, V.K.; Powell, H.M.; et al. Mixed-Species Biofilm Compromises Wound Healing by Disrupting Epidermal Barrier Function. J. Pathol. 2014, 233, 331–343. [Google Scholar] [CrossRef]
- Brandner, J.M.; Zorn-Kruppa, M.; Yoshida, T.; Moll, I.; Beck, L.A.; De Benedetto, A. Epidermal Tight Junctions in Health and Disease. Tissue Barriers 2015, 3, e974451. [Google Scholar] [CrossRef]
- Brettmann, E.A.; de Guzman Strong, C. Recent Evolution of the Human Skin Barrier. Exp. Dermatol. 2018, 27, 859–866. [Google Scholar] [CrossRef]
- He, H.; Bissonnette, R.; Wu, J.; Diaz, A.; Saint-Cyr Proulx, E.; Maari, C.; Jack, C.; Louis, M.; Estrada, Y.; Krueger, J.G.; et al. Tape Strips Detect Distinct Immune and Barrier Profiles in Atopic Dermatitis and Psoriasis. J. Allergy Clin. Immunol. 2021, 147, 199–212. [Google Scholar] [CrossRef] [PubMed]
- Murphrey, M.B.; Miao, J.H.; Zito, P.M. Histology, Stratum Corneum. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- de Benedetto, A. Tight Junctions in the Skin: Still a Lot to Learn. Br. J. Dermatol. 2021, 184, 388–389. [Google Scholar] [CrossRef]
- Wertz, P. Epidermal Lamellar Granules. Skin Pharmacol. Physiol. 2018, 31, 262–268. [Google Scholar] [CrossRef]
- Otani, T.; Furuse, M. Tight Junction Structure and Function Revisited. Trends Cell Biol. 2020, 30, 805–817. [Google Scholar] [CrossRef]
- Zihni, C.; Mills, C.; Matter, K.; Balda, M.S. Tight Junctions: From Simple Barriers to Multifunctional Molecular Gates. Nat. Rev. Mol. Cell Biol. 2016, 17, 564–580. [Google Scholar] [CrossRef]
- Zheng, M.; Sun, S.; Zhou, J.; Liu, M. Virulence Factors Impair Epithelial Junctions during Bacterial Infection. J. Clin. Lab. Anal. 2021, 35, e23627. [Google Scholar] [CrossRef]
- Bin, L.; Leung, D.Y.M. Genetic and Epigenetic Studies of Atopic Dermatitis. Allergy Asthma Clin. Immunol. 2016, 12, 52. [Google Scholar] [CrossRef]
- Freeman, S.C.; Sonthalia, S. Histology, Keratohyalin Granules. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2021. [Google Scholar]
- Drislane, C.; Irvine, A.D. The Role of Filaggrin in Atopic Dermatitis and Allergic Disease. Ann. Allergy Asthma Immunol. 2020, 124, 36–43. [Google Scholar] [CrossRef]
- Lee, A.-Y. Molecular Mechanism of Epidermal Barrier Dysfunction as Primary Abnormalities. Int. J. Mol. Sci. 2020, 21, 1194. [Google Scholar] [CrossRef]
- Leyvraz, C.; Charles, R.-P.; Rubera, I.; Guitard, M.; Rotman, S.; Breiden, B.; Sandhoff, K.; Hummler, E. The Epidermal Barrier Function Is Dependent on the Serine Protease CAP1/Prss8. J. Cell Biol. 2005, 170, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Thyssen, J.P.; Jakasa, I.; Riethmüller, C.; Schön, M.P.; Braun, A.; Haftek, M.; Fallon, P.G.; Wróblewski, J.; Jakubowski, H.; Eckhart, L.; et al. Filaggrin Expression and Processing Deficiencies Impair Corneocyte Surface Texture and Stiffness in Mice. J. Investig. Dermatol. 2020, 140, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Donovan, M.; Salamito, M.; Thomas-Collignon, A.; Simonetti, L.; Desbouis, S.; Rain, J.-C.; Formstecher, E.; Bernard, D. Filaggrin and Filaggrin 2 Processing Are Linked Together through Skin Aspartic Acid Protease Activation. PLoS ONE 2020, 15, e0232679. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.; Lim, K.-M. Skin Barrier Dysfunction and Filaggrin. Arch. Pharm. Res. 2021, 44, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Moch, M.; Schwarz, N.; Windoffer, R.; Leube, R.E. The Keratin-Desmosome Scaffold: Pivotal Role of Desmosomes for Keratin Network Morphogenesis. Cell. Mol. Life Sci. 2020, 77, 543–558. [Google Scholar] [CrossRef]
- Brown, S.J.; McLean, W.H.I. One Remarkable Molecule: Filaggrin. J. Investig. Dermatol. 2012, 132, 751–762. [Google Scholar] [CrossRef]
- Zeeuwen, P.L.J.M. Epidermal Differentiation: The Role of Proteases and Their Inhibitors. Eur. J. Cell Biol. 2004, 83, 761–773. [Google Scholar] [CrossRef]
- Martin, M.J.; Estravís, M.; García-Sánchez, A.; Dávila, I.; Isidoro-García, M.; Sanz, C. Genetics and Epigenetics of Atopic Dermatitis: An Updated Systematic Review. Genes 2020, 11, 442. [Google Scholar] [CrossRef]
- Cui, L.; Jia, Y.; Cheng, Z.-W.; Gao, Y.; Zhang, G.-L.; Li, J.-Y.; He, C.-F. Advancements in the Maintenance of Skin Barrier/Skin Lipid Composition and the Involvement of Metabolic Enzymes. J. Cosmet. Dermatol. 2016, 15, 549–558. [Google Scholar] [CrossRef]
- de Veer, S.J.; Furio, L.; Swedberg, J.E.; Munro, C.A.; Brattsand, M.; Clements, J.A.; Hovnanian, A.; Harris, J.M. Selective Substrates and Inhibitors for Kallikrein-Related Peptidase 7 (KLK7) Shed Light on KLK Proteolytic Activity in the Stratum Corneum. J. Investig. Dermatol. 2017, 137, 430–439. [Google Scholar] [CrossRef]
- Sarri, C.A.; Roussaki-Schulze, A.; Vasilopoulos, Y.; Zafiriou, E.; Patsatsi, A.; Stamatis, C.; Gidarokosta, P.; Sotiriadis, D.; Sarafidou, T.; Mamuris, Z. Netherton Syndrome: A Genotype-Phenotype Review. Mol. Diagn. Ther. 2017, 21, 137–152. [Google Scholar] [CrossRef]
- Proksch, E. PH in Nature, Humans and Skin. J. Dermatol. 2018, 45, 1044–1052. [Google Scholar] [CrossRef] [PubMed]
- Chng, K.R.; Tay, A.S.L.; Li, C.; Ng, A.H.Q.; Wang, J.; Suri, B.K.; Matta, S.A.; McGovern, N.; Janela, B.; Wong, X.F.C.C.; et al. Whole Metagenome Profiling Reveals Skin Microbiome-Dependent Susceptibility to Atopic Dermatitis Flare. Nat. Microbiol. 2016, 1, 16106. [Google Scholar] [CrossRef]
- Jang, H.; Matsuda, A.; Jung, K.; Karasawa, K.; Matsuda, K.; Oida, K.; Ishizaka, S.; Ahn, G.; Amagai, Y.; Moon, C.; et al. Skin PH Is the Master Switch of Kallikrein 5-Mediated Skin Barrier Destruction in a Murine Atopic Dermatitis Model. J. Investig. Dermatol. 2016, 136, 127–135. [Google Scholar] [CrossRef]
- van Smeden, J.; Bouwstra, J.A. Stratum Corneum Lipids: Their Role for the Skin Barrier Function in Healthy Subjects and Atopic Dermatitis Patients. Curr. Probl. Dermatol. 2016, 49, 8–26. [Google Scholar] [CrossRef]
- Bhattacharya, N.; Sato, W.J.; Kelly, A.; Ganguli-Indra, G.; Indra, A.K. Epidermal Lipids: Key Mediators of Atopic Dermatitis Pathogenesis. Trends Mol. Med. 2019, 25, 551–562. [Google Scholar] [CrossRef]
- Li, S.; Teegarden, A.; Bauer, E.M.; Choi, J.; Messaddeq, N.; Hendrix, D.A.; Ganguli-Indra, G.; Leid, M.; Indra, A.K. Transcription Factor CTIP1/ BCL11A Regulates Epidermal Differentiation and Lipid Metabolism During Skin Development. Sci. Rep. 2017, 7, 13427. [Google Scholar] [CrossRef]
- Wang, Z.; Kirkwood, J.S.; Taylor, A.W.; Stevens, J.F.; Leid, M.; Ganguli-Indra, G.; Indra, A.K. Transcription Factor Ctip2 Controls Epidermal Lipid Metabolism and Regulates Expression of Genes Involved in Sphingolipid Biosynthesis during Skin Development. J. Investig. Dermatol. 2013, 133, 668–676. [Google Scholar] [CrossRef] [PubMed]
- Berdyshev, E.; Goleva, E.; Bronova, I.; Dyjack, N.; Rios, C.; Jung, J.; Taylor, P.; Jeong, M.; Hall, C.F.; Richers, B.N.; et al. Lipid Abnormalities in Atopic Skin Are Driven by Type 2 Cytokines. JCI Insight 2018, 3, 98006. [Google Scholar] [CrossRef]
- Harris, I.R.; Farrell, A.M.; Grunfeld, C.; Holleran, W.M.; Elias, P.M.; Feingold, K.R. Permeability Barrier Disruption Coordinately Regulates MRNA Levels for Key Enzymes of Cholesterol, Fatty Acid, and Ceramide Synthesis in the Epidermis. J. Investig. Dermatol. 1997, 109, 783–787. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Cheung Lam, A.H.; Sandoval, N.; Wadhwa, R.; Gilkes, J.; Do, T.Q.; Ernst, W.; Chiang, S.-M.; Kosina, S.; Xu, H.H.; Fujii, G.; et al. Assessment of Free Fatty Acids and Cholesteryl Esters Delivered in Liposomes as Novel Class of Antibiotic. BMC Res. Notes 2016, 9, 337. [Google Scholar] [CrossRef]
- Macheleidt, O.; Kaiser, H.W.; Sandhoff, K. Deficiency of Epidermal Protein-Bound Omega-Hydroxyceramides in Atopic Dermatitis. J. Investig. Dermatol. 2002, 119, 166–173. [Google Scholar] [CrossRef]
- Hato, T.; Dagher, P.C. How the Innate Immune System Senses Trouble and Causes Trouble. Clin. J. Am. Soc. Nephrol. 2015, 10, 1459–1469. [Google Scholar] [CrossRef]
- Herwald, H.; Egesten, A. On PAMPs and DAMPs. J. Innate Immun. 2016, 8, 427–428. [Google Scholar] [CrossRef]
- Kaesler, S.; Volz, T.; Skabytska, Y.; Köberle, M.; Hein, U.; Chen, K.-M.; Guenova, E.; Wölbing, F.; Röcken, M.; Biedermann, T. Toll-like Receptor 2 Ligands Promote Chronic Atopic Dermatitis through IL-4–Mediated Suppression of IL-10. J. Allergy Clin. Immunol. 2014, 134, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Weidinger, S.; Klopp, N.; Rummler, L.; Wagenpfeil, S.; Novak, N.; Baurecht, H.-J.; Groer, W.; Darsow, U.; Heinrich, J.; Gauger, A.; et al. Association of NOD1 Polymorphisms with Atopic Eczema and Related Phenotypes. J. Allergy Clin. Immunol. 2005, 116, 177–184. [Google Scholar] [CrossRef] [PubMed]
- De Benedetto, A.; Agnihothri, R.; McGirt, L.Y.; Bankova, L.G.; Beck, L.A. Atopic Dermatitis: A Disease Caused by Innate Immune Defects? J. Investig. Dermatol. 2009, 129, 14–30. [Google Scholar] [CrossRef]
- Czarnowicki, T.; He, H.; Krueger, J.G.; Guttman-Yassky, E. Atopic Dermatitis Endotypes and Implications for Targeted Therapeutics. J. Allergy Clin. Immunol. 2019, 143, 1–11. [Google Scholar] [CrossRef]
- Renert-Yuval, Y.; Guttman-Yassky, E. New Treatments for Atopic Dermatitis Targeting beyond IL-4/IL-13 Cytokines. Ann. Allergy Asthma Immunol. 2020, 124, 28–35. [Google Scholar] [CrossRef]
- Brunner, P.M.; Guttman-Yassky, E.; Leung, D.Y.M. The Immunology of Atopic Dermatitis and Its Reversibility with Broad-Spectrum and Targeted Therapies. J. Allergy Clin. Immunol. 2017, 139, S65–S76. [Google Scholar] [CrossRef]
- Oliva, M.; Renert-Yuval, Y.; Guttman-Yassky, E. The ‘Omics’ Revolution: Redefining the Understanding and Treatment of Allergic Skin Diseases. Curr. Opin. Allergy Clin. Immunol. 2016, 16, 469–476. [Google Scholar] [CrossRef] [PubMed]
- Grice, E.A.; Kong, H.H.; Conlan, S.; Deming, C.B.; Davis, J.; Young, A.C.; NISC Comparative Sequencing Program; Bouffard, G.G.; Blakesley, R.W.; Murray, P.R.; et al. Topographical and Temporal Diversity of the Human Skin Microbiome. Science 2009, 324, 1190–1192. [Google Scholar] [CrossRef]
- Grice, E.A.; Segre, J.A. The Skin Microbiome. Nat. Rev. Microbiol. 2011, 9, 244–253. [Google Scholar] [CrossRef] [PubMed]
- Paller, A.S.; Kong, H.H.; Seed, P.; Naik, S.; Scharschmidt, T.C.; Gallo, R.L.; Luger, T.; Irvine, A.D. The Microbiome in Patients with Atopic Dermatitis. J. Allergy Clin. Immunol. 2019, 143, 26–35. [Google Scholar] [CrossRef] [PubMed]
- Kong, H.H.; Oh, J.; Deming, C.; Conlan, S.; Grice, E.A.; Beatson, M.A.; Nomicos, E.; Polley, E.C.; Komarow, H.D.; NISC Comparative Sequence Program; et al. Temporal Shifts in the Skin Microbiome Associated with Disease Flares and Treatment in Children with Atopic Dermatitis. Genome Res. 2012, 22, 850–859. [Google Scholar] [CrossRef]
- Brown, M.M.; Horswill, A.R. Staphylococcus Epidermidis—Skin Friend or Foe? PLoS Pathog. 2020, 16, e1009026. [Google Scholar] [CrossRef]
- Cogen, A.L.; Yamasaki, K.; Sanchez, K.M.; Dorschner, R.A.; Lai, Y.; MacLeod, D.T.; Torpey, J.W.; Otto, M.; Nizet, V.; Kim, J.E.; et al. Selective Antimicrobial Action Is Provided by Phenol-Soluble Modulins Derived from Staphylococcus Epidermidis, a Normal Resident of the Skin. J. Investig. Dermatol. 2010, 130, 192–200. [Google Scholar] [CrossRef]
- Otto, M. Staphylococcus Colonization of the Skin and Antimicrobial Peptides. Expert Rev. Dermatol. 2010, 5, 183–195. [Google Scholar] [CrossRef]
- Hon, K.L.; Tsang, Y.C.K.; Pong, N.H.; Leung, T.F.; Ip, M. Exploring Staphylococcus Epidermidis in Atopic Eczema: Friend or Foe? Clin. Exp. Dermatol. 2016, 41, 659–663. [Google Scholar] [CrossRef] [PubMed]
- Totté, J.E.E.; Pardo, L.M.; Fieten, K.B.; Vos, M.C.; Broek, T.J.; Schuren, F.H.J.; Pasmans, S.G.M.A. Nasal and Skin Microbiomes Are Associated with Disease Severity in Paediatric Atopic Dermatitis. Br. J. Dermatol. 2019, 181, 796–804. [Google Scholar] [CrossRef] [PubMed]
- Loomis, K.H.; Wu, S.K.; Ernlund, A.; Zudock, K.; Reno, A.; Blount, K.; Karig, D.K. A Mixed Community of Skin Microbiome Representatives Influences Cutaneous Processes More than Individual Members. Microbiome 2021, 9, 22. [Google Scholar] [CrossRef]
- Byrd, A.L.; Deming, C.; Cassidy, S.K.B.; Harrison, O.J.; Ng, W.-I.; Conlan, S.; NISC Comparative Sequencing Program; Belkaid, Y.; Segre, J.A.; Kong, H.H. Staphylococcus aureus and Staphylococcus epidermidis Strain Diversity Underlying Pediatric Atopic Dermatitis. Sci. Transl. Med. 2017, 9, eaal4651. [Google Scholar] [CrossRef]
- Leung, D.Y.M.; Calatroni, A.; Zaramela, L.S.; LeBeau, P.K.; Dyjack, N.; Brar, K.; David, G.; Johnson, K.; Leung, S.; Ramirez-Gama, M.; et al. The Nonlesional Skin Surface Distinguishes Atopic Dermatitis with Food Allergy as a Unique Endotype. Sci. Transl. Med. 2019, 11, eaav2685. [Google Scholar] [CrossRef]
- Tauber, M.; Balica, S.; Hsu, C.-Y.; Jean-Decoster, C.; Lauze, C.; Redoules, D.; Viodé, C.; Schmitt, A.-M.; Serre, G.; Simon, M.; et al. Staphylococcus aureus Density on Lesional and Nonlesional Skin Is Strongly Associated with Disease Severity in Atopic Dermatitis. J. Allergy Clin. Immunol. 2016, 137, 1272–1274. [Google Scholar] [CrossRef] [PubMed]
- Blicharz, L.; Usarek, P.; Młynarczyk, G.; Skowroński, K.; Rudnicka, L.; Samochocki, Z. Nasal Colonization by Staphylococci and Severity of Atopic Dermatitis. Dermatitis 2020, 31, 215–222. [Google Scholar] [CrossRef]
- Flemming, H.-C.; Wingender, J.; Szewzyk, U.; Steinberg, P.; Rice, S.A.; Kjelleberg, S. Biofilms: An Emergent Form of Bacterial Life. Nat. Rev. Microbiol. 2016, 14, 563–575. [Google Scholar] [CrossRef] [PubMed]
- Rabin, N.; Zheng, Y.; Opoku-Temeng, C.; Du, Y.; Bonsu, E.; Sintim, H.O. Biofilm Formation Mechanisms and Targets for Developing Antibiofilm Agents. Future Med. Chem. 2015, 7, 493–512. [Google Scholar] [CrossRef]
- Koo, H.; Allan, R.N.; Howlin, R.P.; Stoodley, P.; Hall-Stoodley, L. Targeting Microbial Biofilms: Current and Prospective Therapeutic Strategies. Nat. Rev. Microbiol. 2017, 15, 740–755. [Google Scholar] [CrossRef]
- Yin, W.; Wang, Y.; Liu, L.; He, J. Biofilms: The Microbial “Protective Clothing” in Extreme Environments. Int. J. Mol. Sci. 2019, 20, 3423. [Google Scholar] [CrossRef] [PubMed]
- Mukherjee, S.; Bassler, B.L. Bacterial Quorum Sensing in Complex and Dynamically Changing Environments. Nat. Rev. Microbiol. 2019, 17, 371–382. [Google Scholar] [CrossRef]
- Schilcher, K.; Horswill, A.R. Staphylococcal Biofilm Development: Structure, Regulation, and Treatment Strategies. Microbiol. Mol. Biol. Rev. 2020, 84, e00026-19. [Google Scholar] [CrossRef]
- Di Domenico, E.G.; Cavallo, I.; Capitanio, B.; Ascenzioni, F.; Pimpinelli, F.; Morrone, A.; Ensoli, F. Staphylococcus aureus and the Cutaneous Microbiota Biofilms in the Pathogenesis of Atopic Dermatitis. Microorganisms 2019, 7, 301. [Google Scholar] [CrossRef]
- Le, K.Y.; Dastgheyb, S.; Ho, T.V.; Otto, M. Molecular Determinants of Staphylococcal Biofilm Dispersal and Structuring. Front. Cell. Infect. Microbiol. 2014, 4, 167. [Google Scholar] [CrossRef] [PubMed]
- Lee, S.O.; Lee, S.; Lee, J.E.; Song, K.-H.; Kang, C.K.; Wi, Y.M.; San-Juan, R.; López-Cortés, L.E.; Lacoma, A.; Prat, C.; et al. Dysfunctional Accessory Gene Regulator (Agr) as a Prognostic Factor in Invasive Staphylococcus aureus Infection: A Systematic Review and Meta-Analysis. Sci. Rep. 2020, 10, 20697. [Google Scholar] [CrossRef]
- Le, K.Y.; Otto, M. Quorum-Sensing Regulation in Staphylococci—an Overview. Front. Microbiol. 2015, 6, 1174. [Google Scholar] [CrossRef]
- Blicharz, L.; Michalak, M.; Szymanek-Majchrzak, K.; Młynarczyk, G.; Skowroński, K.; Rudnicka, L.; Samochocki, Z. The Propensity to Form Biofilm In Vitro by Staphylococcus aureus Strains Isolated from the Anterior Nares of Patients with Atopic Dermatitis: Clinical Associations. Dermatology 2021, 237, 528–534. [Google Scholar] [CrossRef]
- Foster, T.J.; Geoghegan, J.A.; Ganesh, V.K.; Höök, M. Adhesion, Invasion and Evasion: The Many Functions of the Surface Proteins of Staphylococcus aureus. Nat. Rev. Microbiol. 2014, 12, 49–62. [Google Scholar] [CrossRef]
- Cho, S.-H.; Strickland, I.; Boguniewicz, M.; Leung, D.Y.M. Fibronectin and Fibrinogen Contribute to the Enhanced Binding of Staphylococcus aureus to Atopic Skin. J. Allergy Clin. Immunol. 2001, 108, 269–274. [Google Scholar] [CrossRef]
- Clausen, M.-L.; Edslev, S.M.; Andersen, P.S.; Clemmensen, K.; Krogfelt, K.A.; Agner, T. Staphylococcus aureus Colonization in Atopic Eczema and Its Association with Filaggrin Gene Mutations. Br. J. Dermatol. 2017, 177, 1394–1400. [Google Scholar] [CrossRef] [PubMed]
- Cai, S.C.S.; Chen, H.; Koh, W.-P.; Common, J.E.A.; van Bever, H.P.; McLean, W.H.I.; Lane, E.B.; Giam, Y.C.; Tang, M.B.Y. Filaggrin Mutations Are Associated with Recurrent Skin Infection in Singaporean Chinese Patients with Atopic Dermatitis. Br. J. Dermatol. 2012, 166, 200–203. [Google Scholar] [CrossRef]
- Nakatsuji, T.; Chen, T.H.; Two, A.M.; Chun, K.A.; Narala, S.; Geha, R.S.; Hata, T.R.; Gallo, R.L. Staphylococcus aureus Exploits Epidermal Barrier Defects in Atopic Dermatitis to Trigger Cytokine Expression. J. Investig. Dermatol. 2016, 136, 2192–2200. [Google Scholar] [CrossRef]
- Simpson, E.L.; Villarreal, M.; Jepson, B.; Rafaels, N.; David, G.; Hanifin, J.; Taylor, P.; Boguniewicz, M.; Yoshida, T.; De Benedetto, A.; et al. Patients with Atopic Dermatitis Colonized with Staphylococcus aureus Have a Distinct Phenotype and Endotype. J. Investig. Dermatol. 2018, 138, 2224–2233. [Google Scholar] [CrossRef]
- Feuillie, C.; Vitry, P.; McAleer, M.A.; Kezic, S.; Irvine, A.D.; Geoghegan, J.A.; Dufrêne, Y.F. Adhesion of Staphylococcus aureus to Corneocytes from Atopic Dermatitis Patients Is Controlled by Natural Moisturizing Factor Levels. mBio 2018, 9, e01184-18. [Google Scholar] [CrossRef]
- Towell, A.M.; Feuillie, C.; Vitry, P.; Da Costa, T.M.; Mathelié-Guinlet, M.; Kezic, S.; Fleury, O.M.; McAleer, M.A.; Dufrêne, Y.F.; Irvine, A.D.; et al. Staphylococcus aureus Binds to the N-Terminal Region of Corneodesmosin to Adhere to the Stratum Corneum in Atopic Dermatitis. Proc. Natl. Acad. Sci. USA. 2021, 118, e2014444118. [Google Scholar] [CrossRef]
- Igawa, S.; Ohzono, A.; Pham, P.; Wang, Z.; Nakatsuji, T.; Dokoshi, T.; Di Nardo, A. Sphingosine 1-Phosphate Receptor 2 Is Central to Maintaining Epidermal Barrier Homeostasis. J. Investig. Dermatol. 2021, 141, 1188–1197. [Google Scholar] [CrossRef]
- Lipsky, Z.W.; Marques, C.N.H.; German, G.K. Lipid Depletion Enables Permeation of Staphylococcus aureus Bacteria through Human Stratum Corneum. Tissue Barriers 2020, 8, 1754706. [Google Scholar] [CrossRef]
- Baurecht, H.; Rühlemann, M.C.; Rodríguez, E.; Thielking, F.; Harder, I.; Erkens, A.-S.; Stölzl, D.; Ellinghaus, E.; Hotze, M.; Lieb, W.; et al. Epidermal Lipid Composition, Barrier Integrity, and Eczematous Inflammation Are Associated with Skin Microbiome Configuration. J. Allergy Clin. Immunol. 2018, 141, 1668–1676. [Google Scholar] [CrossRef]
- Cleary, J.M.; Lipsky, Z.W.; Kim, M.; Marques, C.N.H.; German, G.K. Heterogeneous Ceramide Distributions Alter Spatially Resolved Growth of Staphylococcus aureus on Human Stratum Corneum. J. R. Soc. Interface 2018, 15, 20170848. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Villarreal, M.; Stewart, S.; Choi, J.; Ganguli-Indra, G.; Babineau, D.C.; Philpot, C.; David, G.; Yoshida, T.; Boguniewicz, M.; et al. Altered Composition of Epidermal Lipids Correlates with Staphylococcus aureus Colonization Status in Atopic Dermatitis. Br. J. Dermatol. 2017, 177, e125–e127. [Google Scholar] [CrossRef] [PubMed]
- Meisel, J.S.; Sfyroera, G.; Bartow-McKenney, C.; Gimblet, C.; Bugayev, J.; Horwinski, J.; Kim, B.; Brestoff, J.R.; Tyldsley, A.S.; Zheng, Q.; et al. Commensal Microbiota Modulate Gene Expression in the Skin. Microbiome 2018, 6, 20. [Google Scholar] [CrossRef]
- Ohnemus, U.; Kohrmeyer, K.; Houdek, P.; Rohde, H.; Wladykowski, E.; Vidal, S.; Horstkotte, M.A.; Aepfelbacher, M.; Kirschner, N.; Behne, M.J.; et al. Regulation of Epidermal Tight-Junctions (TJ) during Infection with Exfoliative Toxin-Negative Staphylococcus Strains. J. Investig. Dermatol. 2008, 128, 906–916. [Google Scholar] [CrossRef] [PubMed]
- Bäsler, K.; Galliano, M.-F.; Bergmann, S.; Rohde, H.; Wladykowski, E.; Vidal-y-Sy, S.; Guiraud, B.; Houdek, P.; Schüring, G.; Volksdorf, T.; et al. Biphasic Influence of Staphylococcus aureus on Human Epidermal Tight Junctions. Ann. N. Y. Acad. Sci. 2017, 1405, 53–70. [Google Scholar] [CrossRef]
- Wang, B.; McHugh, B.J.; Qureshi, A.; Campopiano, D.J.; Clarke, D.J.; Fitzgerald, J.R.; Dorin, J.R.; Weller, R.; Davidson, D.J. IL-1β-Induced Protection of Keratinocytes against Staphylococcus aureus-Secreted Proteases Is Mediated by Human β-Defensin 2. J. Investig. Dermatol. 2017, 137, 95–105. [Google Scholar] [CrossRef]
- Kuo, I.-H.; Carpenter-Mendini, A.; Yoshida, T.; McGirt, L.Y.; Ivanov, A.I.; Barnes, K.C.; Gallo, R.L.; Borkowski, A.W.; Yamasaki, K.; Leung, D.Y.; et al. Activation of Epidermal Toll-like Receptor 2 Enhances Tight Junction Function: Implications for Atopic Dermatitis and Skin Barrier Repair. J. Investig. Dermatol. 2013, 133, 988–998. [Google Scholar] [CrossRef]
- Martens, K.; Seys, S.F.; Alpizar, Y.A.; Schrijvers, R.; Bullens, D.M.A.; Breynaert, C.; Lebeer, S.; Steelant, B. Staphylococcus aureus Enterotoxin B Disrupts Nasal Epithelial Barrier Integrity. Clin. Exp. Allergy 2021, 51, 87–98. [Google Scholar] [CrossRef]
- Mitamura, Y.; Nunomura, S.; Nanri, Y.; Ogawa, M.; Yoshihara, T.; Masuoka, M.; Tsuji, G.; Nakahara, T.; Hashimoto-Hachiya, A.; Conway, S.J.; et al. The IL-13/Periostin/IL-24 Pathway Causes Epidermal Barrier Dysfunction in Allergic Skin Inflammation. Allergy 2018, 73, 1881–1891. [Google Scholar] [CrossRef] [PubMed]
- Ryu, W.-I.; Lee, H.; Bae, H.C.; Ryu, H.J.; Son, S.W. IL-33 down-Regulates Filaggrin Expression by Inducing STAT3 and ERK Phosphorylation in Human Keratinocytes. J. Dermatol. Sci. 2016, 82, 131–134. [Google Scholar] [CrossRef]
- Imai, Y. Interleukin-33 in Atopic Dermatitis. J. Dermatol. Sci. 2019, 96, 2–7. [Google Scholar] [CrossRef]
- Park, K.-D.; Pak, S.; Park, K.-K. The Pathogenetic Effect of Natural and Bacterial Toxins on Atopic Dermatitis. Toxins 2016, 9, 3. [Google Scholar] [CrossRef]
- Nygaard, U.; van den Bogaard, E.H.; Niehues, H.; Hvid, M.; Deleuran, M.; Johansen, C.; Vestergaard, C. The “Alarmins” HMBG1 and IL-33 Downregulate Structural Skin Barrier Proteins and Impair Epidermal Growth. Acta Derm.-Venereol. 2017, 97, 305–312. [Google Scholar] [CrossRef]
- Kim, B.S.; Choi, J.K.; Jung, H.J.; Park, K.H.; Jang, Y.H.; Lee, W.J.; Lee, S.-J.; Kim, S.-H.; Kang, H.Y.; Kim, J.M.; et al. Effects of Topical Application of a Recombinant Staphylococcal Enterotoxin A on DNCB and Dust Mite Extract-Induced Atopic Dermatitis-like Lesions in a Murine Model. Eur. J. Dermatol. EJD 2014, 24, 186–193. [Google Scholar] [CrossRef]
- Al Kindi, A.; Williams, H.; Matsuda, K.; Alkahtani, A.M.; Saville, C.; Bennett, H.; Alshammari, Y.; Tan, S.Y.; O’Neill, C.; Tanaka, A.; et al. Staphylococcus aureus Second Immunoglobulin-Binding Protein Drives Atopic Dermatitis via IL-33. J. Allergy Clin. Immunol. 2021, 147, 1354–1368. [Google Scholar] [CrossRef]
- Jeon, Y.J.; Gil, C.H.; Won, J.; Jo, A.; Kim, H.J. Symbiotic Microbiome Staphylococcus aureus from Human Nasal Mucus Modulates IL-33-Mediated Type 2 Immune Responses in Allergic Nasal Mucosa. BMC Microbiol. 2020, 20, 301. [Google Scholar] [CrossRef]
- Brauweiler, A.M.; Leung, D.Y.M.; Goleva, E. The Transcription Factor P63 Is a Direct Effector of IL-4- and IL-13-Mediated Repression of Keratinocyte Differentiation. J. Investig. Dermatol. 2021, 141, 770–778. [Google Scholar] [CrossRef]
- Brauweiler, A.M.; Goleva, E.; Leung, D.Y.M. Staphylococcus aureus Lipoteichoic Acid Damages the Skin Barrier through an IL-1-Mediated Pathway. J. Investig. Dermatol. 2019, 139, 1753–1761. [Google Scholar] [CrossRef]
- Simon, M. Effects of Environmental Skin Stressors on Filaggrin Degradation Products: Importance for Eczema. Br. J. Dermatol. 2018, 179, 560–561. [Google Scholar] [CrossRef]
- Son, E.D.; Kim, H.-J.; Park, T.; Shin, K.; Bae, I.-H.; Lim, K.-M.; Cho, E.-G.; Lee, T.R. Staphylococcus aureus Inhibits Terminal Differentiation of Normal Human Keratinocytes by Stimulating Interleukin-6 Secretion. J. Dermatol. Sci. 2014, 74, 64–71. [Google Scholar] [CrossRef]
- Williams, M.R.; Nakatsuji, T.; Sanford, J.A.; Vrbanac, A.F.; Gallo, R.L. Staphylococcus aureus Induces Increased Serine Protease Activity in Keratinocytes. J. Investig. Dermatol. 2017, 137, 377–384. [Google Scholar] [CrossRef]
- Williams, M.R.; Costa, S.K.; Zaramela, L.S.; Khalil, S.; Todd, D.A.; Winter, H.L.; Sanford, J.A.; O’Neill, A.M.; Liggins, M.C.; Nakatsuji, T.; et al. Quorum Sensing between Bacterial Species on the Skin Protects against Epidermal Injury in Atopic Dermatitis. Sci. Transl. Med. 2019, 11, eaat8329. [Google Scholar] [CrossRef] [PubMed]
- Elmwall, J.; Kwiecinski, J.; Na, M.; Ali, A.A.; Osla, V.; Shaw, L.N.; Wang, W.; Sävman, K.; Josefsson, E.; Bylund, J.; et al. Galectin-3 Is a Target for Proteases Involved in the Virulence of Staphylococcus aureus. Infect. Immun. 2017, 85, e00177-17. [Google Scholar] [CrossRef] [PubMed]
- Cau, L.; Williams, M.R.; Butcher, A.M.; Nakatsuji, T.; Kavanaugh, J.S.; Cheng, J.Y.; Shafiq, F.; Higbee, K.; Hata, T.R.; Horswill, A.R.; et al. Staphylococcus Epidermidis Protease EcpA Can Be a Deleterious Component of the Skin Microbiome in Atopic Dermatitis. J. Allergy Clin. Immunol. 2021, 147, 955–966. [Google Scholar] [CrossRef] [PubMed]
- Ridder, M.J.; Daly, S.M.; Triplett, K.D.; Seawell, N.A.; Hall, P.R.; Bose, J.L. Staphylococcus aureus Fatty Acid Kinase FakA Modulates Pathogenesis during Skin Infection via Proteases. Infect. Immun. 2020, 88, e00163-20. [Google Scholar] [CrossRef]
- Pietrocola, G.; Nobile, G.; Rindi, S.; Speziale, P. Staphylococcus aureus Manipulates Innate Immunity through Own and Host-Expressed Proteases. Front. Cell. Infect. Microbiol. 2017, 7, 166. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.S.; Yosipovitch, G. The Skin Microbiota and Itch: Is There a Link? J. Clin. Med. 2020, 9, 1190. [Google Scholar] [CrossRef]
- Cevikbas, F.; Wang, X.; Akiyama, T.; Kempkes, C.; Savinko, T.; Antal, A.; Kukova, G.; Buhl, T.; Ikoma, A.; Buddenkotte, J.; et al. A Sensory Neuron-Expressed IL-31 Receptor Mediates T Helper Cell-Dependent Itch: Involvement of TRPV1 and TRPA1. J. Allergy Clin. Immunol. 2014, 133, 448–460. [Google Scholar] [CrossRef] [PubMed]
- Buhl, T.; Ikoma, A.; Kempkes, C.; Cevikbas, F.; Sulk, M.; Buddenkotte, J.; Akiyama, T.; Crumrine, D.; Camerer, E.; Carstens, E.; et al. Protease-Activated Receptor-2 Regulates Neuro-Epidermal Communication in Atopic Dermatitis. Front. Immunol. 2020, 11, 1740. [Google Scholar] [CrossRef]
- Kempkes, C.; Buddenkotte, J.; Cevikbas, F.; Buhl, T.; Steinhoff, M. Role of PAR-2 in Neuroimmune Communication and Itch. In Itch: Mechanisms and Treatment; Carstens, E., Akiyama, T., Eds.; Frontiers in Neuroscience; CRC Press/Taylor & Francis: Boca Raton, FL, USA, 2014; ISBN 978-1-4665-0543-8. [Google Scholar]
- Williams, M.R.; Nakatsuji, T.; Gallo, R.L. Staphylococcus aureus: Master Manipulator of the Skin. Cell Host Microbe 2017, 22, 579–581. [Google Scholar] [CrossRef] [PubMed]
- Nakamura, Y.; Oscherwitz, J.; Cease, K.B.; Chan, S.M.; Muñoz-Planillo, R.; Hasegawa, M.; Villaruz, A.E.; Cheung, G.Y.C.; McGavin, M.J.; Travers, J.B.; et al. Staphylococcus δ-Toxin Induces Allergic Skin Disease by Activating Mast Cells. Nature 2013, 503, 397–401. [Google Scholar] [CrossRef] [PubMed]
- Chiu, I.M.; Heesters, B.A.; Ghasemlou, N.; Von Hehn, C.A.; Zhao, F.; Tran, J.; Wainger, B.; Strominger, A.; Muralidharan, S.; Horswill, A.R.; et al. Bacteria Activate Sensory Neurons That Modulate Pain and Inflammation. Nature 2013, 501, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Evers, A.W.M.; Schut, C.; Gieler, U.; Spillekom-van Koulil, S.; van Beugen, S. Itch Management: Psychotherapeutic Approach. Curr. Probl. Dermatol. 2016, 50, 64–70. [Google Scholar] [CrossRef]
- Mijouin, L.; Hillion, M.; Ramdani, Y.; Jaouen, T.; Duclairoir-Poc, C.; Follet-Gueye, M.-L.; Lati, E.; Yvergnaux, F.; Driouich, A.; Lefeuvre, L.; et al. Effects of a Skin Neuropeptide (Substance p) on Cutaneous Microflora. PLoS ONE 2013, 8, e78773. [Google Scholar] [CrossRef]
- N’Diaye, A.; Gannesen, A.; Borrel, V.; Maillot, O.; Enaut, J.; Racine, P.-J.; Plakunov, V.; Chevalier, S.; Lesouhaitier, O.; Feuilloley, M.G.J. Substance P and Calcitonin Gene-Related Peptide: Key Regulators of Cutaneous Microbiota Homeostasis. Front. Endocrinol. 2017, 8, 15. [Google Scholar] [CrossRef]
- Park, K.-H.; Greenwood-Quaintance, K.E.; Uhl, J.R.; Cunningham, S.A.; Chia, N.; Jeraldo, P.R.; Sampathkumar, P.; Nelson, H.; Patel, R. Molecular Epidemiology of Staphylococcus aureus Bacteremia in a Single Large Minnesota Medical Center in 2015 as Assessed Using MLST, Core Genome MLST and Spa Typing. PLoS ONE 2017, 12, e0179003. [Google Scholar] [CrossRef]
- Harkins, C.P.; Pettigrew, K.A.; Oravcová, K.; Gardner, J.; Hearn, R.M.R.; Rice, D.; Mather, A.E.; Parkhill, J.; Brown, S.J.; Proby, C.M.; et al. The Microevolution and Epidemiology of Staphylococcus aureus Colonization during Atopic Eczema Disease Flare. J. Investig. Dermatol. 2018, 138, 336–343. [Google Scholar] [CrossRef]
- Fleury, O.M.; McAleer, M.A.; Feuillie, C.; Formosa-Dague, C.; Sansevere, E.; Bennett, D.E.; Towell, A.M.; McLean, W.H.I.; Kezic, S.; Robinson, D.A.; et al. Clumping Factor B Promotes Adherence of Staphylococcus aureus to Corneocytes in Atopic Dermatitis. Infect. Immun. 2017, 85, e00994-16. [Google Scholar] [CrossRef]
- Lone, A.G.; Atci, E.; Renslow, R.; Beyenal, H.; Noh, S.; Fransson, B.; Abu-Lail, N.; Park, J.-J.; Gang, D.R.; Call, D.R. Staphylococcus aureus Induces Hypoxia and Cellular Damage in Porcine Dermal Explants. Infect. Immun. 2015, 83, 2531–2541. [Google Scholar] [CrossRef]
- Di Domenico, E.G.; Cavallo, I.; Bordignon, V.; Prignano, G.; Sperduti, I.; Gurtner, A.; Trento, E.; Toma, L.; Pimpinelli, F.; Capitanio, B.; et al. Inflammatory Cytokines and Biofilm Production Sustain Staphylococcus aureus Outgrowth and Persistence: A Pivotal Interplay in the Pathogenesis of Atopic Dermatitis. Sci. Rep. 2018, 8, 9573. [Google Scholar] [CrossRef]
- Tankersley, A.; Frank, M.B.; Bebak, M.; Brennan, R. Early Effects of Staphylococcus aureus Biofilm Secreted Products on Inflammatory Responses of Human Epithelial Keratinocytes. J. Inflamm. Lond. Engl. 2014, 11, 17. [Google Scholar] [CrossRef]
- Allen, H.B.; Vaze, N.D.; Choi, C.; Hailu, T.; Tulbert, B.H.; Cusack, C.A.; Joshi, S.G. The Presence and Impact of Biofilm-Producing Staphylococci in Atopic Dermatitis. JAMA Dermatol. 2014, 150, 260–265. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.Q.; Tang, Y.; Ju, Y.; Zhang, X.Y.; Yan, J.J.; Wang, C.M.; Yang, Y.; Zhu, C.; Tang, Z.X.; Zhou, Y.; et al. Scratching Damages Tight Junctions through the Akt–Claudin 1 Axis in Atopic Dermatitis. Clin. Exp. Dermatol. 2021, 46, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Watters, C.; Fleming, D.; Bishop, D.; Rumbaugh, K.P. Host Responses to Biofilm. Prog. Mol. Biol. Transl. Sci. 2016, 142, 193–239. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, T.; Stevens, M.L.; Baatyrbek Kyzy, A.; Alarcon, R.; He, H.; Kroner, J.W.; Spagna, D.; Grashel, B.; Sidler, E.; Martin, L.J.; et al. Biofilm Propensity of Staphylococcus aureus Skin Isolates Is Associated with Increased Atopic Dermatitis Severity and Barrier Dysfunction in the MPAACH Pediatric Cohort. Allergy 2021, 76, 302–313. [Google Scholar] [CrossRef]
- Sonesson, A.; Przybyszewska, K.; Eriksson, S.; Mörgelin, M.; Kjellström, S.; Davies, J.; Potempa, J.; Schmidtchen, A. Identification of Bacterial Biofilm and the Staphylococcus aureus Derived Protease, Staphopain, on the Skin Surface of Patients with Atopic Dermatitis. Sci. Rep. 2017, 7, 8689. [Google Scholar] [CrossRef]
- Takai, T. TSLP Expression: Cellular Sources, Triggers, and Regulatory Mechanisms. Allergol. Int. Off. J. Jpn. Soc. Allergol. 2012, 61, 3–17. [Google Scholar] [CrossRef]
- Panchatcharam, B.S.; Cooksley, C.M.; Ramezanpour, M.; Vediappan, R.S.; Bassiouni, A.; Wormald, P.J.; Psaltis, A.J.; Vreugde, S. Staphylococcus aureus Biofilm Exoproteins Are Cytotoxic to Human Nasal Epithelial Barrier in Chronic Rhinosinusitis. Int. Forum Allergy Rhinol. 2020, 10, 871–883. [Google Scholar] [CrossRef]
- Błażewicz, I.; Jaśkiewicz, M.; Piechowicz, L.; Neubauer, D.; Nowicki, R.J.; Kamysz, W.; Barańska-Rybak, W. Activity of Antimicrobial Peptides and Conventional Antibiotics against Superantigen Positive Staphylococcus aureus Isolated from Patients with Atopic Dermatitis. Postepy Dermatol. Alergol. 2018, 35, 74–82. [Google Scholar] [CrossRef]
- Harkins, C.P.; McAleer, M.A.; Bennett, D.; McHugh, M.; Fleury, O.M.; Pettigrew, K.A.; Oravcová, K.; Parkhill, J.; Proby, C.M.; Dawe, R.S.; et al. The Widespread Use of Topical Antimicrobials Enriches for Resistance in Staphylococcus aureus Isolated from Patients with Atopic Dermatitis. Br. J. Dermatol. 2018, 179, 951–958. [Google Scholar] [CrossRef]
- Majewski, S.; Bhattacharya, T.; Asztalos, M.; Bohaty, B.; Durham, K.C.; West, D.P.; Hebert, A.A.; Paller, A.S. Sodium Hypochlorite Body Wash in the Management of Staphylococcus aureus–Colonized Moderate-to-severe Atopic Dermatitis in Infants, Children, and Adolescents. Pediatr. Dermatol. 2019, 36, 442–447. [Google Scholar] [CrossRef] [PubMed]
- Maarouf, M.; Shi, V.Y. Bleach for Atopic Dermatitis. Dermatitis 2018, 29, 120–126. [Google Scholar] [CrossRef]
- Eriksson, S.; van der Plas, M.J.A.; Mörgelin, M.; Sonesson, A. Antibacterial and Antibiofilm Effects of Sodium Hypochlorite against Staphylococcus aureus Isolates Derived from Patients with Atopic Dermatitis. Br. J. Dermatol. 2017, 177, 513–521. [Google Scholar] [CrossRef]
- Nakatsuji, T.; Chen, T.H.; Narala, S.; Chun, K.A.; Two, A.M.; Yun, T.; Shafiq, F.; Kotol, P.F.; Bouslimani, A.; Melnik, A.V.; et al. Antimicrobials from Human Skin Commensal Bacteria Protect against Staphylococcus aureus and Are Deficient in Atopic Dermatitis. Sci. Transl. Med. 2017, 9, eaah4680. [Google Scholar] [CrossRef]
- Myles, I.A.; Earland, N.J.; Anderson, E.D.; Moore, I.N.; Kieh, M.D.; Williams, K.W.; Saleem, A.; Fontecilla, N.M.; Welch, P.A.; Darnell, D.A.; et al. First-in-Human Topical Microbiome Transplantation with Roseomonas Mucosa for Atopic Dermatitis. JCI Insight 2018, 3, 120608. [Google Scholar] [CrossRef] [PubMed]
- Shimamori, Y.; Mitsunaka, S.; Yamashita, H.; Suzuki, T.; Kitao, T.; Kubori, T.; Nagai, H.; Takeda, S.; Ando, H. Staphylococcal Phage in Combination with Staphylococcus Epidermidis as a Potential Treatment for Staphylococcus aureus-Associated Atopic Dermatitis and Suppressor of Phage-Resistant Mutants. Viruses 2020, 13, 7. [Google Scholar] [CrossRef] [PubMed]
- Totté, J.; de Wit, J.; Pardo, L.; Schuren, F.; van Doorn, M.; Pasmans, S. Targeted Anti-Staphylococcal Therapy with Endolysins in Atopic Dermatitis and the Effect on Steroid Use, Disease Severity and the Microbiome: Study Protocol for a Randomized Controlled Trial (MAAS Trial). Trials 2017, 18, 404. [Google Scholar] [CrossRef]
- Dawgul, M.; Baranska-Rybak, W.; Piechowicz, L.; Bauer, M.; Neubauer, D.; Nowicki, R.; Kamysz, W. The Antistaphylococcal Activity of Citropin 1.1 and Temporin A against Planktonic Cells and Biofilms Formed by Isolates from Patients with Atopic Dermatitis: An Assessment of Their Potential to Induce Microbial Resistance Compared to Conventional Antimicrobials. Pharmaceuticals 2016, 9, 30. [Google Scholar] [CrossRef]
- Niemeyer-van der Kolk, T.; van der Wall, H.; Hogendoorn, G.K.; Rijneveld, R.; Luijten, S.; van Alewijk, D.C.J.G.; van den Munckhof, E.H.A.; de Kam, M.L.; Feiss, G.L.; Prens, E.P.; et al. Pharmacodynamic Effects of Topical Omiganan in Patients With Mild to Moderate Atopic Dermatitis in a Randomized, Placebo-Controlled, Phase II Trial. Clin. Transl. Sci. 2020, 13, 994–1003. [Google Scholar] [CrossRef]
- Olesen, C.M.; Ingham, A.C.; Thomsen, S.F.; Clausen, M.-L.; Andersen, P.S.; Edslev, S.M.; Yüksel, Y.T.; Guttman-Yassky, E.; Agner, T. Changes in Skin and Nasal Microbiome and Staphylococcal Species Following Treatment of Atopic Dermatitis with Dupilumab. Microorganisms 2021, 9, 1487. [Google Scholar] [CrossRef]
- Lossius, A.H.; Sundnes, O.; Ingham, A.C.; Edslev, S.M.; Bjørnholt, J.V.; Lilje, B.; Bradley, M.; Asad, S.; Haraldsen, G.; Skytt-Andersen, P.; et al. Shifts in the Skin Microbiota after UVB Treatment in Adult Atopic Dermatitis. Dermatology 2021, 1–12. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).