
The Role of Mitochondrial Dysfunction in Atrial Fibrillation: Translation to Druggable Target and Biomarker Discovery


 ,
,  and
and 
Abstract
1. Introduction
2. Cardiac Mitochondrial Physiology
2.1. ATP Synthesis by Fatty Acid and Carbohydrate Oxidation
2.2. Reactive Oxygen Species Generation during Oxidative Phosphorylation
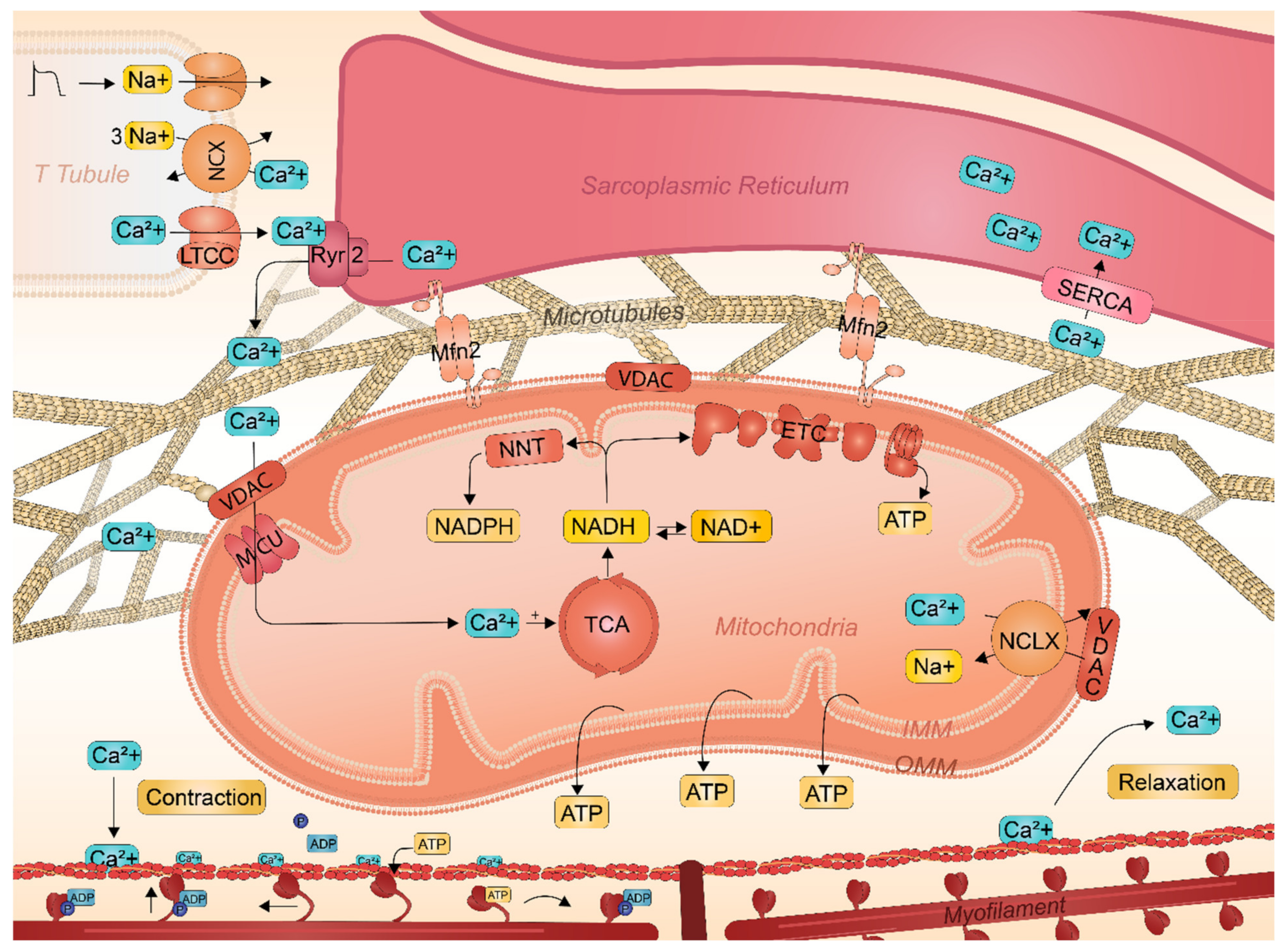
2.3. Key Role of SR-Mitochondrial Ca2+ Handling in Atrial Cardiomyocyte Contractile Function
2.4. Interactions between Sarcoplasmic Reticulum and Mitochondria Safeguards Cardiac Function
2.5. Mitochondrial Heat Shock Proteins Critical for Mitochondrial Gene Expression and Function
3. The Role of Mitochondrial Dysfunction in the Pathophysiology of AF
3.1. Mitochondrial NAD+ Depletion Confers Cardiomyocyte Dysfunction in AF
3.2. Alternations in the Microtubule Network Contribute to AF
4. Potential Mitochondrial Markers for AF Diagnostics
4.1. Oxidative Stress Marker 8-hydroxy-2′-deoxyguanosine in AF
4.2. Circulating Cell-Free Mitochondrial DNA in AF
4.3. Mitochondrial Heat Shock Proteins in AF
5. Mitochondria as a Target for Therapeutic Interventions
5.1. Pharmaceuticals to Conserve Mitochondrial Function
5.2. Nutraceuticals to Conserve Mitochondrial Function
6. Clinical and Future Perspectives
7. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Zoni-Berisso, M.; Lercari, F.; Carazza, T.; Domenicucci, S. Epidemiology of atrial fibrillation: European perspective. Clin. Epidemiol. 2014, 6, 213. [Google Scholar] [CrossRef] [PubMed]
- Stewart, S.; Hart, C.L.; Hole, D.J.; McMurray, J.J.V. A population-based study of the long-term risks associated with atrial fibrillation: 20-year follow-up of the Renfrew/Paisley study. Am. J. Med. 2002, 113, 359–364. [Google Scholar] [CrossRef]
- Wolf, P.A.; Abbott, R.D.; Kannel, W.B. Atrial fibrillation as an independent risk factor for stroke: The Framingham Study. Stroke 1991, 22, 983–988. [Google Scholar] [CrossRef]
- Krijthe, B.P.; Kunst, A.; Benjamin, E.J.; Lip, G.Y.H.; Franco, O.H.; Hofman, A.; Witteman, J.C.M.; Stricker, B.H.; Heeringa, J. Projections on the number of individuals with atrial fibrillation in the European Union, from 2000 to 2060. Eur. Heart J. 2013, 34, 2746–2751. [Google Scholar] [CrossRef]
- Van Marion, D.M.S.; Lanters, E.A.H.; Wiersma, M.; Allessie, M.A.; Brundel, B.B.; de Groot, N.M.S. Diagnosis and therapy of atrial fibrillation: The past, the present and the future. J. Atr. Fibrillation 2015, 8, 1216. [Google Scholar] [PubMed]
- Zhang, D.; Wu, C.T.; Qi, X.; Meijering, R.A.; Hoogstra-Berends, F.; Tadevosyan, A.; Cubukcuoglu Deniz, G.; Durdu, S.; Akar, A.R.; Sibon, O.C.; et al. Activation of histone deacetylase-6 induces contractile dysfunction through derailment of alpha-tubulin proteostasis in experimental and human atrial fibrillation. Circulation 2014, 129, 346–358. [Google Scholar] [CrossRef]
- Zhang, D.; Hu, X.; Li, J.; Liu, J.; Baks-te Bulte, L.; Wiersma, M.; van Marion, D.M.S.; Tolouee, M.; Hoogstra-Berends, F.; Lanters, E.A.H. DNA damage-induced PARP1 activation confers cardiomyocyte dysfunction through NAD+ depletion in experimental atrial fibrillation. Nat. Commun. 2019, 10, 1–17. [Google Scholar] [CrossRef] [PubMed]
- De Groot, N.M.S.; Houben, R.P.M.; Smeets, J.L.; Boersma, E.; Schotten, U.; Schalij, M.J.; Crijns, H.; Allessie, M.A. Electropathological substrate of longstanding persistent atrial fibrillation in patients with structural heart disease: Epicardial breakthrough. Circulation 2010, 122, 1674–1682. [Google Scholar] [CrossRef]
- Hindricks, G.; Potpara, T.; Dagres, N.; Arbelo, E.; Bax, J.J.; Blomström-Lundqvist, C.; Boriani, G.; Castella, M.; Dan, G.-A.; Dilaveris, P.E. 2020 ESC Guidelines for the diagnosis and management of atrial fibrillation developed in collaboration with the European Association for Cardio-Thoracic Surgery (EACTS) The Task Force for the diagnosis and management of atrial fibrillation of the European Society of Cardiology (ESC) Developed with the special contribution of the European Heart Rhythm Association (EHRA) of the ESC. Eur. Heart J. 2021, 42, 373–498. [Google Scholar] [PubMed]
- Palaniyandi, S.S.; Qi, X.; Yogalingam, G.; Ferreira, J.C.B.; Mochly-Rosen, D. Regulation of mitochondrial processes: A target for heart failure. Drug Discov. Today Dis. Mech. 2010, 7, e95–e102. [Google Scholar] [CrossRef]
- Murphy, E.; Ardehali, H.; Balaban, R.S.; DiLisa, F.; Dorn, G.W.; Kitsis, R.N.; Otsu, K.; Ping, P.; Rizzuto, R.; Sack, M.N. Mitochondrial function, biology, and role in disease: A scientific statement from the American Heart Association. Circ. Res. 2016, 118, 1960–1991. [Google Scholar] [CrossRef] [PubMed]
- Kolwicz Jr, S.C.; Purohit, S.; Tian, R. Cardiac metabolism and its interactions with contraction, growth, and survival of cardiomyocytes. Circ. Res. 2013, 113, 603–616. [Google Scholar] [CrossRef]
- Neely, J.R.; Morgan, H.E. Relationship between carbohydrate and lipid metabolism and the energy balance of heart muscle. Annu. Rev. Physiol. 1974, 36, 413–459. [Google Scholar] [CrossRef] [PubMed]
- Bing, R.J.; Siegel, A.; Ungar, I.; Gilbert, M. Metabolism of the human heart: II. Studies on fat, ketone and amino acid metabolism. Am. J. Med. 1954, 16, 504–515. [Google Scholar] [CrossRef]
- Chen, Q.; Vazquez, E.J.; Moghaddas, S.; Hoppel, C.L.; Lesnefsky, E.J. Production of reactive oxygen species by mitochondria central role of complex III. J. Biol. Chem. 2003, 278, 36027–36031. [Google Scholar] [CrossRef] [PubMed]
- Sovari, A.A.; Dudley Jr, S.C. Reactive oxygen species-targeted therapeutic interventions for atrial fibrillation. Front. Physiol. 2012, 3, 311. [Google Scholar] [CrossRef]
- Taverne, Y.J.; Bogers, A.J.; Duncker, D.J.; Merkus, D. Reactive oxygen species and the cardiovascular system. Oxid Med. Cell Longev 2013, 2013, 862423. [Google Scholar] [CrossRef] [PubMed]
- Taverne, Y.J.; Merkus, D.; Bogers, A.J.; Halliwell, B.; Duncker, D.J.; Lyons, T.W. Reactive Oxygen Species: Radical Factors in the Evolution of Animal Life: A molecular timescale from Earth’s earliest history to the rise of complex life. Bioessays 2018, 40, 1700158. [Google Scholar] [CrossRef] [PubMed]
- Gambardella, J.; Sorriento, D.; Ciccarelli, M.; Del Giudice, C.; Fiordelisi, A.; Napolitano, L.; Trimarco, B.; Iaccarino, G.; Santulli, G. Functional role of mitochondria in arrhythmogenesis. Adv. Exp. Med. Biol. 2017, 982, 191–202. [Google Scholar]
- Lin, P.-H.; Lee, S.-H.; Su, C.-P.; Wei, Y.-H. Oxidative damage to mitochondrial DNA in atrial muscle of patients with atrial fibrillation. Free Radic. Biol. Med. 2003, 35, 1310–1318. [Google Scholar] [CrossRef]
- Bukowska, A.; Schild, L.; Keilhoff, G.; Hirte, D.; Neumann, M.; Gardemann, A.; Neumann, K.H.; Röhl, F.-W.; Huth, C.; Goette, A. Mitochondrial dysfunction and redox signaling in atrial tachyarrhythmia. Exp. Biol. Med. 2008, 233, 558–574. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Santulli, G.; Reiken, S.R.; Yuan, Q.; Osborne, B.W.; Chen, B.-X.; Marks, A.R. Mitochondrial oxidative stress promotes atrial fibrillation. Sci. Rep. 2015, 5, 1–11. [Google Scholar] [CrossRef]
- Wiersma, M.; van Marion, D.; Wüst, R.C.I.; Houtkooper, R.H.; Zhang, D.; de Groot, N.; Henning, R.H.; Brundel, B.J.J.M. Mitochondrial dysfunction underlies cardiomyocyte remodeling in experimental and clinical atrial fibrillation. Cells 2019, 8, 1202. [Google Scholar] [CrossRef]
- Houten, S.M.; Wanders, R.J.A. A general introduction to the biochemistry of mitochondrial fatty acid β-oxidation. J. Inherit. Metab. Dis. 2010, 33, 469–477. [Google Scholar] [CrossRef]
- Abel, E.D.; Doenst, T. Mitochondrial adaptations to physiological vs. pathological cardiac hypertrophy. Cardiovasc. Res. 2011, 90, 234–242. [Google Scholar] [CrossRef]
- McGarry, J.D.; Brown, N.F. The mitochondrial carnitine palmitoyltransferase system—From concept to molecular analysis. Eur. J. Biochem. 1997, 244, 1–14. [Google Scholar] [CrossRef]
- Nolfi-Donegan, D.; Braganza, A.; Shiva, S. Mitochondrial Electron Transport: Oxidative Phosphorylation, Mitochondrial Oxidant Production, and Methods of Measurement. Redox Biol. 2020, 37, 101674. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, R.R.; Gandour, R.D.; van der Leij, F.R. Molecular enzymology of carnitine transfer and transport. Biochim. Biophys. Acta (BBA)-Protein Struct. Mol. Enzymol. 2001, 1546, 21–43. [Google Scholar] [CrossRef]
- Mitchell, P. Coupling of phosphorylation to electron and hydrogen transfer by a chemi-osmotic type of mechanism. Nature 1961, 191, 144–148. [Google Scholar] [CrossRef]
- McCommis, K.S.; Finck, B.N. Mitochondrial pyruvate transport: A historical perspective and future research directions. Biochem. J. 2015, 466, 443–454. [Google Scholar] [CrossRef] [PubMed]
- Huizing, M.; Ruitenbeek, W.; Thinnes, F.P.; Depinto, V.; Wendel, U.; Trijbels, F.J.M.; Smit, L.M.E.; Ter Laak, H.J.; van den Heuvel, L.P. Deficiency of the voltage-dependent anion channel: A novel cause of mitochondriopathy. Pediatric Res. 1996, 39, 760–765. [Google Scholar] [CrossRef]
- Emelyanova, L.; Ashary, Z.; Cosic, M.; Negmadjanov, U.; Ross, G.; Rizvi, F.; Olet, S.; Kress, D.; Sra, J.; Tajik, A.J.; et al. Selective downregulation of mitochondrial electron transport chain activity and increased oxidative stress in human atrial fibrillation. Am. J. Physiol Heart Circ. Physiol 2016, 311, H54–H63. [Google Scholar] [CrossRef] [PubMed]
- Brown, S.M.; Larsen, N.K.; Thankam, F.G.; Agrawal, D.K. Fetal cardiomyocyte phenotype, ketone body metabolism, and mitochondrial dysfunction in the pathology of atrial fibrillation. Mol. Cell. Biochem. 2021, 476, 1165–1178. [Google Scholar] [CrossRef] [PubMed]
- Dröge, W. Free radicals in the physiological control of cell function. Physiol. Rev. 2002, 82, 47–95. [Google Scholar] [CrossRef] [PubMed]
- Kudryavtseva, A.V.; Krasnov, G.S.; Dmitriev, A.A.; Alekseev, B.Y.; Kardymon, O.L.; Sadritdinova, A.F.; Fedorova, M.S.; Pokrovsky, A.V.; Melnikova, N.V.; Kaprin, A.D. Mitochondrial dysfunction and oxidative stress in aging and cancer. Oncotarget 2016, 7, 44879. [Google Scholar] [CrossRef]
- Newsholme, P.; Haber, E.P.; Hirabara, S.M.; Rebelato, E.L.O.; Procópio, J.; Morgan, D.; Oliveira--Emilio, H.C.; Carpinelli, A.R.; Curi, R. Diabetes associated cell stress and dysfunction: Role of mitochondrial and non--mitochondrial ROS production and activity. J. Physiol. 2007, 583, 9–24. [Google Scholar] [CrossRef] [PubMed]
- Mason, F.E.; Pronto, J.R.D.; Alhussini, K.; Maack, C.; Voigt, N. Cellular and mitochondrial mechanisms of atrial fibrillation. Basic Res. Cardiol. 2020, 115, 1–16. [Google Scholar] [CrossRef]
- Yang, K.-C.; Dudley Jr, S.C. Oxidative stress and atrial fibrillation: Finding a missing piece to the puzzle. Am. Heart Assoc. 2013, 128, 1724–1726. [Google Scholar] [CrossRef]
- Harada, M.; Melka, J.; Sobue, Y.; Nattel, S. Metabolic considerations in atrial fibrillation―Mechanistic insights and therapeutic opportunities―. Circ. J. 2017, 81, 1749–1757. [Google Scholar] [CrossRef] [PubMed]
- Korantzopoulos, P.; Letsas, K.; Fragakis, N.; Tse, G.; Liu, T. Oxidative stress and atrial fibrillation: An update. Free Radic. Res. 2018, 52, 1199–1209. [Google Scholar] [CrossRef]
- Bers, D.M. Cardiac excitation–contraction coupling. Nature 2002, 415, 198–205. [Google Scholar] [CrossRef]
- Dedkova, E.N.; Blatter, L.A. Calcium signaling in cardiac mitochondria. J. Mol. Cell Cardiol 2013, 58, 125–133. [Google Scholar] [CrossRef]
- Bers, D.M. Calcium fluxes involved in control of cardiac myocyte contraction. Circ. Res. 2000, 87, 275–281. [Google Scholar] [CrossRef]
- Konstantinidis, K.; Lederer, W.J.; Rizzuto, R.; Kitsis, R.N. Mitofusin 2 joins the sarcoplasmic reticulum and mitochondria at the hip to sustain cardiac energetics. Circ. Res. 2012, 111, 821–823. [Google Scholar] [CrossRef] [PubMed]
- Bertero, E.; Maack, C. Calcium signaling and reactive oxygen species in mitochondria. Circ. Res. 2018, 122, 1460–1478. [Google Scholar] [CrossRef] [PubMed]
- Bridge, J.H.; Spitzer, K.W. The relationship between charge movements associated with ICa and INa-Ca in cardiac myocytes. Science 1990, 248, 376–378. [Google Scholar] [CrossRef]
- Balaban, R.S.; Bose, S.; French, S.A.; Territo, P.R. Role of calcium in metabolic signaling between cardiac sarcoplasmic reticulum and mitochondria in vitro. Am. J. Physiol Cell Physiol 2003, 284, C285–C293. [Google Scholar] [CrossRef]
- Richards, M.A.; Clarke, J.D.; Saravanan, P.; Voigt, N.; Dobrev, D.; Eisner, D.A.; Trafford, A.W.; Dibb, K.M. Transverse tubules are a common feature in large mammalian atrial myocytes including human. Am. J. Physiol. Heart Circ. Physiol. 2011, 301, H1996–H2005. [Google Scholar] [CrossRef]
- Santulli, G.; Xie, W.; Reiken, S.R.; Marks, A.R. Mitochondrial calcium overload is a key determinant in heart failure. Proc. Natl. Acad. Sci. USA 2015, 112, 11389–11394. [Google Scholar] [CrossRef]
- Franzini--Armstrong, C.; Protasi, F.; Tijskens, P. The assembly of calcium release units in cardiac muscle. Ann. N. Y. Acad. Sci. 2005, 1047, 76–85. [Google Scholar] [CrossRef] [PubMed]
- Gu, Y.; Gorelik, J.; Spohr, H.A.; Shevchuk, A.; Lab, M.J.; Harding, S.E.; Vodyanoy, I.; Klenerman, D.; Korchev, Y.E. High--resolution scanning patch--clamp: New insights into cell function. FASEB J. 2002, 16, 748–750. [Google Scholar] [CrossRef]
- Brette, F.; Orchard, C. T-tubule function in mammalian cardiac myocytes. Circ. Res. 2003, 92, 1182–1192. [Google Scholar] [CrossRef] [PubMed]
- Hammond, J.W.; Cai, D.; Verhey, K.J. Tubulin modifications and their cellular functions. Curr. Opin. Cell Biol. 2008, 20, 71–76. [Google Scholar] [CrossRef] [PubMed]
- Rostovtseva, T.K.; Sheldon, K.L.; Hassanzadeh, E.; Monge, C.; Saks, V.; Bezrukov, S.M.; Sackett, D.L. Tubulin binding blocks mitochondrial voltage-dependent anion channel and regulates respiration. Proc. Natl. Acad. Sci. USA 2008, 105, 18746–18751. [Google Scholar] [CrossRef]
- Murley, A.; Nunnari, J. The emerging network of mitochondria-organelle contacts. Mol. Cell 2016, 61, 648–653. [Google Scholar] [CrossRef]
- Dorn, G.W., 2nd; Maack, C. SR and mitochondria: Calcium cross-talk between kissing cousins. J. Mol. Cell. Cardiol. 2013, 55, 42–49. [Google Scholar] [CrossRef]
- Zizi, M.; Forte, M.; Blachly-Dyson, E.; Colombini, M. NADH regulates the gating of VDAC, the mitochondrial outer membrane channel. J. Biol. Chem. 1994, 269, 1614–1616. [Google Scholar] [CrossRef]
- Rostovtseva, T.; Colombini, M. ATP flux is controlled by a voltage-gated channel from the mitochondrial outer membrane. J. Biol. Chem. 1996, 271, 28006–28008. [Google Scholar] [CrossRef]
- Gincel, D.; Silberberg, S.D.; Shoshan-Barmatz, V. Modulation of the voltage-dependent anion channel (VDAC) by Glutamate1. J. Bioenerg. Biomembr. 2000, 32, 571–583. [Google Scholar] [CrossRef]
- Naon, D.; Zaninello, M.; Giacomello, M.; Varanita, T.; Grespi, F.; Lakshminaranayan, S.; Serafini, A.; Semenzato, M.; Herkenne, S.; Hernández-Alvarez, M.I. Critical reappraisal confirms that Mitofusin 2 is an endoplasmic reticulum–mitochondria tether. Proc. Natl. Acad. Sci. USA 2016, 113, 11249–11254. [Google Scholar] [CrossRef]
- De Brito, O.M.; Scorrano, L. Mitofusin 2 tethers endoplasmic reticulum to mitochondria. Nature 2008, 456, 605–610. [Google Scholar] [CrossRef]
- Chen, Y.; Csordas, G.; Jowdy, C.; Schneider, T.G.; Csordas, N.; Wang, W.; Liu, Y.; Kohlhaas, M.; Meiser, M.; Bergem, S.; et al. Mitofusin 2-containing mitochondrial-reticular microdomains direct rapid cardiomyocyte bioenergetic responses via interorganelle Ca(2+) crosstalk. Circ. Res. 2012, 111, 863–875. [Google Scholar] [CrossRef] [PubMed]
- Cardenas, C.; Miller, R.A.; Smith, I.; Bui, T.; Molgo, J.; Muller, M.; Vais, H.; Cheung, K.H.; Yang, J.; Parker, I.; et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 2010, 142, 270–283. [Google Scholar] [CrossRef]
- Hatefi, Y.; Yamaguchi, M. Nicotinamide nucleotide transhydrogenase: A model for utilization of substrate binding energy for proton translocation. FASEB J. 1996, 10, 444–452. [Google Scholar] [CrossRef] [PubMed]
- Kohlhaas, M.; Liu, T.; Knopp, A.; Zeller, T.; Ong, M.F.; Böhm, M.; O’Rourke, B.; Maack, C. Elevated cytosolic Na+ increases mitochondrial formation of reactive oxygen species in failing cardiac myocytes. Circulation 2010, 121, 1606–1613. [Google Scholar] [CrossRef] [PubMed]
- Csordas, G.; Varnai, P.; Golenar, T.; Roy, S.; Purkins, G.; Schneider, T.G.; Balla, T.; Hajnoczky, G. Imaging interorganelle contacts and local calcium dynamics at the ER-mitochondrial interface. Mol. Cell 2010, 39, 121–132. [Google Scholar] [CrossRef] [PubMed]
- Papanicolaou, K.N.; Khairallah, R.J.; Ngoh, G.A.; Chikando, A.; Luptak, I.; O’Shea, K.M.; Riley, D.D.; Lugus, J.J.; Colucci, W.S.; Lederer, W.J.; et al. Mitofusin-2 maintains mitochondrial structure and contributes to stress-induced permeability transition in cardiac myocytes. Mol. Cell. Biol. 2011, 31, 1309–1328. [Google Scholar] [CrossRef]
- Dibb, K.M.; Clarke, J.D.; Horn, M.A.; Richards, M.A.; Graham, H.K.; Eisner, D.A.; Trafford, A.W. Characterization of an extensive transverse tubular network in sheep atrial myocytes and its depletion in heart failure. Circ. Heart Fail. 2009, 2, 482–489. [Google Scholar] [CrossRef]
- Denham, N.C.; Pearman, C.M.; Caldwell, J.L.; Madders, G.W.P.; Eisner, D.A.; Trafford, A.W.; Dibb, K.M. Calcium in the pathophysiology of atrial fibrillation and heart failure. Front. Physiol. 2018, 9, 1380. [Google Scholar] [CrossRef]
- Ali, A.T.; Boehme, L.; Carbajosa, G.; Seitan, V.C.; Small, K.S.; Hodgkinson, A. Nuclear genetic regulation of the human mitochondrial transcriptome. eLife 2019, 8, e41927. [Google Scholar] [CrossRef] [PubMed]
- Kummer, E.; Ban, N. Mechanisms and regulation of protein synthesis in mitochondria. Nat. Rev. Mol. Cell Biol 2021, 22, 307–325. [Google Scholar] [CrossRef]
- Mercer, T.R.; Neph, S.; Dinger, M.E.; Crawford, J.; Smith, M.A.; Shearwood, A.-M.J.; Haugen, E.; Bracken, C.P.; Rackham, O.; Stamatoyannopoulos, J.A. The human mitochondrial transcriptome. Cell 2011, 146, 645–658. [Google Scholar] [CrossRef] [PubMed]
- Koll, H.; Guiard, B.; Rassow, J.; Ostermann, J.; Horwich, A.L.; Neupert, W.; Hartl, F.U. Antifolding activity of hsp60 couples protein import into the mitochondrial matrix with export to the intermembrane space. Cell 1992, 68, 1163–1175. [Google Scholar] [CrossRef]
- Hohfeld, J.; Hartl, F.U. Role of the chaperonin cofactor Hsp10 in protein folding and sorting in yeast mitochondria. J. Cell Biol. 1994, 126, 305–315. [Google Scholar] [CrossRef] [PubMed]
- Lin, K.M.; Lin, B.; Lian, I.Y.; Mestril, R.; Scheffler, I.E.; Dillmann, W.H. Combined and individual mitochondrial HSP60 and HSP10 expression in cardiac myocytes protects mitochondrial function and prevents apoptotic cell deaths induced by simulated ischemia-reoxygenation. Circulation 2001, 103, 1787–1792. [Google Scholar] [CrossRef]
- Fan, F.; Duan, Y.; Yang, F.; Trexler, C.; Wang, H.; Huang, L.; Li, Y.; Tang, H.; Wang, G.; Fang, X. Deletion of heat shock protein 60 in adult mouse cardiomyocytes perturbs mitochondrial protein homeostasis and causes heart failure. Cell Death Differ. 2020, 27, 587–600. [Google Scholar] [CrossRef] [PubMed]
- Hu, X.; Li, J.; van Marion, D.M.S.; Zhang, D.; Brundel, B.J.J.M. Heat shock protein inducer GGA*-59 reverses contractile and structural remodeling via restoration of the microtubule network in experimental Atrial Fibrillation. J. Mol. Cell. Cardiol. 2019, 134, 86–97. [Google Scholar] [CrossRef] [PubMed]
- Ozcan, C.; Li, Z.; Kim, G.; Jeevanandam, V.; Uriel, N. Molecular Mechanism of the Association Between Atrial Fibrillation and Heart Failure Includes Energy Metabolic Dysregulation Due to Mitochondrial Dysfunction. J. Card Fail. 2019, 25, 911–920. [Google Scholar] [CrossRef]
- Xiao, J.; Zhang, H.; Liang, D.; Liu, Y.; Liu, Y.; Zhao, H.; Li, J.; Peng, L.; Chen, Y.H. Taxol, a microtubule stabilizer, prevents atrial fibrillation in in vitro atrial fibrillation models using rabbit hearts. Med. Sci Monit. 2010, 16, BR353–BR360. [Google Scholar] [PubMed]
- Lushchak, V.I. Free radicals, reactive oxygen species, oxidative stress and its classification. Chem. Biol. Interact. 2014, 224, 164–175. [Google Scholar] [CrossRef]
- Chang, J.-P.; Chen, M.-C.; Liu, W.-H.; Yang, C.-H.; Chen, C.-J.; Chen, Y.-L.; Pan, K.-L.; Tsai, T.-H.; Chang, H.-W. Atrial myocardial nox2 containing NADPH oxidase activity contribution to oxidative stress in mitral regurgitation: Potential mechanism for atrial remodeling. Cardiovasc. Pathol. 2011, 20, 99–106. [Google Scholar] [CrossRef]
- Kim, Y.M.; Guzik, T.J.; Zhang, Y.H.; Zhang, M.H.; Kattach, H.; Ratnatunga, C.; Pillai, R.; Channon, K.M.; Casadei, B. A myocardial Nox2 containing NAD (P) H oxidase contributes to oxidative stress in human atrial fibrillation. Circ. Res. 2005, 97, 629–636. [Google Scholar] [CrossRef]
- Youn, J.-Y.; Zhang, J.; Zhang, Y.; Chen, H.; Liu, D.; Ping, P.; Weiss, J.N.; Cai, H. Oxidative stress in atrial fibrillation: An emerging role of NADPH oxidase. J. Mol. Cell. Cardiol. 2013, 62, 72–79. [Google Scholar] [CrossRef]
- Ren, X.; Wang, X.; Yuan, M.; Tian, C.; Li, H.; Yang, X.; Li, X.; Li, Y.; Yang, Y.; Liu, N.; et al. Mechanisms and Treatments of Oxidative Stress in Atrial Fibrillation. Curr. Pharm. Des. 2018, 24, 3062–3071. [Google Scholar] [CrossRef]
- Kim, Y.M.; Kattach, H.; Ratnatunga, C.; Pillai, R.; Channon, K.M.; Casadei, B. Association of atrial nicotinamide adenine dinucleotide phosphate oxidase activity with the development of atrial fibrillation after cardiac surgery. J. Am. Coll. Cardiol. 2008, 51, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Yoo, S.; Pfenniger, A.; Hoffman, J.; Zhang, W.; Ng, J.; Burrell, A.; Johnson, D.A.; Gussak, G.; Waugh, T.; Bull, S.; et al. Attenuation of Oxidative Injury With Targeted Expression of NADPH Oxidase 2 Short Hairpin RNA Prevents Onset and Maintenance of Electrical Remodeling in the Canine Atrium: A Novel Gene Therapy Approach to Atrial Fibrillation. Circulation 2020, 142, 1261–1278. [Google Scholar] [CrossRef] [PubMed]
- Boland, M.L.; Chourasia, A.H.; Macleod, K.F. Mitochondrial dysfunction in cancer. Front. Oncol. 2013, 3, 292. [Google Scholar] [CrossRef] [PubMed]
- Kowaltowski, A.J.; de Souza-Pinto, N.C.; Castilho, R.F.; Vercesi, A.E. Mitochondria and reactive oxygen species. Free Radic. Biol. Med. 2009, 47, 333–343. [Google Scholar] [CrossRef] [PubMed]
- Ramos, K.S.; Brundel, B.J.J.M. DNA damage, an innocent bystander in atrial fibrillation and other cardiovascular diseases? Front. Cardiovasc. Med. 2020, 7, 67. [Google Scholar] [CrossRef]
- Hassa, P.O.; Hottiger, M.O. The diverse biological roles of mammalian PARPS, a small but powerful family of poly-ADP-ribose polymerases. Front. Biosci. 2008, 13, 3046–3082. [Google Scholar] [CrossRef]
- Du, L.; Zhang, X.; Han, Y.Y.; Burke, N.A.; Kochanek, P.M.; Watkins, S.C.; Graham, S.H.; Carcillo, J.A.; Szabó, C.; Clark, R.S.B. Intra-mitochondrial poly (ADP-ribosylation) contributes to NAD+ depletion and cell death induced by oxidative stress. J. Biol. Chem. 2003, 278, 18426–18433. [Google Scholar] [CrossRef]
- Pillai, J.B.; Isbatan, A.; Imai, S.-i.; Gupta, M.P. Poly (ADP-ribose) polymerase-1-dependent cardiac myocyte cell death during heart failure is mediated by NAD+ depletion and reduced Sir2α deacetylase activity. J. Biol. Chem. 2005, 280, 43121–43130. [Google Scholar] [CrossRef] [PubMed]
- Pacher, P.; Szabo, C. Role of poly(ADP-ribose) polymerase 1 (PARP-1) in cardiovascular diseases: The therapeutic potential of PARP inhibitors. Cardiovasc. Drug Rev. 2007, 25, 235–260. [Google Scholar] [CrossRef]
- Ruiz-Meana, M.; Fernandez-Sanz, C.; Garcia-Dorado, D. The SR-mitochondria interaction: A new player in cardiac pathophysiology. Cardiovasc. Res. 2010, 88, 30–39. [Google Scholar] [CrossRef] [PubMed]
- Hubbert, C.; Guardiola, A.; Shao, R.; Kawaguchi, Y.; Ito, A.; Nixon, A.; Yoshida, M.; Wang, X.F.; Yao, T.P. HDAC6 is a microtubule-associated deacetylase. Nature 2002, 417, 455–458. [Google Scholar] [CrossRef] [PubMed]
- Haggarty, S.J.; Koeller, K.M.; Wong, J.C.; Grozinger, C.M.; Schreiber, S.L. Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc. Natl. Acad. Sci. USA 2003, 100, 4389–4394. [Google Scholar] [CrossRef] [PubMed]
- Cheng, K.C.; Cahill, D.S.; Kasai, H.; Nishimura, S.; Loeb, L.A. 8-Hydroxyguanine, an abundant form of oxidative DNA damage, causes GT and AC substitutions. J. Biol. Chem. 1992, 267, 166–172. [Google Scholar] [CrossRef]
- Li, J.; Zhang, D.; Ramos, K.S.; Baks, L.; Wiersma, M.; Lanters, E.A.H.; Bogers, A.; de Groot, N.M.S.; Brundel, B. Blood-based 8-hydroxy-2′-deoxyguanosine level: A potential diagnostic biomarker for atrial fibrillation. Heart Rhythm 2021, 18, 271–277. [Google Scholar] [CrossRef] [PubMed]
- Montaigne, D.; Marechal, X.; Lefebvre, P.; Modine, T.; Fayad, G.; Dehondt, H.; Hurt, C.; Coisne, A.; Koussa, M.; Remy-Jouet, I. Mitochondrial dysfunction as an arrhythmogenic substrate: A translational proof-of-concept study in patients with metabolic syndrome in whom post-operative atrial fibrillation develops. J. Am. Coll. Cardiol. 2013, 62, 1466–1473. [Google Scholar] [CrossRef]
- Toyama, K.; Yamabe, H.; Uemura, T.; Nagayoshi, Y.; Morihisa, K.; Koyama, J.; Kanazawa, H.; Hoshiyama, T.; Ogawa, H. Analysis of oxidative stress expressed by urinary level of 8-hydroxy-2′-deoxyguanosine and biopyrrin in atrial fibrillation: Effect of sinus rhythm restoration. Int. J. Cardiol. 2013, 168, 80–85. [Google Scholar] [CrossRef]
- Wiersma, M.; van Marion, D.; Bouman, E.J.; Li, J.; Zhang, D.; Ramos, K.S.; Lanters, E.A.H.; de Groot, N.; Brundel, B.J.J.M. Cell-free circulating mitochondrial DNA: A potential blood-based marker for atrial fibrillation. Cells 2020, 9, 1159. [Google Scholar] [CrossRef] [PubMed]
- Van Wijk, S.W.; Ramos, K.S.; Brundel, B. Cardioprotective Role of Heat Shock Proteins in Atrial Fibrillation: From Mechanism of Action to Therapeutic and Diagnostic Target. Int. J. Mol. Sci 2021, 22, 442. [Google Scholar] [CrossRef] [PubMed]
- Van Marion, D.M.; Hu, X.; Zhang, D.; Hoogstra-Berends, F.; Seerden, J.-P.G.; Loen, L.; Heeres, A.; Steen, H.; Henning, R.H.; Brundel, B.J.J.M. Screening of novel HSP-inducing compounds to conserve cardiomyocyte function in experimental atrial fibrillation. Drug Des. Dev. Ther. 2019, 13, 345. [Google Scholar] [CrossRef]
- Schäfler, A.E.; Kirmanoglou, K.; Balbach, J.; Pecher, P.; Hannekum, A.; Schumacher, B. The expression of heat shock protein 60 in myocardium of patients with chronic atrial fibrillation. Basic Res. Cardiol. 2002, 97, 258–261. [Google Scholar] [CrossRef]
- Schäfler, A.E.; Kirmanoglou, K.; Pecher, P.; Hannekum, A.; Schumacher, B. Overexpression of heat shock protein 60/10 in myocardium of patients with chronic atrial fibrillation. Ann. Thorac. Surg. 2002, 74, 767–770. [Google Scholar] [CrossRef]
- Decker, R.S.; Decker, M.L.; Nakamura, S.; Zhao, Y.-S.; Hedjbeli, S.; Harris, K.R.; Klocke, F.J. HSC73-tubulin complex formation during low-flow ischemia in the canine myocardium. Am. J. Physiol. Heart Circ. Physiol. 2002, 283, H1322–H1333. [Google Scholar] [CrossRef]
- Marion, D.; Lanters, E.A.H.; Ramos, K.S.; Li, J.; Wiersma, M.; Baks-te Bulte, L.; Jqm Muskens, A.; Boersma, E.; de Groot, N.; Brundel, B.J.J.M. Evaluating Serum Heat Shock Protein Levels as Novel Biomarkers for Atrial Fibrillation. Cells 2020, 9, 2105. [Google Scholar] [CrossRef]
- Yang, M.; Tan, H.; Cheng, L.; He, M.; Wei, Q.; Tanguay, R.M.; Wu, T. Expression of heat shock proteins in myocardium of patients with atrial fibrillation. Cell Stress Chaperones 2007, 12, 142. [Google Scholar] [CrossRef]
- Birk, A.V.; Liu, S.; Soong, Y.; Mills, W.; Singh, P.; Warren, J.D.; Seshan, S.V.; Pardee, J.D.; Szeto, H.H. The mitochondrial-targeted compound SS-31 re-energizes ischemic mitochondria by interacting with cardiolipin. J. Am. Soc. Nephrol. 2013, 24, 1250–1261. [Google Scholar] [CrossRef]
- De J García--Rivas, G.; Carvajal, K.; Correa, F.; Zazueta, C. Ru360, a specific mitochondrial calcium uptake inhibitor, improves cardiac post--ischaemic functional recovery in rats in vivo. Br. J. Pharmacol. 2006, 149, 829–837. [Google Scholar] [CrossRef] [PubMed]
- Xie, A.; Song, Z.; Liu, H.; Zhou, A.; Shi, G.; Wang, Q.; Gu, L.; Liu, M.; Xie, L.H.; Qu, Z.; et al. Mitochondrial Ca(2+) Influx Contributes to Arrhythmic Risk in Nonischemic Cardiomyopathy. J. Am. Heart Assoc. 2018, 7, 007805. [Google Scholar] [CrossRef]
- Machiraju, P.; Wang, X.; Sabouny, R.; Huang, J.; Zhao, T.; Iqbal, F.; King, M.; Prasher, D.; Lodha, A.; Jimenez-Tellez, N.; et al. SS-31 Peptide Reverses the Mitochondrial Fragmentation Present in Fibroblasts From Patients With DCMA, a Mitochondrial Cardiomyopathy. Front. Cardiovasc. Med. 2019, 6, 167. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Brundel, B.; Zhang, D. Preservation of the microtubule network to beat atrial fibrillation: Role of SR-mitochondrial contacts. Eur. Heart J. 2020, 41, 946. [Google Scholar] [CrossRef]
- Brundel, B.J.J.M.; Henning, R.H.; Ke, L.; van Gelder, I.C.; Crijns, H.J.G.M.; Kampinga, H.H. Heat shock protein upregulation protects against pacing-induced myolysis in HL-1 atrial myocytes and in human atrial fibrillation. J. Mol. Cell. Cardiol. 2006, 41, 555–562. [Google Scholar] [CrossRef]
- Brundel, B.J.J.M.; Shiroshita-Takeshita, A.; Qi, X.; Yeh, Y.-H.; Chartier, D.; van Gelder, I.C.; Henning, R.H.; Kampinga, H.H.; Nattel, S. Induction of heat shock response protects the heart against atrial fibrillation. Circ. Res. 2006, 99, 1394–1402. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Ke, L.; Mackovicova, K.; van der Want, J.J.; Sibon, O.C.; Tanguay, R.M.; Morrow, G.; Henning, R.H.; Kampinga, H.H.; Brundel, B.J. Effects of different small HSPB members on contractile dysfunction and structural changes in a Drosophila melanogaster model for Atrial Fibrillation. J. Mol. Cell Cardiol 2011, 51, 381–389. [Google Scholar] [CrossRef] [PubMed]
- Van Marion, D.M.S.; Dorsch, L.; Hoogstra-Berends, F.; Kakuchaya, T.; Bockeria, L.; de Groot, N.M.S.; Brundel, B. Oral geranylgeranylacetone treatment increases heat shock protein expression in human atrial tissue. Heart Rhythm 2020, 17, 115–122. [Google Scholar] [CrossRef]
- Matasic, D.S.; Brenner, C.; London, B. Emerging potential benefits of modulating NAD+ metabolism in cardiovascular disease. Am. J. Physiol. Heart Circ. Physiol. 2018, 314, H839–H852. [Google Scholar] [CrossRef]
- Hosseini, L.; Vafaee, M.S.; Mahmoudi, J.; Badalzadeh, R. Nicotinamide adenine dinucleotide emerges as a therapeutic target in aging and ischemic conditions. Biogerontology 2019, 20, 381–395. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Shen, Y.; Zhou, L.; Sangwung, P.; Fujioka, H.; Zhang, L.; Liao, X. Short-term administration of Nicotinamide Mononucleotide preserves cardiac mitochondrial homeostasis and prevents heart failure. J. Mol. Cell. Cardiol. 2017, 112, 64–73. [Google Scholar] [CrossRef]
- Xu, W.; Le Li, L.Z. NAD+ Metabolism as an Emerging Therapeutic Target for Cardiovascular Diseases Associated With Sudden Cardiac Death. Front. Physiol. 2020, 11, 901. [Google Scholar] [CrossRef] [PubMed]
- Bogan, K.L.; Brenner, C. Nicotinic acid, nicotinamide, and nicotinamide riboside: A molecular evaluation of NAD+ precursor vitamins in human nutrition. Annu. Rev. Nutr. 2008, 28, 115–130. [Google Scholar] [CrossRef] [PubMed]
- Trammell, S.A.J.; Schmidt, M.S.; Weidemann, B.J.; Redpath, P.; Jaksch, F.; Dellinger, R.W.; Li, Z.; Abel, E.D.; Migaud, M.E.; Brenner, C. Nicotinamide riboside is uniquely and orally bioavailable in mice and humans. Nat. Commun. 2016, 7, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Scalise, M.; Pochini, L.; Galluccio, M.; Console, L.; Indiveri, C. Glutamine Transport and Mitochondrial Metabolism in Cancer Cell Growth. Front. Oncol 2017, 7, 306. [Google Scholar] [CrossRef]
- Fan, J.; Kamphorst, J.J.; Mathew, R.; Chung, M.K.; White, E.; Shlomi, T.; Rabinowitz, J.D. Glutamine-driven oxidative phosphorylation is a major ATP source in transformed mammalian cells in both normoxia and hypoxia. Mol. Syst. Biol. 2013, 9, 712. [Google Scholar] [CrossRef]
- Wischmeyer, P.E. Glutamine and heat shock protein expression. Nutrition 2002, 18, 225–228. [Google Scholar] [CrossRef]
- Leite, J.S.M.; Cruzat, V.F.; Krause, M.; de Bittencourt, P.I.H. Physiological regulation of the heat shock response by glutamine: Implications for chronic low-grade inflammatory diseases in age-related conditions. Nutrire 2016, 41, 1–34. [Google Scholar] [CrossRef]
- Turkez, H.; Geyikoglu, F.; Yousef, M.I.; Celik, K.; Bakir, T.O. Ameliorative effect of supplementation with L-glutamine on oxidative stress, DNA damage, cell viability and hepatotoxicity induced by 2,3,7,8-tetrachlorodibenzo-p-dioxin in rat hepatocyte cultures. Cytotechnology 2012, 64, 687–699. [Google Scholar] [CrossRef][Green Version]
- Cao, Y.; Kennedy, R.; Klimberg, V.S. Glutamine protects against doxorubicin-induced cardiotoxicity. J. Surg. Res. 1999, 85, 178–182. [Google Scholar] [CrossRef]
- Starreveld, R.; Ramos, K.S.; Muskens, A.; Brundel, B.; de Groot, N.M.S. Daily Supplementation of L-Glutamine in Atrial Fibrillation Patients: The Effect on Heat Shock Proteins and Metabolites. Cells 2020, 9, 1729. [Google Scholar] [CrossRef]
- Bentinger, M.; Brismar, K.; Dallner, G. The antioxidant role of coenzyme Q. Mitochondrion 2007, 7, S41–S50. [Google Scholar] [CrossRef] [PubMed]
- Schmelzer, C.; Döring, F. Micronutrient special issue: Coenzyme Q10 requirements for DNA damage prevention. Mutat. Res. Fundam. Mol. Mech. Mutagenesis 2012, 733, 61–68. [Google Scholar] [CrossRef]
- Kumar, A.; Kaur, H.; Devi, P.; Mohan, V. Role of coenzyme Q10 (CoQ10) in cardiac disease, hypertension and Meniere-like syndrome. Pharmacol. Ther. 2009, 124, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Folkers, K.; Vadhanavikit, S.; Mortensen, S.A. Biochemical rationale and myocardial tissue data on the effective therapy of cardiomyopathy with coenzyme Q10. Proc. Natl. Acad. Sci. USA 1985, 82, 901–904. [Google Scholar] [CrossRef] [PubMed]
- Niklowitz, P.; Sonnenschein, A.; Janetzky, B.; Andler, W.; Menke, T. Enrichment of coenzyme Q10 in plasma and blood cells: Defense against oxidative damage. Int. J. Biol. Sci. 2007, 3, 257. [Google Scholar] [CrossRef]
- Migliore, L.; Molinu, S.; Naccarati, A.; Mancuso, M.; Rocchi, A.; Siciliano, G. Evaluation of cytogenetic and DNA damage in mitochondrial disease patients: Effects of coenzyme Q10 therapy. Mutagenesis 2004, 19, 43–49. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Q.; Kebbati, A.H.; Zhang, Y.; Tang, Y.; Okello, E.; Huang, C. Effect of coenzyme Q10 on the incidence of atrial fibrillation in patients with heart failure. J. Investig. Med. 2015, 63, 735–739. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Author | Outcome |
|---|---|
| Emelyanova et al. (2016) [32] | AF is associated with a selective reduction in the mitochondrial ETC activity and increased oxidative stress in humans which may contribute to the progression of the substrate for AF. |
| Hu et al. (2019) [77] | HSP inducer GGA, reverses contractile and structural remodeling via restoration of the microtubule network in experimental AF. |
| Ozcan et al. (2019) [78] | The pathogenesis of AF is associated with energy deficit and metabolic dysregulation in human and mice atria due to mitochondrial dysfunction. |
| Wiersma et al. (2019) [23] | Mitochondrial dysfunction is involved in AF promotion, and compounds directed at the conservation of mitochondrial function protect against contractile dysfunction in Drosophila models for AF. |
| Xiao et al. (2010) [79] | Taxol, a microtubule stabilizer, prevents AF in an in vitro AF model using rabbit hearts. The microtubule stabilizer most likely prevents AF by reducing the level of ROS. |
| Xie et al. (2015) [22] | Mitochondrial-derived ROS oxidize atrial RyR2 in human cardiomyocytes. This leads to increased intracellular Ca2+ leak and impaired mitochondrial function, contributing to the pathogenesis of AF. Interestingly, reduced mitochondrial ROS production attenuates SR Ca2+ leak and prevents AF. |
| Zhang et al. (2014) [7] | Patients with AF show increased HDAC6 activity. Activation of HDAC6 induces the degradation of the microtubule network and contractile dysfunction in experimental and human AF. Drugs directed at the conservation of the microtubule network attenuate AF in a dog model for AF. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pool, L.; Wijdeveld, L.F.J.M.; de Groot, N.M.S.; Brundel, B.J.J.M. The Role of Mitochondrial Dysfunction in Atrial Fibrillation: Translation to Druggable Target and Biomarker Discovery. Int. J. Mol. Sci. 2021, 22, 8463. https://doi.org/10.3390/ijms22168463
Pool L, Wijdeveld LFJM, de Groot NMS, Brundel BJJM. The Role of Mitochondrial Dysfunction in Atrial Fibrillation: Translation to Druggable Target and Biomarker Discovery. International Journal of Molecular Sciences. 2021; 22(16):8463. https://doi.org/10.3390/ijms22168463
Chicago/Turabian StylePool, Lisa, Leonoor F. J. M. Wijdeveld, Natasja M. S. de Groot, and Bianca J. J. M. Brundel. 2021. "The Role of Mitochondrial Dysfunction in Atrial Fibrillation: Translation to Druggable Target and Biomarker Discovery" International Journal of Molecular Sciences 22, no. 16: 8463. https://doi.org/10.3390/ijms22168463
APA StylePool, L., Wijdeveld, L. F. J. M., de Groot, N. M. S., & Brundel, B. J. J. M. (2021). The Role of Mitochondrial Dysfunction in Atrial Fibrillation: Translation to Druggable Target and Biomarker Discovery. International Journal of Molecular Sciences, 22(16), 8463. https://doi.org/10.3390/ijms22168463