
An Emerging Role for Sigma-1 Receptors in the Treatment of Developmental and Epileptic Encephalopathies

 , ,
, ,
Abstract
1. Introduction
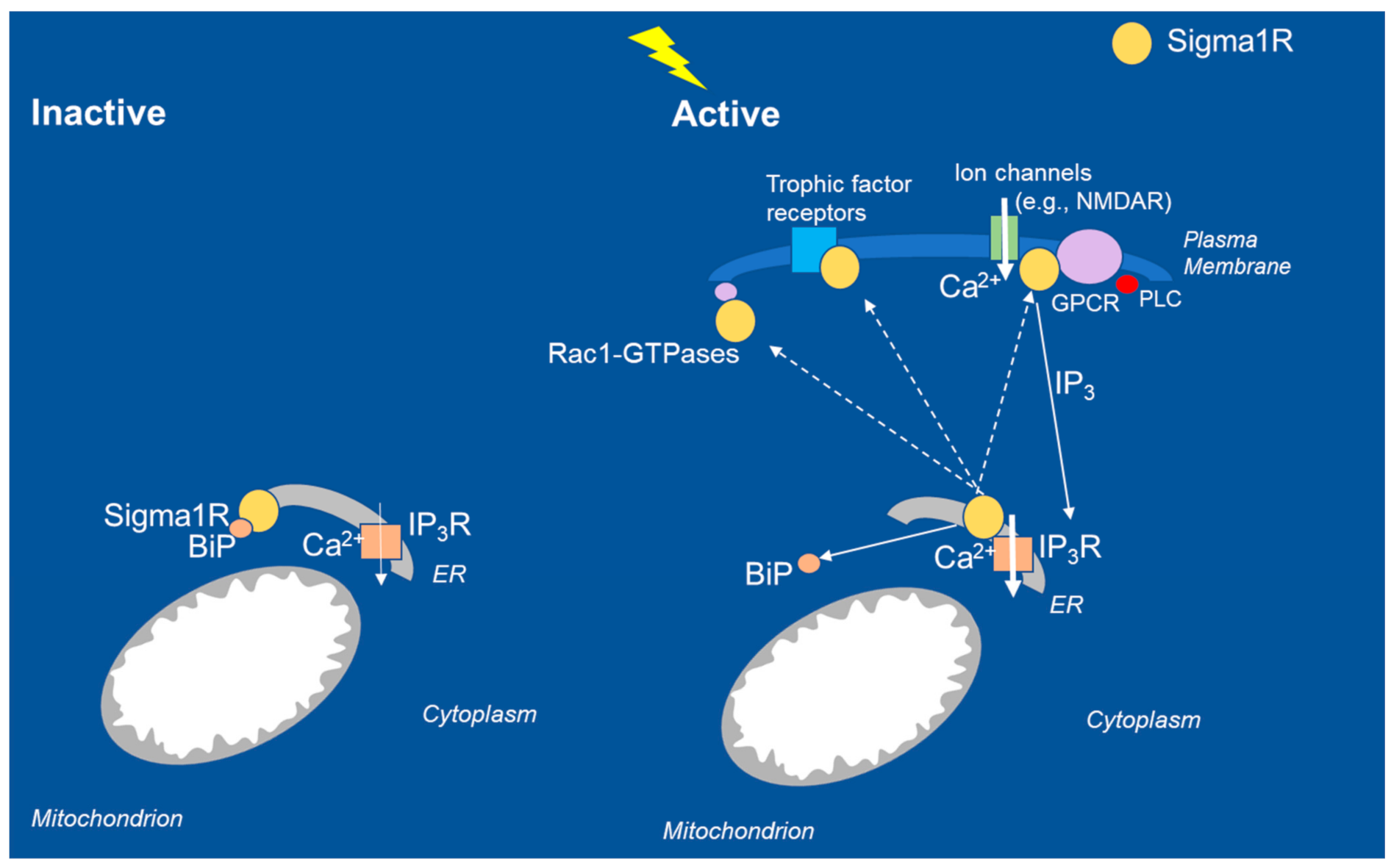
2. Function of Sigma1R
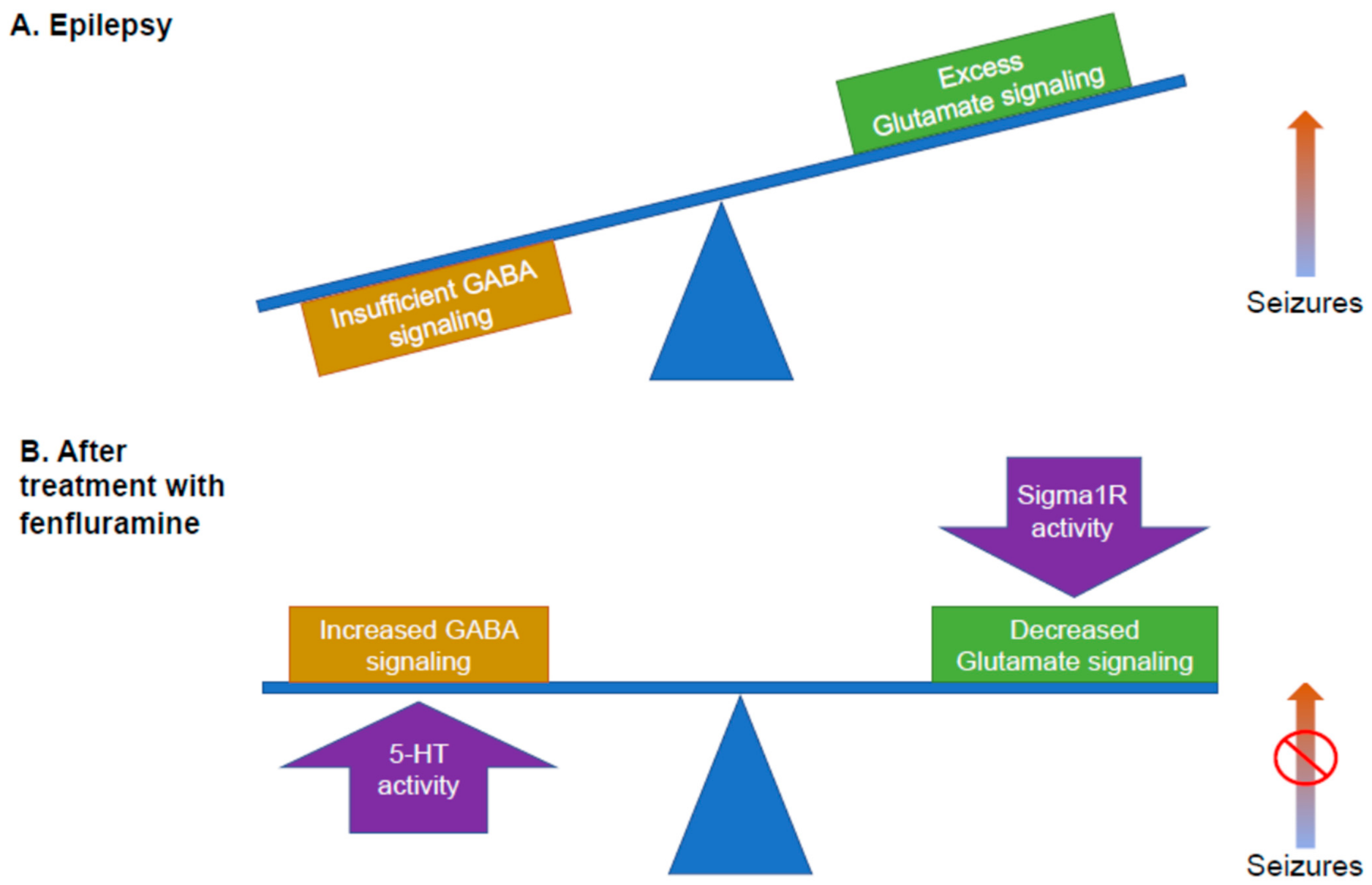
3. A Role for Sigma1R in Seizures and Epileptogenesis and GABAergic Signaling
4. Fenfluramine: From Serotonergic Uptake Inhibitor to Sigma1R Positive Modulator
4.1. In Vitro and Ex Vivo Binding and Functional Assays Demonstrate Fenfluramine Is a Positive Modulator of Sigma1R
4.2. In Vivo Functional Assay Demonstrates Fenfluramine Is a Positive Modulator of Sigma1R
4.3. Positive Modulators of Sigma1R Have Demonstrated Efficacy across a Spectrum of In Vivo Seizure Models
4.4. 5-HT Receptors and Sigma1R
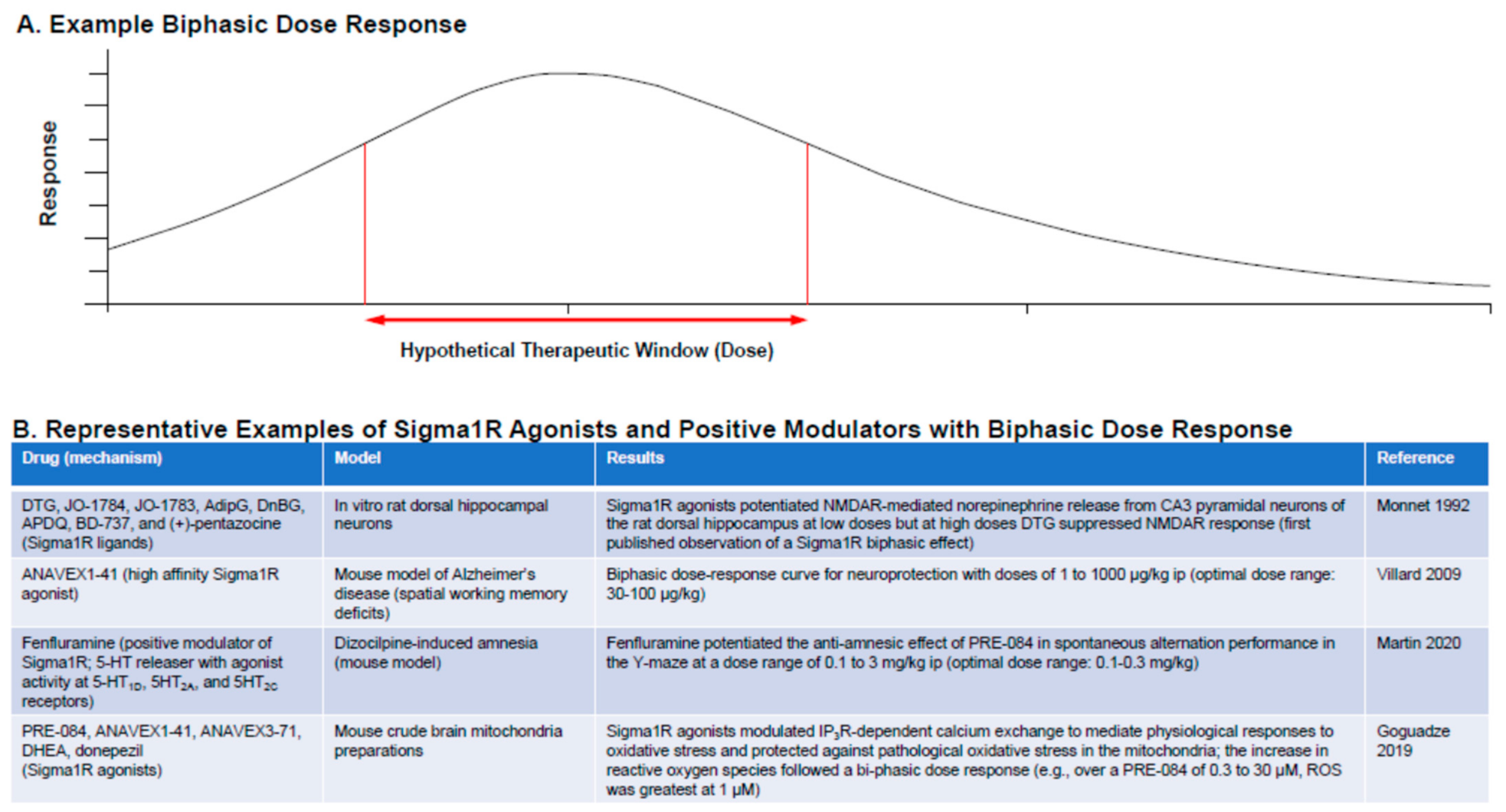
4.5. Sigma1R Ligands: Dose Considerations
5. Epileptogenesis, Neuroactive Steroids, and Sigma1R
6. A Role for Sigma1R in Non-Seizure Comorbidities of DEEs
6.1. Neuroplasticity and Connectivity: Sigma1Rs in Maintaining Dendritic Arborization
6.2. Neurodegeneration
6.3. Cognitive Deficits: Learning and Memory
6.4. Behavior: Hyperactivity, Social Interaction, and Autistic-like Behavior
6.5. Emotional Regulation: Anxiety-like Symptoms or Depression-like Symptoms
7. Limitations and Avenues for Future Research
7.1. The Sigma1R Interactome
7.2. The Distribution of Sigma1R across Cellular Compartments
7.3. Sigma1R and Epilepsy
7.4. The Role of Sigma1R in SUDEP
8. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Abbreviations
| 5-HT | serotonin (5-hydroxytryptamine) |
| ALS | amyotrophic lateral sclerosis |
| ASM | antiseizure medication |
| BDNF | brain-derived neurotrophic factor |
| BIC | bicuculline |
| BiP | immunoglobulin-binding protein |
| BRIEF | Behavior Rating Index of Executive Function |
| CA3 | cornu ammonis subfield 3 |
| CaM | calmodulin |
| CDD | CDKL5-associated disorder |
| CNS | central nervous system |
| DEE | developmental and epileptic encephalopathy |
| DHEA | dehydroepiandrosterone |
| DHEAS | dehydroepiandrosterone sulfate |
| EEG | electroencephalogram/electroencephalographic |
| ER | endoplasmic reticulum |
| FTLD | frontotemporal lobar dementia |
| GABA | γ-aminobutyric acid |
| GTC | generalized tonic-clonic |
| GTPase | hydrolase enzyme of guanosine triphosphate |
| HINT1 | histidine triad nucleotide binding protein 1 (HINT1) |
| i.c.v. | intracerebroventricular |
| iNOS | inducible nitric oxide synthase |
| IP3R | inositol triphosphate receptor |
| LFP | local field potential |
| MAM | mitochondria-associated membrane |
| mTOR | mechanistic target of rapamycin |
| NFκB | nuclear factor κB |
| NMDA | N-methyl-d-aspartate |
| NMDAR | N-methyl-d-aspartate receptor |
| OCD | obsessive-compulsive disorder |
| PNS | peripheral nervous system |
| PREGS | pregnenolone sulfate |
| PTZ | pentylenetetrazol |
| ROS | reactive oxygen species |
| Sigma1R | sigma-1 receptor |
| SNRI | serotonin-norepinephrine reuptake inhibitor |
| SSRI | selective serotonin reuptake inhibitor |
| SUDEP | sudden unexpected death in epilepsy |
| TRP | transient receptor potential |
| UTR | untranslated region |
References
- Dravet, C. The core Dravet syndrome phenotype. Epilepsia 2011, 52, 3–9. [Google Scholar] [CrossRef] [PubMed]
- Eschbach, K.; Scarbro, S.; Juarez-Colunga, E.; Allen, V.; Hsu, S.; Knupp, K. Growth and endocrine function in children with Dravet syndrome. Seizure 2017, 52, 117–122. [Google Scholar] [CrossRef] [PubMed]
- Gataullina, S.; Lemaire, E.; Wendling, F.; Kaminska, A.; Watrin, F.; Riquet, A.; Ville, D.; Moutard, M.L.; de Saint Martin, A.; Napuri, S.; et al. Epilepsy in young Tsc1(+/−) mice exhibits age-dependent expression that mimics that of human tuberous sclerosis complex. Epilepsia 2016, 57, 648–659. [Google Scholar] [CrossRef] [PubMed]
- Baumann, M.H.; Bulling, S.; Benaderet, T.S.; Saha, K.; Ayestas, M.A.; Partilla, J.S.; Ali, S.F.; Stockner, T.; Rothman, R.B.; Sandtner, W.; et al. Evidence for a role of transporter-mediated currents in the depletion of brain serotonin induced by serotonin transporter substrates. Neuropsychopharmacology 2014, 39, 1355–1365. [Google Scholar] [CrossRef]
- Fitzgerald, L.W.; Burn, T.C.; Brown, B.S.; Patterson, J.P.; Corjay, M.H.; Valentine, P.A.; Sun, J.H.; Link, J.R.; Abbaszade, I.; Hollis, J.M.; et al. Possible role of valvular serotonin 5-HT2B receptors in the cardiopathy associated with fenfluramine. Mol. Pharmacol. 2000, 57, 75–81. [Google Scholar]
- Porter, R.H.; Benwell, K.R.; Lamb, H.; Malcolm, C.S.; Allen, N.H.; Revell, D.F.; Adams, D.R.; Sheardown, M.J. Functional characterization of agonists at recombinant human 5-HT2A, 5-HT2B and 5-HT2C receptors in CHO-K1 cells. Br. J. Pharmacol. 1999, 128, 13–20. [Google Scholar] [CrossRef]
- Knupp, K.G.; Scheffer, I.E.; Ceulemans, B.; Sullivan, J.; Nickels, K.C.; Miller, I.; Lagae, L.; Guerrini, R.; Zuber, S.M.; Nabbout, R.; et al. Efficacy and safety of FINTEPLA (fenfluramine) for the treatment of seizures associated with Lennox-Gastaut syndrome: A randomized, double-blind, placebo-controlled clinical trial. In Proceedings of the Virtual American Epilepsy Society (AES) Annual Meeting, Online, 4–8 December 2020. [Google Scholar]
- Lagae, L.; Sullivan, J.; Knupp, K.; Laux, L.; Polster, T.; Nikanorova, M.; Devinsky, O.; Cross, J.H.; Guerrini, R.; Talwar, D.; et al. Fenfluramine hydrochloride for the treatment of seizures in Dravet syndrome: A randomised, double-blind, placebo-controlled trial. Lancet 2020, 394, 2243–2254. [Google Scholar] [CrossRef]
- Nabbout, R.; Mistry, A.; Zuberi, S.; Villeneuve, N.; Gil-Nagel, A.; Sanchez-Carpintero, R.; Stephani, U.; Laux, L.; Wirrell, E.; Knupp, K.; et al. Fenfluramine for treatment-resistant seizures in patients with Dravet syndrome receiving stiripentol-inclusive regimens: A randomized clinical trial. JAMA Neurol. 2020, 77, 300–308. [Google Scholar] [CrossRef]
- Sullivan, J.; Lagae, L.; Cross, J.H.; Devinsky, O.; Guerrini, R.; Knupp, K.G.; Laux, L.; Miller, I.; Nikanorova, M.; Polster, T.; et al. Fenfluramine (FINTEPLA) in Dravet syndrome: Results of a third randomized, placebo-controlled clinical trial (Study 3). In Proceedings of the American Epilepsy Society 2020, Online, 4–8 December 2020. [Google Scholar]
- Sullivan, J.; Scheffer, I.E.; Lagae, L.; Nabbout, R.; Pringsheim, M.; Talwar, D.; Polster, T.; Galer, B.; Lock, M.; Agarwal, A.; et al. Fenfluramine HCl (Fintepla®) provides long-term clinically meaningful reduction in seizure frequency: Analysis of an ongoing open-label extension study. Epilepsia 2020, 61, 2396–2404. [Google Scholar] [CrossRef]
- Bishop, K.I.; Isquith, P.K.; Gioia, G.A.; Gammaitoni, A.R.; Farfel, G.; Galer, B.S.; Nabbout, R.; Wirrell, E.C.; Polster, T.; Sullivan, J. Improved everyday executive functioning following profound reduction in seizure frequency with fenfluramine: Analysis from a phase 3 long-term extension study in children/young adults with Dravet syndrome. Epilepsy Behav. 2021, 121, 108024. [Google Scholar] [CrossRef]
- Cross, J.H.; Galer, B.S.; Gil-Nagel, A.; Devinsky, O.; Ceulemans, B.; Lagae, L.; Schoonjans, A.S.; Donner, E.; Wirrell, E.; Kothare, S.; et al. Impact of Fintepla (fenfluramine) on the expected incidence rate of SUDEP in patients with Dravet syndrome. In Proceedings of the Academy of Managed Care Pharmacy (AMCP), Online, 12–16 April 2021. [Google Scholar]
- Bishop, K.I.; Isquith, P.K.; Gioia, G.A.; Knupp, K.G.; Scheffer, I.E.; Sullivan, J.; Nabbout, R.; Farfel, G.; Galer, B.S.; Shore, S.; et al. FINTEPLA (fenfluramine) treatment improves everyday executive functioning in patients with Lennox-Gastaut syndrome: Analysis from a phase 3 clinical trial. In Proceedings of the American Academy of Neurology Annual Meeting, Online, 17–22 April 2021. [Google Scholar]
- Devinsky, O.; King, L.; Schwartz, D.; Conway, E.; Price, D. Effect of fenfluramine on convulsive seizures in CDKL5 deficiency disorder. Epilepsia 2021, 62, e98–e102. [Google Scholar] [CrossRef]
- Thiele, E.A.; Bruno, P.L.; Vu, U.; Geenen, G.; Doshi, S.P.; Patel, S.; Sourbron, J. Safety and efficacy of add-on ZX008 (fenfluramine HCl oral solution) in sunflower syndrome: An open-label pilot study of 5 patients. In Proceedings of the American Academy of Neurology Annual Meeting, Online, 25 April–1 May 2020. [Google Scholar]
- Geenen, K.R.; Doshi, S.P.; Patel, S.; Sourbron, J.; Falk, A.; Morgan, A.; Vu, U.; Bruno, P.L.; Thiele, E.A. Fenfluramine for seizures associated with Sunflower syndrome. Dev. Med. Child. Neurol. 2021. [Google Scholar] [CrossRef]
- Bagdy, G.; Kecskemeti, V.; Riba, P.; Jakus, R. Serotonin and epilepsy. J. Neurochem. 2007, 100, 857–873. [Google Scholar] [CrossRef]
- Gharedaghi, M.H.; Seyedabadi, M.; Ghia, J.E.; Dehpour, A.R.; Rahimian, R. The role of different serotonin receptor subtypes in seizure susceptibility. Exp. Brain Res. 2014, 232, 347–367. [Google Scholar] [CrossRef]
- Favale, E.; Audenino, D.; Cocito, L.; Albano, C. The anticonvulsant effect of citalopram as an indirect evidence of serotonergic impairment in human epileptogenesis. Seizure 2003, 12, 316–318. [Google Scholar] [CrossRef]
- Tolete, P.; Knupp, K.; Karlovich, M.; De Carlo, E.; Bluvstein, J.; Conway, E.; Friedman, D.; Dugan, P.; Devinsky, O. Lorcaserin therapy for severe epilepsy of childhood onset: A case series. Neurology 2018, 91, 837–839. [Google Scholar] [CrossRef]
- Martin, P.; de Witte, P.A.M.; Maurice, T.; Gammaitoni, A.; Farfel, G.; Galer, B. Fenfluramine acts as a positive modulator of sigma-1 receptors. Epilepsy Behav. 2020, 105, 106989. [Google Scholar] [CrossRef]
- Sourbron, J.; Smolders, I.; de Witte, P.; Lagae, L. Pharmacological analysis of the anti-epileptic mechanisms of fenfluramine in scn1a mutant zebrafish. Front. Pharmacol. 2017, 8, 191. [Google Scholar] [CrossRef]
- Rodriguez-Munoz, M.; Sanchez-Blazquez, P.; Garzon, J. Fenfluramine diminishes NMDA receptor-mediated seizures via its mixed activity at serotonin 5HT2A and type 1 sigma receptors. Oncotarget 2018, 9, 23373–23389. [Google Scholar] [CrossRef]
- Faingold, C.L.; Randall, M.; Zeng, C.; Peng, S.; Long, X.; Feng, H.-J. Serotonergic agents act on 5-HT3 receptors in the brain to block seizure-induced respiratory arrest in the DBA/1 mouse model of SUDEP. Epilepsy Behav. 2016, 64, 166–170. [Google Scholar] [CrossRef]
- Faingold, C.L.; Tupal, S. The action of fenfluramine to prevent seizure-induced death in the DBA/1 mouse SUDEP model is selectively blocked by an antagonist or enhanced by an agonist for the serotonin 5-HT4 receptor. In Proceedings of the American Epilepsy Society Annual Meeting, Baltimore, MD, USA, 6–10 December 2019. Abstract 3.292. [Google Scholar]
- Cagnotto, A.; Bastone, A.; Mennini, T. [3H](+)-pentazocine binding to rat brain δ1 receptors. Eur. J. Pharmacol. 1994, 266, 131–138. [Google Scholar] [CrossRef]
- Tiraboschi, E.; Martina, S.; van der Ent, W.; Grzyb, K.; Gawel, K.; Cordero-Maldonado, M.L.; Poovathingal, S.K.; Heintz, S.; Satheesh, S.V.; Brattespe, J.; et al. New insights into the early mechanisms of epileptogenesis in a zebrafish model of Dravet syndrome. Epilepsia 2020, 61, 549–560. [Google Scholar] [CrossRef] [PubMed]
- Yu, F.H.; Mantegazza, M.; Westenbroek, R.E.; Robbins, C.A.; Kalume, F.; Burton, K.A.; Spain, W.J.; McKnight, G.S.; Scheuer, T.; Catterall, W.A. Reduced sodium current in GABAergic interneurons in a mouse model of severe myoclonic epilepsy in infancy. Nat. Neurosci. 2006, 9, 1142–1149. [Google Scholar] [CrossRef] [PubMed]
- Sourbron, J.; Schneider, H.; Kecskés, A.; Liu, Y.; Buening, E.M.; Lagae, L.; Smolders, I.; de Witte, P.A.M. Serotonergic modulation as effective treatment for dravet syndrome in a zebrafish mutant model. ACS Chem. Neurosci. 2016, 7, 588–598. [Google Scholar] [CrossRef]
- Liu, S.; Jin, Z.; Zhang, Y.; Rong, S.; He, W.; Sun, K.; Wan, D.; Huo, J.; Xiao, L.; Li, X.; et al. The glucagon-like peptide-1 analogue liraglutide reduces seizures susceptibility, cognition dysfunction and neuronal apoptosis in a mouse model of dravet syndrome. Front. Pharmacol. 2020, 11, 136. [Google Scholar] [CrossRef]
- Stein, R.E.; Kaplan, J.S.; Li, J.; Catterall, W.A. Hippocampal deletion of NaV1.1 channels in mice causes thermal seizures and cognitive deficit characteristic of Dravet syndrome. Proc. Natl. Acad. Sci. USA 2019, 116, 16571–16576. [Google Scholar] [CrossRef]
- Han, S.; Tai, C.; Westenbroek, R.E.; Yu, F.H.; Cheah, C.S.; Potter, G.B.; Rubenstein, J.L.; Scheuer, T.; de la Iglesia, H.O.; Catterall, W.A. Autistic behavior in Scn1a+/- mice and rescue by enhanced GABAergic transmission. Nature 2012, 489, 385–390. [Google Scholar] [CrossRef]
- Kaplan, J.S.; Stella, N.; Catterall, W.A.; Westenbroek, R.E. Cannabidiol attenuates seizures and social deficits in a mouse model of Dravet syndrome. Proc. Natl. Acad. Sci. USA 2017, 114, 11229–11234. [Google Scholar] [CrossRef]
- Kalume, F.; Yu, F.H.; Westenbroek, R.E.; Scheuer, T.; Catterall, W.A. Reduced sodium current in Purkinje neurons from Nav1.1 mutant mice: Implications for ataxia in severe myoclonic epilepsy in infancy. J. Neurosci. 2007, 27, 11065–11074. [Google Scholar] [CrossRef]
- Bahceci, D.; Anderson, L.L.; Occelli Hanbury Brown, C.V.; Zhou, C.; Arnold, J.C. Adolescent behavioral abnormalities in a Scn1a(+/−) mouse model of Dravet syndrome. Epilepsy Behav. 2020, 103, 106842. [Google Scholar] [CrossRef]
- Kalume, F.; Oakley, J.C.; Westenbroek, R.E.; Gile, J.; de la Iglesia, H.O.; Scheuer, T.; Catterall, W.A. Sleep impairment and reduced interneuron excitability in a mouse model of Dravet syndrome. Neurobiol. Dis. 2015, 77, 141–154. [Google Scholar] [CrossRef]
- Kalume, F.; Westenbroek, R.E.; Cheah, C.S.; Yu, F.H.; Oakley, J.C.; Scheuer, T.; Catterall, W.A. Sudden unexpected death in a mouse model of Dravet syndrome. J. Clin. Investig. 2013, 123, 1798–1808. [Google Scholar] [CrossRef]
- Vavers, E.; Zvejniece, B.; Stelfa, G.; Svalbe, B.; Vilks, K.; Kupats, E.; Mezapuke, R.; Lauberte, L.; Dambrova, M.; Zvejniece, L. Genetic inactivation of the sigma-1 chaperone protein results in decreased expression of the R2 subunit of the GABA-B receptor and increased susceptibility to seizures. Neurobiol. Dis. 2021, 150, 105244. [Google Scholar] [CrossRef]
- Vavers, E.; Svalbe, B.; Lauberte, L.; Stonans, I.; Misane, I.; Dambrova, M.; Zvejniece, L. The activity of selective sigma-1 receptor ligands in seizure models in vivo. Behav. Brain Res. 2017, 328, 13–18. [Google Scholar] [CrossRef]
- Rebowe, N.; Sprouse, J.; Brunner, D.; Kabitzke, P.; Velisek, L.; Benson, M.D.; Veliskova, J.; Missling, C. ANAVEX 2-73 as a potential treatment for Rett syndome and other pediatric or infantile disorders with seizure pathology. In Proceedings of the Rett Syndrome Symposium, Itasca, IL, USA, 22–24 June 2016. [Google Scholar]
- Rebowe, N.; Missling, C.U. Novel anti-seizure compound ANAVEX 2-73, a Sigma-1 receptor agonist, in multiple seizure models. In Proceedings of theAmerican Epilepsy Society Annual Meeting, Philadelphia, PA, USA, 4–8 December 2015. [Google Scholar]
- Guo, L.; Chen, Y.; Zhao, R.; Wang, G.; Friedman, E.; Zhang, A.; Zhen, X. Allosteric modulation of sigma-1 receptors elicits anti-seizure activities. Br. J. Pharmacol. 2015, 172, 4052–4065. [Google Scholar] [CrossRef]
- Chaki, S.; Okuyama, S.; Ogawa, S.; Tanaka, M.; Muramatsu, M.; Nakazato, A.; Tomisawa, K. Solubilization and characterization of binding sites for [3H]NE-100, a novel and potent sigma 1 ligand, from guinea pig brain. Life Sci. 1996, 59, 1331–1340. [Google Scholar] [CrossRef]
- Cobos, E.J.; Baeyens, J.M.; Del Pozo, E. Phenytoin differentially modulates the affinity of agonist and antagonist ligands for sigma 1 receptors of guinea pig brain. Synapse 2005, 55, 192–195. [Google Scholar] [CrossRef]
- DeHaven-Hudkins, D.L.; Ford-Rice, F.Y.; Allen, J.T.; Hudkins, R.L. Allosteric modulation of ligand binding to [3H](+)pentazocine-defined sigma recognition sites by phenytoin. Life Sci. 1993, 53, 41–48. [Google Scholar] [CrossRef]
- Jones, G.L.; Amato, R.J.; Wimbish, G.H.; Peyton, G.A. Comparison of anticonvulsant potencies of cyheptamide, carbamazepine, and phenytoin. J. Pharm. Sci. 1981, 70, 618–620. [Google Scholar] [CrossRef]
- Edmonds, H.L., Jr.; Jiang, Y.D.; Zhang, P.Y.; Shank, R.P. Anticonvulsant activity of topiramate and phenytoin in a rat model of ischemia-induced epilepsy. Life Sci. 1996, 59, PL127–PL131. [Google Scholar] [CrossRef]
- Hayashi, T.; Tsai, S.Y.; Mori, T.; Fujimoto, M.; Su, T.P. Targeting ligand-operated chaperone sigma-1 receptors in the treatment of neuropsychiatric disorders. Expert Opin. Ther. Targets 2011, 15, 557–577. [Google Scholar] [CrossRef]
- Rousseaux, C.G.; Greene, S.F. Sigma receptors [sRs]: Biology in normal and diseased states. J. Recept. Signal. Transduct. Res. 2016, 36, 327–388. [Google Scholar]
- Vavers, E.; Zvejniece, L.; Maurice, T.; Dambrova, M. Allosteric modulators of sigma-1 receptor: A review. Front. Pharmacol. 2019, 10, 223. [Google Scholar] [CrossRef]
- Nabbout, R.; Chemaly, N.; Chipaux, M.; Barcia, G.; Bouis, C.; Dubouch, C.; Leunen, D.; Jambaqué, I.; Dulac, O.; Dellatolas, G.; et al. Encephalopathy in children with Dravet syndrome is not a pure consequence of epilepsy. Orphanet J. Rare Dis. 2013, 8, 176. [Google Scholar] [CrossRef]
- Cross, J.H.; Lagae, L. The concept of disease modification. Eur. J. Paediatr. Neurol. 2020, 24, 43–46. [Google Scholar] [CrossRef]
- Kaufmann, W.E.; Sprouse, J.; Rebowe, N.; Hanania, T.; Klamer, D.; Missling, C.U. ANAVEX(R)2-73 (blarcamesine), a Sigma-1 receptor agonist, ameliorates neurologic impairments in a mouse model of Rett syndrome. Pharmacol. Biochem. Behav. 2019, 187, 172796. [Google Scholar] [CrossRef]
- Najjar, A.; Najjar, A.; Karaman, R. Newly developed prodrugs and prodrugs in development; an insight of the recent years. Molecules 2020, 25, 884. [Google Scholar] [CrossRef]
- Ye, N.; Qin, W.; Tian, S.; Xu, Q.; Wold, E.A.; Zhou, J.; Zhen, X.C. Small molecules selectively targeting sigma-1 receptor for the treatment of neurological diseases. J. Med. Chem. 2020, 63, 15187–15217. [Google Scholar] [CrossRef]
- Hampel, H.; Afshar, M.; Etcheto, A.; Goodsaid, F.; Missling, C. Longitudinal 148-week extension study for ANAVEX®2-73 phase 2a Alzheimer’s disease demonstrates maintained activities of daily living score (ADCSADL) and reduced cognitive decline (MMSE) for patient cohort on higher drug concentration and confirms role of patient selection biomarkers. In Proceedings of the Clinical Trials on Alzheimer’s Disease (CTAD) Conference, San Diego, CA, USA, 4–7 December 2019. [Google Scholar]
- Martin, W.R.; Eades, C.G.; Thompson, J.A.; Huppler, R.E.; Gilbert, P.E. The effects of morphine- and nalorphine-like drugs in the nondependent and morphine-dependent chronic spinal dog. J. Pharmacol. Exp. Ther. 1976, 197, 517–532. [Google Scholar]
- Hanner, M.; Moebius, F.F.; Flandorfer, A.; Knaus, H.G.; Striessnig, J.; Kempner, E.; Glossmann, H. Purification, molecular cloning, and expression of the mammalian sigma1-binding site. Proc. Natl. Acad. Sci. USA 1996, 93, 8072–8077. [Google Scholar] [CrossRef]
- Voronin, M.V.; Vakhitova, Y.V.; Seredenin, S.B. Chaperone Sigma1R and antidepressant effect. Int. J. Mol. Sci. 2020, 21, 7088. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, M.; Kamata, E.; Hirose, A.; Takahashi, M.; Morita, T.; Ema, M. In silico assessment of chemical mutagenesis in comparison with results of Salmonella microsome assay on 909 chemicals. Mutat. Res. 2005, 588, 129–135. [Google Scholar] [CrossRef] [PubMed]
- Hayashi, T.; Su, T.P. Sigma-1 receptor chaperones at the ER-mitochondrion interface regulate Ca2+ signaling and cell survival. Cell 2007, 131, 596–610. [Google Scholar] [CrossRef] [PubMed]
- Omi, T.; Tanimukai, H.; Kanayama, D.; Sakagami, Y.; Tagami, S.; Okochi, M.; Morihara, T.; Sato, M.; Yanagida, K.; Kitasyoji, A.; et al. Fluvoxamine alleviates ER stress via induction of Sigma-1 receptor. Cell Death Dis. 2014, 5, e1332. [Google Scholar] [CrossRef]
- Rosen, D.A.; Seki, S.M.; Fernández-Castañeda, A.; Beiter, R.M.; Eccles, J.D.; Woodfolk, J.A.; Gaultier, A. Modulation of the Sigma-1 receptor-IRE1 pathway is beneficial in preclinical models of inflammation and sepsis. Sci. Transl. Med. 2019, 11, 478. [Google Scholar] [CrossRef]
- Cortes-Montero, E.; Sanchez-Blazquez, P.; Onetti, Y.; Merlos, M.; Garzon, J. Ligands exert biased activity to regulate Sigma 1 receptor interactions with cationic TRPA1, TRPV1, and TRPM8 channels. Front. Pharmacol. 2019, 10, 634. [Google Scholar] [CrossRef]
- Tsai, S.Y.A.; Su, T.P. Sigma-1 receptors fine-tune the neuronal networks. In Advances in Experimental Medicine and Biology. Sigma Receptors: Their Role in Disease and as Therapeutic Targets; Smith, S.B., Su, T.P., Eds.; Springer International Publishing AG: Cham, Switzerland, 2017; Volume 964, pp. 79–83. [Google Scholar]
- Hayashi, T.; Su, T. The sigma receptor: Evolution of the concept in neuropsychopharmacology. Curr. Neuropharmacol. 2005, 3, 267–280. [Google Scholar] [CrossRef]
- Brunklaus, A.; Zuberi, S.M. Dravet syndrome—From epileptic encephalopathy to channelopathy. Epilepsia 2014, 55, 979–984. [Google Scholar] [CrossRef]
- Ryskamp, D.A.; Korban, S.; Zhemkov, V.; Kraskovskaya, N.; Bezprozvanny, I. Neuronal sigma-1 receptors: Signaling functions and protective roles in neurodegenerative diseases. Front. Neurosci. 2019, 13, 862. [Google Scholar] [CrossRef]
- Watanabe, S.; Ilieva, H.; Tamada, H.; Nomura, H.; Komine, O.; Endo, F.; Jin, S.; Mancias, P.; Kiyama, H.; Yamanaka, K. Mitochondria-associated membrane collapse is a common pathomechanism in SIGMAR1- and SOD1-linked ALS. EMBO Mol. Med. 2016, 8, 1421–1437. [Google Scholar] [CrossRef]
- Al-Saif, A.; Al-Mohanna, F.; Bohlega, S. A mutation in sigma-1 receptor causes juvenile amyotrophic lateral sclerosis. Ann. Neurol. 2011, 70, 913–919. [Google Scholar] [CrossRef]
- Ullah, M.I.; Ahmad, A.; Raza, S.I.; Amar, A.; Ali, A.; Bhatti, A.; John, P.; Mohyuddin, A.; Ahmad, W.; Hassan, M.J. In silico analysis of SIGMAR1 variant (rs4879809) segregating in a consanguineous Pakistani family showing amyotrophic lateral sclerosis without frontotemporal lobar dementia. Neurogenetics 2015, 16, 299–306. [Google Scholar] [CrossRef]
- Li, X.; Hu, Z.; Liu, L.; Xie, Y.; Zhan, Y.; Zi, X.; Wang, J.; Wu, L.; Xia, K.; Tang, B.; et al. A SIGMAR1 splice-site mutation causes distal hereditary motor neuropathy. Neurology 2015, 84, 2430–2437. [Google Scholar] [CrossRef]
- Horga, A.; Tomaselli, P.J.; Gonzalez, M.A.; Laurà, M.; Muntoni, F.; Manzur, A.Y.; Hanna, M.G.; Blake, J.C.; Houlden, H.; Züchner, S.; et al. SIGMAR1 mutation associated with autosomal recessive Silver-like syndrome. Neurology 2016, 87, 1607–1612. [Google Scholar] [CrossRef]
- Luty, A.A.; Kwok, J.B.; Dobson-Stone, C.; Loy, C.T.; Coupland, K.G.; Karlstrom, H.; Sobow, T.; Tchorzewska, J.; Maruszak, A.; Barcikowska, M.; et al. Sigma nonopioid intracellular receptor 1 mutations cause frontotemporal lobar degeneration-motor neuron disease. Ann. Neurol. 2010, 68, 639–649. [Google Scholar] [CrossRef]
- Claes, L.; Del-Favero, J.; Ceulemans, B.; Lagae, L.; Van Broeckhoven, C.; De Jonghe, P. De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am. J. Hum. Genet. 2001, 68, 1327–1332. [Google Scholar] [CrossRef]
- Ceulemans, B.; Schoonjans, A.S.; Marchau, F.; Paelinck, B.P.; Lagae, L. Five-year extended follow-up status of 10 patients with Dravet syndrome treated with fenfluramine. Epilepsia 2016, 57, e129–e134. [Google Scholar] [CrossRef]
- Arzimanoglou, A. Dravet syndrome: From electroclinical characteristics to molecular biology. Epilepsia 2009, 50, 3–9. [Google Scholar] [CrossRef]
- Lagae, L.; Schoonjans, A.S.; Gammaitoni, A.R.; Galer, B.S.; Ceulemans, B. A pilot, open-label study of the effectiveness and tolerability of low-dose ZX008 (fenfluramine HCl) in Lennox-Gastaut syndrome. Epilepsia 2018, 59, 1881–1888. [Google Scholar] [CrossRef]
- Silenieks, L.B.; Carroll, N.K.; Van Niekerk, A.; Van Niekerk, E.; Taylor, C.; Upton, N.; Higgins, G.A. Evaluation of selective 5-HT2C agonists in acute seizure models. ACS Chem. Neurosci. 2019, 10, 3284–3295. [Google Scholar] [CrossRef]
- Ribot, R.; Ouyang, B.; Kanner, A.M. The impact of antidepressants on seizure frequency and depressive and anxiety disorders of patients with epilepsy: Is it worth investigating? Epilepsy Behav. 2017, 70, 5–9. [Google Scholar] [CrossRef]
- Tunnicliff, G. Basis of the antiseizure action of phenytoin. Gen. Pharmacol. 1996, 27, 1091–1097. [Google Scholar] [CrossRef]
- Maurice, T. Protection by sigma-1 receptor agonists is synergic with donepezil, but not with memantine, in a mouse model of amyloid-induced memory impairments. Behav. Brain Res. 2016, 296, 270–278. [Google Scholar] [CrossRef]
- Maurice, T.; Hiramatsu, M.; Itoh, J.; Kameyama, T.; Hasegawa, T.; Nabeshima, T. Behavioral evidence for a modulating role of s ligands in memory processes. I. Attenuation of dizocilpine (MK-801)-induced amnesia. Brain Res. 1994, 647, 44–56. [Google Scholar] [CrossRef]
- Maurice, T.; Phan, V.L.; Urani, A.; Guillemain, I. Differential involvement of the sigma1 (s1) receptor in the anti-amnesic effect of neuroactive steroids, as demonstrated using an in vivo antisense strategy in the mouse. Br. J. Pharmacol. 2001, 134, 1731–1741. [Google Scholar] [CrossRef]
- Maurice, T.; Su, T.P.; Privat, A. Sigma1 (s 1) receptor agonists and neurosteroids attenuate B25-35-amyloid peptide-induced amnesia in mice through a common mechanism. Neuroscience 1998, 83, 413–428. [Google Scholar] [CrossRef]
- Meunier, J.; Ieni, J.; Maurice, T. The anti-amnesic and neuroprotective effects of donepezil against amyloid b25-35 peptide-induced toxicity in mice involve an interaction with the s1 receptor. Br. J. Pharmacol. 2006, 149, 998–1012. [Google Scholar] [CrossRef]
- Sourbron, J.; Partoens, M.; Scheldeman, C.; Zhang, Y.; Lagae, L.; de Witte, P. Drug repurposing for Dravet syndrome in scn1Lab(-/-) mutant zebrafish. Epilepsia 2019, 60, e8–e13. [Google Scholar] [CrossRef]
- Vicente-Sánchez, A.; Sánchez-Blázquez, P.; Rodriguez-Muñoz, M.; Garzón, J. HINT1 protein cooperates with cannabinoid 1 receptor to negatively regulate glutamate NMDA receptor activity. Mol. Brain 2013, 6, 42. [Google Scholar] [CrossRef]
- Rodriguez-Munoz, M.; Onetti, Y.; Cortes-Montero, E.; Garzon, J.; Sanchez-Blazquez, P. Cannabidiol enhances morphine antinociception, diminishes NMDA-mediated seizures and reduces stroke damage via the sigma 1 receptor. Mol. Brain 2018, 11, 51. [Google Scholar] [CrossRef]
- Honda, M.; Uchida, K.; Tanabe, M.; Ono, H. Fluvoxamine, a selective serotonin reuptake inhibitor, exerts its antiallodynic effects on neuropathic pain in mice via 5-HT2A/2C receptors. Neuropharmacology 2006, 51, 866–872. [Google Scholar] [CrossRef] [PubMed]
- Ago, Y.; Hasebe, S.; Hiramatsu, N.; Hashimoto, H.; Takuma, K.; Matsuda, T. Psychopharmacology of combined activation of the serotonin1A and s1 receptors. Eur. J. Pharmacol. 2017, 809, 172–177. [Google Scholar] [CrossRef] [PubMed]
- Ago, Y.; Yano, K.; Hiramatsu, N.; Takuma, K.; Matsuda, T. Fluvoxamine enhances prefrontal dopaminergic neurotransmission in adrenalectomized/castrated mice via both 5-HT reuptake inhibition and s1 receptor activation. Psychopharmacology 2011, 217, 377–386. [Google Scholar] [CrossRef] [PubMed]
- Alhaj, M.W.; Zaitone, S.A.; Moustafa, Y.M. Fluvoxamine alleviates seizure activity and downregulates hippocampal GAP-43 expression in pentylenetetrazole-kindled mice: Role of 5-HT3 receptors. Behav. Pharmacol. 2015, 26, 369–382. [Google Scholar] [CrossRef]
- Jamali, H.; Heydari, A. Effect of dextromethorphan/quinidine on pentylenetetrazole- induced clonic and tonic seizure thresholds in mice. Neurosci. Lett. 2020, 729, 134988. [Google Scholar] [CrossRef]
- Monnet, F.P.; Debonnel, G.; de Montigny, C. In vivo electrophysiological evidence for a selective modulation of N-methyl-D-aspartate-induced neuronal activation in rat CA3 dorsal hippocampus by sigma ligands. J. Pharmacol. Exp. Ther. 1992, 261, 123–130. [Google Scholar]
- Maurice, T. Bi-phasic dose response in the preclinical and clinical developments of sigma-1 receptor ligands for the treatment of neurodegenerative disorders. Expert Opin. Drug Discov. 2021, 16, 373–389. [Google Scholar] [CrossRef]
- Villard, V.; Espallergues, J.; Keller, E.; Alkam, T.; Nitta, A.; Yamada, K.; Nabeshima, T.; Vamvakides, A.; Maurice, T. Antiamnesic and neuroprotective effects of the aminotetrahydrofuran derivative ANAVEX1-41 against amyloid beta(25-35)-induced toxicity in mice. Neuropsychopharmacology 2009, 34, 1552–1566. [Google Scholar] [CrossRef]
- Goguadze, N.; Zhuravliova, E.; Morin, D.; Mikeladze, D.; Maurice, T. Sigma-1 receptor agonists induce oxidative stress in mitochondria and enhance complex I activity in physiological condition but protect against pathological oxidative stress. Neurotox. Res. 2019, 35, 1–18. [Google Scholar] [CrossRef]
- Longone, P.; di Michele, F.; D’Agati, E.; Romeo, E.; Pasini, A.; Rupprecht, R. Neurosteroids as neuromodulators in the treatment of anxiety disorders. Front. Endocrinol. 2011, 2, 55. [Google Scholar] [CrossRef]
- Monnet, F.P.; Maurice, T. The sigma1 protein as a target for the non-genomic effects of neuro(active)steroids: Molecular, physiological, and behavioral aspects. J. Pharmacol. Sci. 2006, 100, 93–118. [Google Scholar] [CrossRef]
- Guo, X.; Williams, A.; Miller, D.; Milad, M.P. Evaluation of a bundled checklist in decreasing hysterectomy surgical site infections. Am. J. Obstet. Gynecol. 2018, 218, S921. [Google Scholar] [CrossRef]
- Hawkins, N.A.; Anderson, L.L.; Gertler, T.S.; Laux, L.; George, A.L., Jr.; Kearney, J.A. Screening of conventional anticonvulsants in a genetic mouse model of epilepsy. Ann. Clin. Transl. Neurol. 2017, 4, 326–339. [Google Scholar] [CrossRef]
- Hawkins, N.A.; Lewis, M.; Hammond, R.S.; Doherty, J.J.; Kearney, J.A. The synthetic neuroactive steroid SGE-516 reduces seizure burden and improves survival in a Dravet syndrome mouse model. Sci. Rep. 2017, 7, 5327. [Google Scholar]
- Maurice, T.; Gammaitoni, A.R.; Galer, B.S.; Reeder, T.; Martin, P. Neurosteroids modulate the in vivo responses to fenfluramine at the sigma-1 receptor in mice. In Proceedings of the Society for Neuroscience, Chicago, IL, USA, 19–23 October 2019. [Google Scholar]
- Tsai, S.Y.; Hayashi, T.; Harvey, B.K.; Wang, Y.; Wu, W.W.; Shen, R.F.; Zhang, Y.; Becker, K.G.; Hoffer, B.J.; Su, T.P. Sigma-1 receptors regulate hippocampal dendritic spine formation via a free radical-sensitive mechanism involving Rac1xGTP pathway. Proc. Natl. Acad. Sci. USA 2009, 106, 22468–22473. [Google Scholar] [CrossRef]
- Pozdnyakova, N.; Krisanova, N.; Dudarenko, M.; Vavers, E.; Zvejniece, L.; Dambrova, M.; Borisova, T. Inhibition of sigma-1 receptors substantially modulates GABA and glutamate transport in presynaptic nerve terminals. Exp. Neurol. 2020, 333, 113434. [Google Scholar] [CrossRef]
- Yasui, Y.; Su, T.P. Potential molecular mechanisms on the role of the Sigma-1 receptor in the action of cocaine and methamphetamine. J. Drug Alcohol Res. 2016, 5, 235970. [Google Scholar] [CrossRef][Green Version]
- Matsumoto, R.R.; Gilmore, D.L.; Pouw, B.; Bowen, W.D.; Williams, W.; Kausar, A.; Coop, A. Novel analogs of the δ receptor ligand BD1008 attenuate cocaine-induced toxicity in mice. Eur. J. Pharmacol. 2004, 492, 21–26. [Google Scholar] [CrossRef]
- Smith, K.; Hopp, M.; Mundin, G.; Leyendecker, P.; Bailey, P.; Grothe, B.; Uhl, R.; Reimer, K. Single- and multiple-dose pharmacokinetic evaluation of oxycodone and naloxone in an opioid agonist/antagonist prolonged-release combination in healthy adult volunteers. Clin. Ther. 2008, 30, 2051–2068. [Google Scholar] [CrossRef]
- Hindmarch, I.; Hashimoto, K. Cognition and depression: The effects of fluvoxamine, a sigma-1 receptor agonist, reconsidered. Hum. Psychopharmacol. 2010, 25, 193–200. [Google Scholar] [CrossRef]
- Wu, Z.; Li, L.; Zheng, L.T.; Xu, Z.; Guo, L.; Zhen, X. Allosteric modulation of sigma-1 receptors by SKF83959 inhibits microglia-mediated inflammation. J. Neurochem. 2015, 134, 904–914. [Google Scholar] [CrossRef]
- Xiang, C.; Wang, Y.; Zhang, H.; Han, F. The role of endoplasmic reticulum stress in neurodegenerative disease. Apoptosis 2017, 22, 1–26. [Google Scholar] [CrossRef]
- Brunklaus, A.; Ellis, R.; Reavey, E.; Forbes, G.H.; Zuberi, S.M. Prognostic, clinical and demographic features in SCN1A mutation-positive Dravet syndrome. Brain 2012, 135, 2329–2336. [Google Scholar] [CrossRef]
- Lagae, L.; Brambilla, I.; Mingorance, A.; Gibson, E.; Battersby, A. Quality of life and comorbidities associated with Dravet syndrome severity: A multinational cohort survey. Dev. Med. Child. Neurol. 2018, 60, 63–72. [Google Scholar] [CrossRef]
- Villas, N.; Meskis, M.A.; Goodliffe, S. Dravet syndrome: Characteristics, comorbidities, and caregiver concerns. Epilepsy Behav. 2017, 74, 81–86. [Google Scholar] [CrossRef]
- Lagae, L. Cognitive side effects of anti-epileptic drugs. The relevance in childhood epilepsy. Seizure 2006, 15, 235–241. [Google Scholar] [CrossRef][Green Version]
- Park, S.P.; Kwon, S.H. Cognitive effects of antiepileptic drugs. J. Clin. Neurol. 2008, 4, 99–106. [Google Scholar] [CrossRef]
- Aman, M.G.; Kern, R.A. Review of fenfluramine in the treatment of the developmental disabilities. J. Am. Acad. Child. Adolesc. Psychiatry 1989, 28, 549–565. [Google Scholar] [CrossRef] [PubMed]
- Aman, M.G.; Kern, R.A.; McGhee, D.E.; Arnold, L.E. Fenfluramine and methylphenidate in children with mental retardation and attention deficit hyperactivity disorder: Laboratory effects. J. Autism Dev. Disord. 1993, 23, 491–506. [Google Scholar] [CrossRef] [PubMed]
- Murphy, S.E.; Wright, L.C.; Browning, M.; Cowen, P.J.; Harmer, C.J. A role for 5-HT4 receptors in human learning and memory. Psychol. Med. 2020, 50, 2722–2730. [Google Scholar] [CrossRef] [PubMed]
- Niitsu, T.; Iyo, M.; Hashimoto, K. Sigma-1 receptor agonists as therapeutic drugs for cognitive impairment in neuropsychiatric diseases. Curr. Pharm. Des. 2012, 18, 875–883. [Google Scholar] [CrossRef]
- Cross, J.H.; Auvin, S.; Falip, M.; Striano, P.; Arzimanoglou, A. Expert opinion on the management of Lennox-Gastaut syndrome: Treatment algorithms and practical considerations. Front. Neurol. 2017, 8, 505. [Google Scholar] [CrossRef]
- Ouss, L.; Leunen, D.; Laschet, J.; Chemaly, N.; Barcia, G.; Losito, E.M.; Aouidad, A.; Barrault, Z.; Desguerre, I.; Breuillard, D.; et al. Autism spectrum disorder and cognitive profile in children with Dravet syndrome: Delineation of a specific phenotype. Epilepsia Open 2018, 4, 40–53. [Google Scholar] [CrossRef]
- Wang, H.; Hu, L.; Liu, C.; Su, Z.; Wang, L.; Pan, G.; Guo, Y.; He, J. 5-HT2 receptors mediate functional modulation of GABAa receptors and inhibitory synaptic transmissions in human iPS-derived neurons. Sci. Rep. 2016, 6, 20033. [Google Scholar] [CrossRef]
- Carcolé, M.; Zamanillo, D.; Merlos, M.; Fernández-Pastor, B.; Cabañero, D.; Maldonado, R. Blockade of the sigma-1 receptor relieves cognitive and emotional impairments associated to chronic osteoarthritis pain. Front. Pharmacol. 2019, 10, 468. [Google Scholar] [CrossRef]
- Sveinsson, O.; Andersson, T.; Carlsson, S.; Tomson, T. The incidence of SUDEP: A nationwide population-based cohort study. Neurology 2017, 89, 170–177. [Google Scholar] [CrossRef]
- Sörman, E.; Wang, D.; Hajos, M.; Kocsis, B. Control of hippocampal theta rhythm by serotonin: Role of 5-HT2c receptors. Neuropharmacology 2011, 61, 489–494. [Google Scholar] [CrossRef]
- Hantsoo, L.; Epperson, C.N. Premenstrual dysphoric disorder: Epidemiology and treatment. Curr. Psychiatry Rep. 2015, 17, 87. [Google Scholar] [CrossRef]
- Parry, B.L.; Meliska, C.J.; Martínez, L.F.; López, A.M.; Sorenson, D.L.; Hauger, R.L.; Elliott, J.A. Late, but not early, wake therapy reduces morning plasma melatonin: Relationship to mood in premenstrual dysphoric disorder. Psychiatry Res. 2008, 161, 76–86. [Google Scholar] [CrossRef][Green Version]
- Tupal, S.; Faingold, C.L. Evidence supporting a role of serotonin in modulation of sudden death induced by seizures in DBA/2 mice. Epilepsia 2006, 47, 21–26. [Google Scholar] [CrossRef]
- Tupal, S.; Faingold, C.L. Fenfluramine, a serotonin-releasing drug, prevents seizure-induced respiratory arrest and is anticonvulsant in the DBA/1 mouse model of SUDEP. Epilepsia 2019, 60, 485–494. [Google Scholar] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Phenotype | DEE Model | Molecular Mechanism | Reference |
|---|---|---|---|
| Seizures | Scn1a−/− and +/− mice | Reduced Na currents in GABA interneurons led to hyperexcitability of glutamatergic neurons in the hippocampus | Yu 2006 [29] |
| Zebrafish scn1Lab model of Dravet syndrome | Epileptiform activity occurred with reduced neural serotonin expression | Sourbron 2016 [30] | |
| scn1lab−/− zebrafish model of Dravet syndrome | GABAergic neuronal loss and astrogliosis (attenuated by fenfluramine) were noted | Tiraboschi 2020 [28] | |
| Neurodegeneration, cognitive deficits | Scn1a−/− mice | Cortical necrosis with cognitive deficits was evident in Scn1a−/− mice (rescued by liraglutide, a GLP-1 analogue) | Liu 2020 [31] |
| Cognitive defects, learning and memory | Cre-Lox Scn1a deletion localized to hippocampus in mice | Local loss of Nav1.1 function in the hippocampus selectively reduced inhibitory neurotransmission, resulting in thermally evoked seizures and spatial learning deficits | Stein 2019 [32] |
| Scn1a−/− and +/− mice | Reduced sodium currents in forebrain GABAergic neurons led to context-dependent spatial memory (rescued by clonazepam, a positive allosteric modulator of GABAA receptors) | Han 2017 [33] | |
| Hyperactive behavior | Zebrafish scn1a mutant model of Dravet syndrome | Sigma1R agonists abolished FFA-mediated inhibition and hyperlocomotion | Sourbron 2017 [23] |
| Autistic-like behavior | Scn1a−/− and +/− mice | Reduced sodium currents in forebrain GABAergic neurons led to social interaction deficits and stereotyped behaviors (rescued by clonazepam, a positive allosteric modulator of GABAA receptors) | Han 2017 [33] |
| Scn1a−/− and +/− mice | Reduced sodium currents in forebrain GABAergic neurons led to autistic-like social interaction deficits (rescued by CBD by antagonism at lipid-activated GPR55) | Kaplan Catterall 2017 [34] | |
| Motor: Ataxia | Scn1a−/− and +/− mice | Reduced sodium currents in Purkinje cerebellar neurons led to ataxia | Kalume 2007 [35] |
| Emotional regulation | Scn1a +/− mice | Anxiety-related thigmotactic behavior and atypical fear expression were noted | Bahceci 2020 [36] |
| Disrupted sleep architecture | Scn1a−/− and +/− mice | Reduced sodium currents in the GABAergic reticular nucleus of the thalamus resulted in disrupted sleep architecture | Kalume 2015 [37] |
| SUDEP | Scn1a +/− mice and conditional brain- and cardiac-specific KO mice | SUDEP following GTCs in Scn1a+/− mice was caused by parasympathetic CNS hyperactivity, leading to lethal bradycardia | Kalume 2013 [38] |
| Mouse SUDEP model | Seizure-induced respiratory arrest (attenuated by fluoxetine at 5-HT3 without affecting seizures) occurred | Faingold 2016 [25] | |
| In vivo mouse DBA-1 model of SUDEP | Seizure-induced respiratory arrest (inhibited by FFA activity at 5-HT4) occurred | Faingold 2019 [26] |
| Model | Molecular Mechanism | Reference |
|---|---|---|
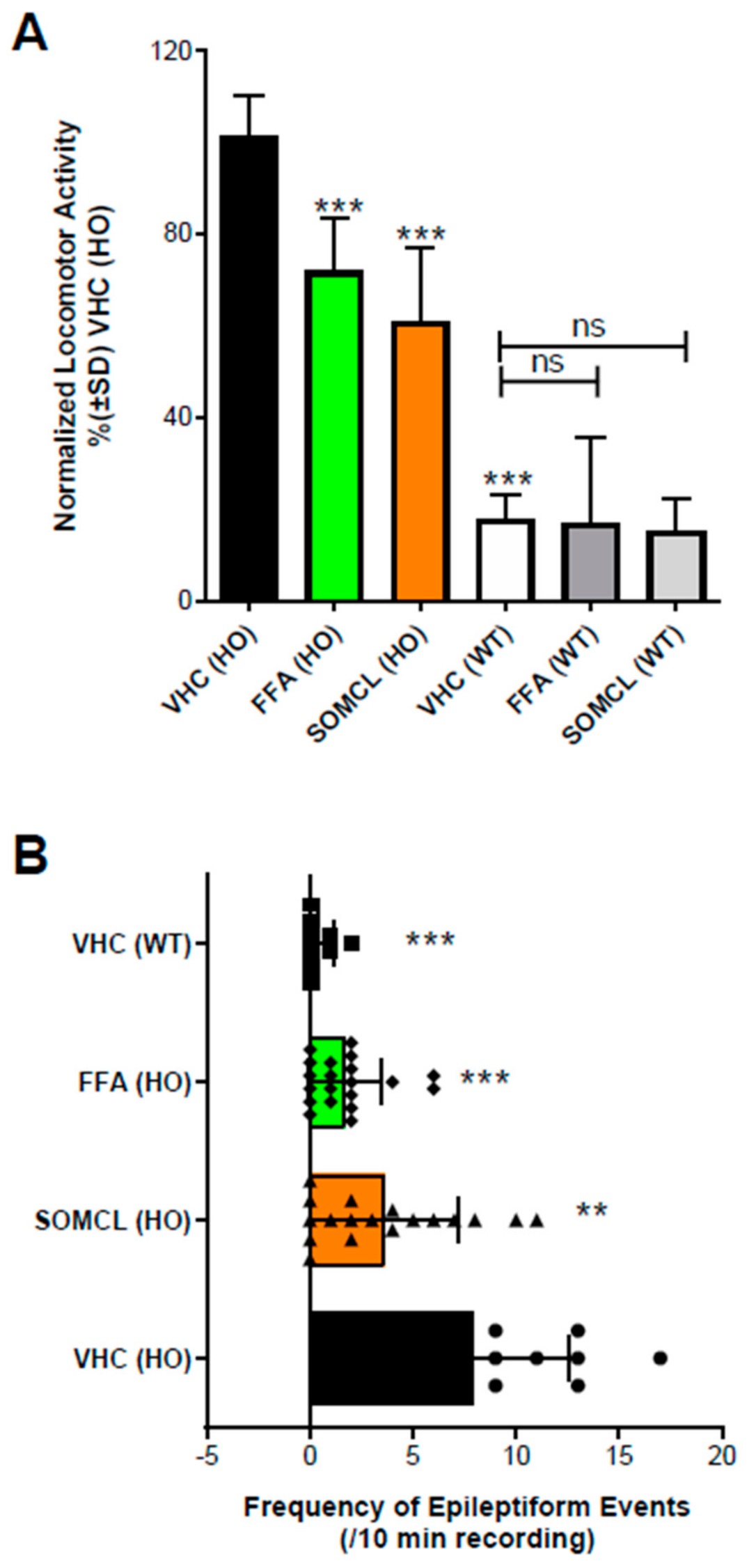
| Dravet zebrafish model | Epileptogenic activity was inhibited by administration of the Sigma1R positive allosteric modulator SOMCL-668 | Reported here |
| Zebrafish scn1a mutant model of Dravet syndrome | Sigma1R agonist PRE-084 and 5-HT1D and 5-HT2C antagonists reduced FFA-mediated inhibition of epileptiform activity and completely abolished FFA-mediated hyperlocomotion | Sourbron 2017 [23] |
| Sigma1R knockout mice | Susceptibility (reduced threshold) to pentylenetetrazol (PTZ) and (+)-bicuculline (BIC) infusion-induced acute seizures was increased | Vavers 2021 [39] |
| NMDA-kindled seizures in mice | Sigma1R/HINT1 association with NR1 was disrupted by fenfluramine, ameliorating seizures | Rodriguez-Munoz, 2018 [24] |
| PTZ- and BIC-induced seizure model in mice | Seizure activity was inhibited by administration of the Sigma1R positive allosteric modulator E1R; antiseizure activity of E1R was blocked by the Sigma1R antagonist BD1047 | Vavers 2017 [40] |
| Phase 1 studies of ANAVEX 2-73 (blarcamesine) for infantile spasms (West syndrome) | Sigma1R agonist was granted Orphan Drug Designation for treatment of infantile spasms; preclinical validation for potential treatment for Rett syndrome and other pediatric or infantile disorders with seizure pathology included seizure reduction in rodent chemoconvulsant models and synergistic activity with the antiseizure medications ethosuximide, valproate, and gabapentin | Rebowe 2016 [41] |
| Maximal electroshock seizure (MES) model in mice PTZ-induced tonic seizure model in mice | Tonic seizure activity was inhibited by administration of ANAVEX2-73, a Sigma1R agonist (pluripotent modulator) with muscarinic activity; administration of subtherapeutic doses of ANAVEX2-73 showed synergistic antiseizure activity in combination with the ASMs ethosuximide, valproate, and gabapentin in the MES model | Rebowe 2015 [42] Rebowe 2015 [42] |
| SCZ-induced tonic seizure model in mice | Rebowe 2015 [42] | |
| MES model in mice | Guo 2015 [43] | |
| PTZ-induced seizure model in mice Kainate acid−induced status epilepticus model in mice | Seizure activity was inhibited by administration of the Sigma1R positive allosteric modulators SKF83959 and SOMCL-668; antiseizure activity of SKF83959 and SOMCL-668 was blocked by the Sigma1R antagonist BD1047 | Guo 2015 [43] Guo 2015 [43] |
| Rat hippocampal slices (ex vivo) | Epileptiform local field potentials were attenuated by the Sigma 1R positive allosteric modulators SKF83959 and its derivative SOMCL-668 | Guo 2015 [43] |
| In vitro guinea pig brain homogenates | The antiseizure medication phenytoin acted as a potent positive allosteric modulator of Sigma1R activity by augmenting binding of the Sigma1R agonists (+) pentazocine, (+)-SKF-10,047, PRE-084, and (+)-3-(3-hydroxyphenyl)-N-(1-propyl) piperidine hydrochloride to solubilized Sigma1R binding sites in homogenized guinea pig brain without affecting binding of the tritiated antagonist NE-100 | Chaki 1996 [44] Cobos 2005 [45] DeHaven 1993 [46] |
| MES model in mice | The positive allosteric Sigma1R modulator and antiseizure medication phenytoin (5−20 mg/kg i.p.) induced potent antiseizure activity via inhibition of voltage-gated sodium channels Phenytoin (20 or 40 mg/kg/day orally) inhibited tonic extension seizures, clonic seizures, and wild running in rats with ED50 of 5.0, 10.8, and 20.7 mg/kg, respectively | Jones 1981 [47] Vavers 2019 [51] Edmonds 1996 [48] |
| Ischemia-induced epilepsy model in rats |
| Phenotype | Model | Molecular Mechanism | Reference |
|---|---|---|---|
| Inflammatory stress | Sigma1R knockout mice | Hypercytokinemia was induced by acute inflammation with restricted endonuclease activity of IRE1, an ER stress sensor | Rosen 2019 [64] |
| Memory | Sigma1R knockout in APPSwe Alzheimer’s disease mouse model | Memory impairment was exacerbated with increased oxidative stress in the hippocampus | Ryskamp 2019 [69] |
| Phase 2a clinical trials (phase 2b/3 ongoing) | ANAVEX 2-73 (blarcamesine), a Sigma1R agonist with some muscarinic modulatory activity, reduced cognitive decline in a longitudinal 148-week study of patients with Alzheimer’s disease | Hampel 2019 [57]; NCT03790709 | |
| Fmr1-KO2 mouse model of fragile X-autism-related disorders model | ANAVEX 2-73 (blarcamesine) reversed Fmr1-KO2 deficit in learning and memory as measured by percent freezing during observation | Rebowe 2016 [41] | |
| Mood | Sigma1R knockout mice | Depression-like phenotype was noted | Hayashi 2011 [49] |
| Motor, cognitive deficiency | Cellular models of Huntington’s disease | mHtt (huntingtin fragment) protein downregulated Sigma1R expression in cultured PC6.3 neurons | Ryskamp 2019 [69] |
| Hyperactivity | Fmr1 KO2 mouse model of Fragile X-autism-related disorders model | ANAVEX 2-73 (blarcamesine) reversed Fmr1-KO2 hyperactivity in locomotor assays (number of squares crossed per unit time) | Rebowe 2016 [41] |
| Motor | SOD1/Sigma1R mouse model of ALS | ALS was exacerbated by disrupting MAM components, dysregulating calcium, and causing mitochondrial dysfunction leading to neuronal degradation | Watanabe 2016 [70] |
| Motor/Neuroprotection | Genetic linkage analysis and cellular expression of mutant protein | Sigma1R genetic variant was associated with ALS; expression of motor neuron cell line model resulted in loss of resistance to ER stress-induced apoptosis | Al Saif 2011 [71] |
| Motor/Neuroprotection | Genetic linkage analysis | Sigma1R mutations were associated with non-juvenile ALS without FTLD | Ulla 2015 [72] |
| Motor/Neuroprotection | Genetic linkage analysis | Sigma1R mutations were associated with nuclear aggregates, ER stress, and increased apoptosis | Li 2015 [73] |
| Motor | Genetic linkage analysis | Homozygous missense variant of SigmaR1 resulted in distal hereditary motor neuropathy and lower limb spasticity (Silver-like syndrome) | Horga 2016 [74] |
| Motor | Genetic linkage analysis | Non-polymorphic mutation in Sigma1R was associated with FTLD and ALS | Luty 2010 [75] |
| Motor | Sigma1R knockout mice | Vulnerability of S1R knockout mice to nigrostriatal axonal degradation was increased in a model of Parkinson’s disease | Ryskamp 2019 [69] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Martin, P.; Reeder, T.; Sourbron, J.; de Witte, P.A.M.; Gammaitoni, A.R.; Galer, B.S. An Emerging Role for Sigma-1 Receptors in the Treatment of Developmental and Epileptic Encephalopathies. Int. J. Mol. Sci. 2021, 22, 8416. https://doi.org/10.3390/ijms22168416
Martin P, Reeder T, Sourbron J, de Witte PAM, Gammaitoni AR, Galer BS. An Emerging Role for Sigma-1 Receptors in the Treatment of Developmental and Epileptic Encephalopathies. International Journal of Molecular Sciences. 2021; 22(16):8416. https://doi.org/10.3390/ijms22168416
Chicago/Turabian StyleMartin, Parthena, Thadd Reeder, Jo Sourbron, Peter A. M. de Witte, Arnold R. Gammaitoni, and Bradley S. Galer. 2021. "An Emerging Role for Sigma-1 Receptors in the Treatment of Developmental and Epileptic Encephalopathies" International Journal of Molecular Sciences 22, no. 16: 8416. https://doi.org/10.3390/ijms22168416
APA StyleMartin, P., Reeder, T., Sourbron, J., de Witte, P. A. M., Gammaitoni, A. R., & Galer, B. S. (2021). An Emerging Role for Sigma-1 Receptors in the Treatment of Developmental and Epileptic Encephalopathies. International Journal of Molecular Sciences, 22(16), 8416. https://doi.org/10.3390/ijms22168416