
Kidney Response to Chemotherapy-Induced Heart Failure: mRNA Analysis in Normotensive and Ren-2 Transgenic Hypertensive Rats

 , , ,
, , ,
Abstract
:1. Introduction
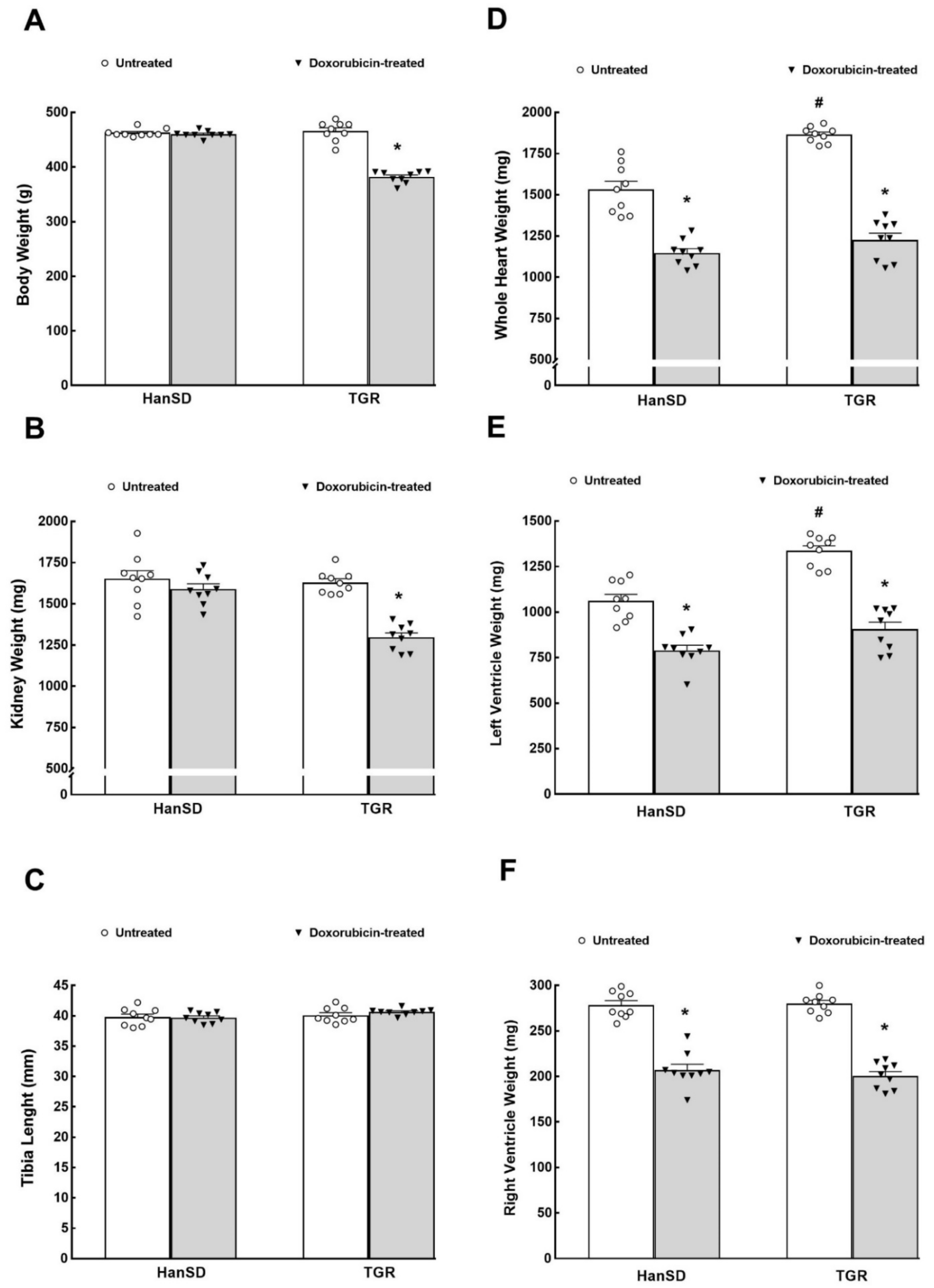
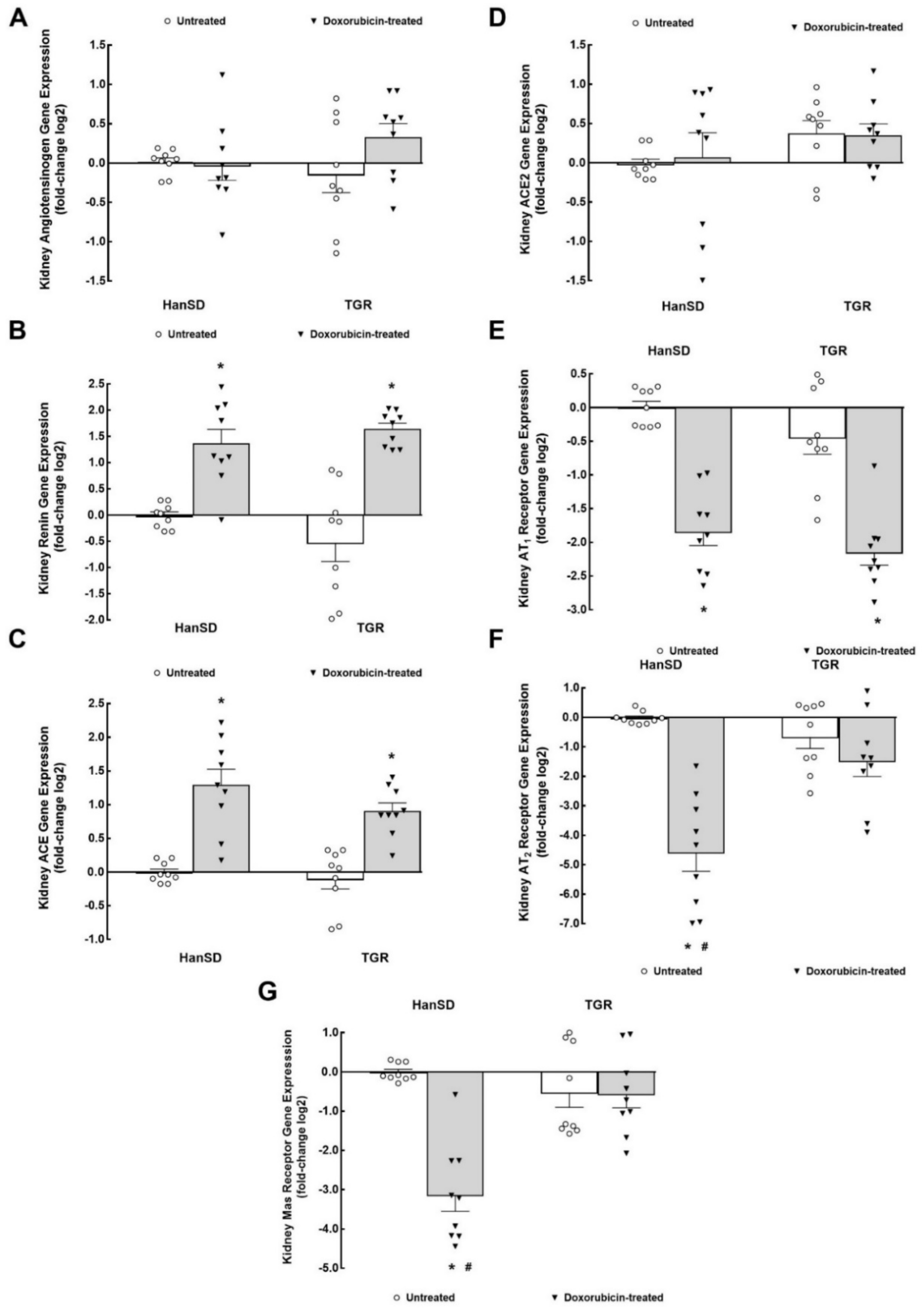
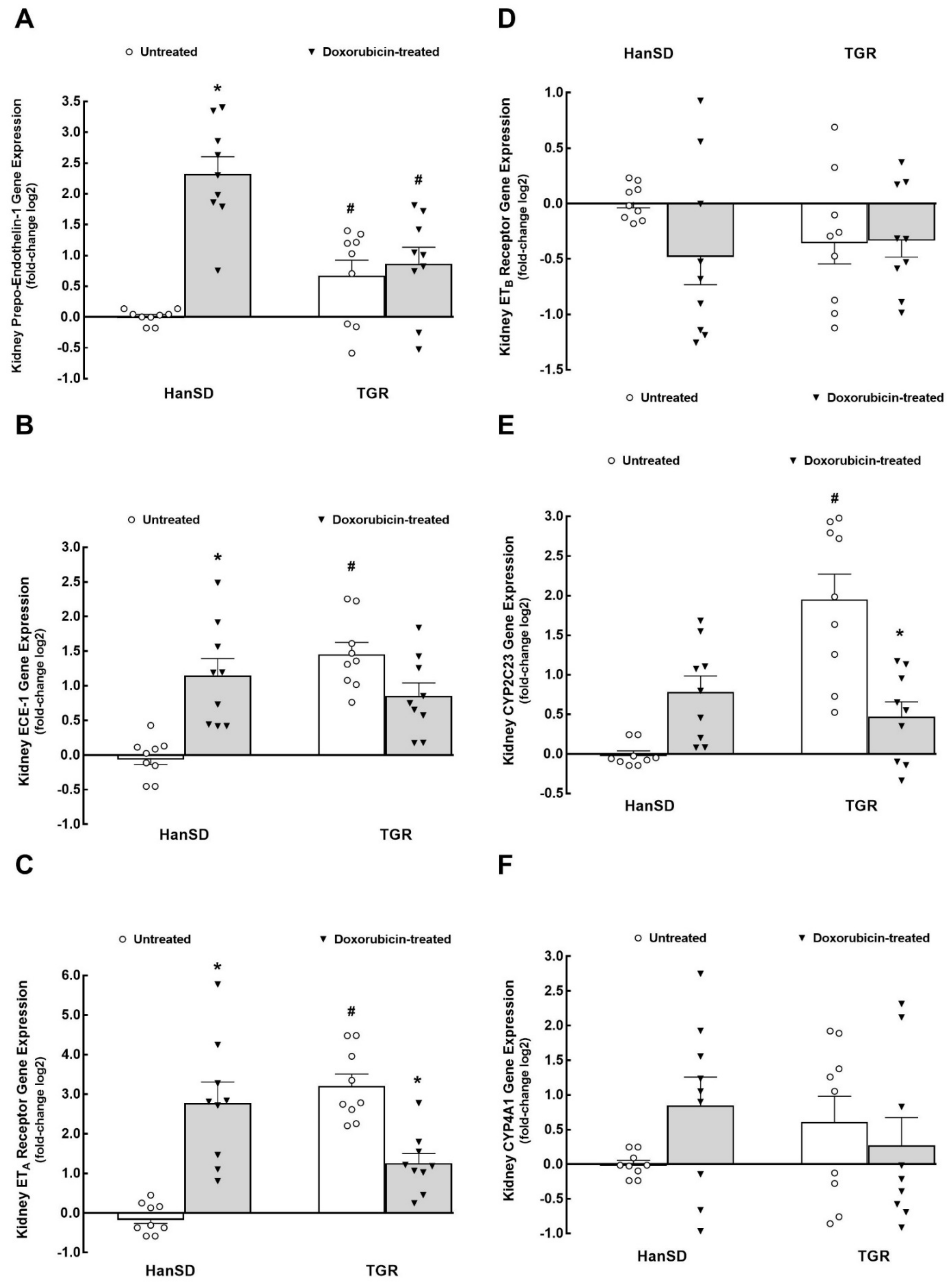
2. Results
3. Discussion
3.1. RAAS System
3.2. ET System
3.3. CYP-Derived Metabolites of Arachidonic Acid (AA)
3.4. Adrenergic System
3.5. Potential Clinical Implication and Limitations of the Study
3.6. Conclusions
4. Methods
4.1. Ethical Approval, Animals and HF Induction
4.2. Detailed Experimental Design
4.2.1. Assessment of the Effects of DOX on Kidney mRNA Expression
- HanSD rats + vehicle
- TGR + vehicle
- HanSD rats + DOX
- TGR + DOX
4.2.2. Relative Gene Expression Calculation
4.3. Statement of Ethics
4.4. Statistical Analysis
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| AA | arachidonic acid |
| ACEi | angiotensin converting enzyme inhibitor |
| ACE | angiotensin converting enzyme |
| ACE2 | angiotensin converting enzyme type 2 |
| ACF | aorto-caval fistula |
| ANG II | angiotensin II |
| ANG 1-7 | angiotensin 1-7 |
| AT1 | angiotensin II type 1 receptor |
| AT2 | angiotensin II type 2 receptor |
| CYP | cytochrome P-450 enzyme |
| CYP2C23 | cytochrome P-450 enzyme subfamily 2C23 |
| CYP4A1 | cytochrome P-450 enzyme subfamily 4A1 |
| DHETEs | dihydroxyeicosatrienoic acids (DHETEs) |
| DOX | doxorubicin |
| ECE-1 | endothelin-converting enzyme type 1 |
| EETs | epoxyeicosatrienoic acids |
| ETA | endothelin type A receptor |
| ETB | endothelin type B receptor |
| ET-1 | endothelin-1 |
| HanSD | normotensive, transgene-negative, Hannover Sprague–Dawley rats |
| HF | heart failure |
| HFrEF | heart failure with reduced ejection fraction |
| IKEM | Institute for Clinical and Experimental Medicine |
| MI | myocardial infarction |
| mRNA | messenger ribonucleic acid |
| NE | norepinephrine |
| RAAS | renin-angiotensin-aldosterone system |
| ROS | reactive oxygen species |
| RSNA | renal sympathetic nerve activity |
| sEH | soluble epoxide hydrolase |
| TGR | Ren2 renin transgenic, hypertensive rats |
| 20-HETE | 20-hydroxyeicosatrienoic acid |
References
- Ponikowski, P.; Voors, A.A.; Anker, S.D.; Bueno, H.; Cleland, J.G.F.; Coats, A.J.S.; Falk, V.; González-Juanatey, J.R.; Harjola, V.-P.; Jankowska, E.A.; et al. 2016 ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure: The Task Force for the diagnosis and treatment of acute and chronic heart failure of the European Society of Cardiology (ESC). Developed with the special contribution of the Heart Failure Association (HFA) of the ESC. Eur. Heart J. 2016, 37, 2129–2200. [Google Scholar] [PubMed]
- Benjamin, E.J.; Blaha, M.J.; Chiuve, S.E. Heart disease and stroke statistics-2017 update: A report from the American Heart Association. Circulation 2017, 135, e146–e603. [Google Scholar] [CrossRef] [PubMed]
- Bulluck, H.; Yellon, D.M.; Hausenloy, D.J. Reducing myocardial infarct size: Challenges and future opportunities. Heart 2016, 102, 341–348. [Google Scholar] [CrossRef] [PubMed]
- Kassi, M.; Hannawi, B.; Trachtenberg, B. Recent advances in heart failure. Curr. Opin. Cardiol. 2018, 33, 249–256. [Google Scholar] [CrossRef] [PubMed]
- Mullens, W.; Verbrugge, F.H.; Nijst, P.; Tang, W.H.W. Renal sodium avidity in heart failure: From pathophysiology to treatment strategies. Eur. Heart J. 2017, 38, 1872–1882. [Google Scholar] [CrossRef]
- Mullens, W.; Damman, K.; Testani, J.M.; Martens, P.; Mueller, C.; Lassus, J.; Tang, W.H.; Skuri, H.; Verbrugge, F.H.; Orso, F.; et al. Evaluation of kidney function throughout the heart failure trajectory—A position statement from the Heart Failure Association of the European Society of Cardiology. Eur. J. Heart Fail. 2020, 22, 584–603. [Google Scholar] [CrossRef]
- Rangawwami, J.; Bhalla, V.; Blair, J.E.A.; Chang, T.I.; Costa, S.; Lentine, K.L.; Lerma, E.V.; Mezeu, K.; Molitch, M.; Mullens, W.; et al. American Heart Asssociation Council on the Kidney in Cardiovascular Disease and Council on Clinical Cardiology. Cardiorenal syndrome: Classification, pathophysiology, diagnosis, and treatment strategies. A scientific statement from the American Heart Association. Circulation 2019, 139, e840–e878. [Google Scholar]
- Khayyat-Kholghi, M.; Oparil, S.; Davis, B.R.; Tereshchenko, L.G. Worsening kidney function is the major mechanism of heart failure in hypertension. The ALLHAT study. JACC Heart Fail. 2021, 9, 100–111. [Google Scholar] [CrossRef]
- Houser, S.R.; Margulies, K.B.; Murphy, A.M.; Spinale, F.G.; Francis, G.S.; Prabhu, S.D.; Rockman, H.A.; Kass, D.A.; Molkentin, J.D.; Sussman, M.A.; et al. Animal models of heart failure: A scientific statement from the American Heart Association. Circ. Res. 2012, 111, 131–150. [Google Scholar] [CrossRef] [Green Version]
- Riehle, C.; Bauersachs, J. Small animal models of heart failure. Cardiovas Res. 2019, 115, 1838–1849. [Google Scholar] [CrossRef]
- Abassi, Z.; Goltsmna, I.; Karram, T.; Winaver, J.; Horrman, A. Aortocaval fistula in rat: A unique model of volume-overload congestive heart failure and cardiac hypertrophy. J. Biomed. Biotechnol. 2011, 2011, 729497. [Google Scholar] [CrossRef] [Green Version]
- Honetschlagerová, Z.; Gawrys, O.; Jíchová, Š.; Škaroupková, P.; Kikerlová, S.; Vaňourková, Z.; Husková, Z.; Melenovský, V.; Kompanowska-Jezierska, E.; Sadowski, J.; et al. Renal sympathetic denervation attenuates congestive heart failure in angiotensin II-dependent hypertension: Studies with Ren-2 transgenic hypertensive rats with aorto-caval fistula. Kidney Blood Press. Res. 2021, 46, 95–113. [Google Scholar] [CrossRef]
- Turcani, M.; Rupp, H. Heart failure development in rats with ascending aortic constriction and angiotensin-converting enzyme inhibition. Br. J. Pharmacol. 2000, 130, 1671–1677. [Google Scholar] [CrossRef]
- Pfeffer, M.A.; Pfeffer, J.M.; Steinberg, C.; Finn, P. Survival after an experimental myocardial infarction: Beneficial effects of long-term therapy with captopril. Circulation 1985, 72, 406–412. [Google Scholar] [CrossRef] [Green Version]
- Pfeffer, J.M. Progressive ventricular dilatation in experimental myocardial infarction and its attenuation by angiotensin-converting enzyme inhibition. Am. J. Cardiol. 1991, 68, 17D–25D. [Google Scholar] [CrossRef]
- CONSENSUS Trial Study Group. Effects of enalapril on mortality in severe congestive heart failure. Results of the Cooperative North Scandinavian Enalapril Survival Study (CONSENSUS). N. Engl. J. Med. 1987, 316, 1429–1435. [Google Scholar] [CrossRef] [PubMed]
- SOLVD Investigators; Yusuf, S.; Pitt, B.; Davis, C.E.; Hood, W.B., Jr.; Cohn, J.N. Effects of enalapril on mortality and the development of heart failure in asymptomatic patients with reduced left ventricular ejection fraction. N. Engl. J. Med. 1992, 327, 658–691. [Google Scholar]
- Trachtenberg, B.H. Future Directions in Cardio-Oncology. Methodist Debakey Cardiovasc. J. 2019, 15, 300–302. [Google Scholar] [PubMed]
- Lenneman, C.G.; Sawyer, D.B. Cardio-Oncology. An updated on cardiotoxicity of cancer-related treatment. Circ. Res. 2016, 118, 1008–1020. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bansal, N.; Blanco, J.G.; Sharma, U.C.; Pokharel, S.; Shisler, S.; Lipshult, S.E. Cardiovascular diseases in survivors of childhood cancer. Cancer Metastasis Rev. 2020, 39, 55–68. [Google Scholar] [CrossRef]
- Moslehi, J.; Zhang, Q.; Moore, K.J. Crosstalk between the heart and cancer. Beyond drug toxicity. Circulation 2020, 142, 684–687. [Google Scholar] [CrossRef] [PubMed]
- Zamorano, J.L.; Lancellotti, P.; Munoz, R.D.; Aboyans, V.; Asteggiano, R.; Galderisi, M.; Habib, G.; Lenihan, D.J.; Lip, G.Y.H.; Lyon, A.R.; et al. 2016 ESC position paper on cancer treatments and cardiovascular toxicity developed under auspices of the ESC Committee for Practice Guildelines. Eur. Heart J. 2016, 37, 2768–2801. [Google Scholar] [CrossRef]
- Hassen, L.J.; Lenihan, D.J.; Baliga, R.R. Hypertension in the cardio-oncology clinic. Heart Fail. Clin. 2019, 15, 487–495. [Google Scholar] [CrossRef]
- Kalyanaraman, B. Teaching the basic of the mechanism of doxorubicin-induced cardiotoxicity: Have we been barking up the wrong tree? Redox Biol. 2020, 29, 101394. [Google Scholar] [CrossRef]
- Wallace, K.B.; Sardao, V.A.; Oliverira, P.J. Mitochondrial determinants of doxorubicin-induced cardiomyopathy. Circ. Res. 2020, 126, 926–941. [Google Scholar] [CrossRef]
- Jeyaprakash, P.; Bmed, M.D.; Sukhmandeep, S.; Ellenberger, K.; Sivapathan, S.; Pathan, F.; Negishi, K. Cardiotoxic effect of modern anthracyclines dosing on left ventricular ejection fraction: A systematic review and meta-analysis of placebo arms from randomized controlled trials. J. Am. Heart Assoc. 2021, 10, e018802. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, T.; Tanimoto, T.; Petrov, A.D.; Ishikawa, K.; Strauss, H.W.; Narula, J. Rat model of cardiotoxic drug-induced cardiomyopathy. In Experimental Models of Cardiovascular Diseases: Methods and Protocols; Ishikawa, K., Ed.; Springer + Business Media, Part of Springer Nature Humana Press: New York, NY, USA, 2018; Volume 1816, pp. 221–232. [Google Scholar]
- Hahn, V.S.; Zhang, K.W.; Sun, L.; Narayan, V.; Lenihan, D.J.; Ky, B. Heart failure with target cancer therapies. Mechanisms and Cardioprotection. Circ. Res. 2021, 128, 1576–1593. [Google Scholar] [CrossRef] [PubMed]
- Medeiros-Lima, D.J.M.; Carvalho, J.J.; Tibirica, E.; Borges, J.P.; Matsuura, C. Time course of cardiomyopathy induced by doxorubicin in rats. Pharmacol. Rep. 2019, 71, 583–590. [Google Scholar] [CrossRef] [PubMed]
- Babaei, H.; Razmaraii, N.; Assadnassab, G.H.; Mohajjel Nayebi, A.; Azarmi, Y.; Mohammadnejad, D.; Azami, A. Ultrastructural and echocardiographic assessment of chronic doxorubicin-induced cardiotoxicity in rats. Arch. Razi Inst. 2020, 75, 55–62. [Google Scholar] [PubMed]
- Ching, C.; Gustafson, D.; Thavendiranathan, P.; Fisch, J.E. Cancer therapy-related cardiac dysfunction: Is endothelial dysfunction at the heart of the matter? Clin. Sci. 2021, 135, 1467–1503. [Google Scholar] [CrossRef]
- Asnani, A.; Moslehi, J.J.; Adhikari, B.B.; Baik, A.H.; Beyer, A.M.; de Boer, R.A.; Ghigo, A.; Grumbach, I.M.; Jain, S.; Zhu, H. Preclinical models of cancer therapy-associated cardiovascular toxicity. A scientific statement from the American Heart Association. Circ. Res. 2021, 129, e21–e34. [Google Scholar] [CrossRef]
- Husková, Z.; Kramer, H.J.; Vaňourková, Z.; Červenka, L. Effects of changes in sodium balance on plasma and kidney angiotensin II levels in anesthetized and conscious Ren-2 transgenic rats. J. Hypertens. 2006, 24, 517–527. [Google Scholar] [CrossRef] [PubMed]
- Weinberg, L.E.; Singal, P.K. Refractory heart failure and age-related differences in adriamycin-induced myocardial changes in rats. Can. J. Physiol. Pharmacol. 1987, 65, 1957–1965. [Google Scholar] [CrossRef]
- Kala, P.; Bartušková, H.; Piťha, J.; Vaňourková, Z.; Kikerlová, S.; Jíchová, Š.; Melenovský, V.; Hošková, L.; Veselka, J.; Kompanowska-Jezierska, E.; et al. Deleterious effects of hyperactivity of the renin-angiotensin system and hypertension on the course of chemotherapy-induced heart failure after doxorubicin administration: A study in Ren-2 transgenic rats. Int. J. Mol. Sci. 2020, 21, 9337. [Google Scholar] [CrossRef]
- Savira, F.; Magaye, R.; Liew, D.; Reid, C.; Kelly, D.J.; Kompa, A.R.; Sangaralingham, S.J.; Burnet, J.C., Jr.; Kaye, D.; Wang, B.H. Cardiorenal syndrome: Multi-organ dysfunction involving the heart, kidney and vasculature. Br. J. Pharmacol. 2020, 177, 2906–2922. [Google Scholar] [CrossRef]
- Schirone, L.; Forte, M.; Palmerio, S.; Yee, S.; Nocella, C.; Angelini, F.; Pagano, F.; Schiavon, S.; Bordin, A.; Carrizzo, A.; et al. A review of the molecular mechanisms underlying the development and progression of cardiac remodeling. Oxid. Med. Cell. Longev. 2017, 2017, 3920195. [Google Scholar] [CrossRef]
- Mishra, S.; Kass, D.A. Cellular and molecular pathobiology of heart failure with preserved ejection fraction. Natl. Rev. Cardiol. 2021, 18, 400–423. [Google Scholar] [CrossRef]
- Burkhoff, D.; Topkara, V.K.; Sayer, G.; Uriel, N. Reverse remodeling with left ventricular assist devices. Circ. Res. 2021, 128, 1594–1612. [Google Scholar] [CrossRef] [PubMed]
- Driesen, R.B.; Verheyen, F.K.; Debie, W.; Blaauw, E.; Babiker, F.A.; Cornelussen, R.N.M.; Ausma, J.; Lenders, M.-H.; Borges, M.; Chaponnier, C.; et al. Re-expression of alpha skeletal actin as a marker for dedifferentiation in cardiac pathologies. J. Cell. Mol. Med. 2009, 13, 896–908. [Google Scholar] [CrossRef] [Green Version]
- Kala, P.; Sedláková, L.; Škaroupková, P.; Kopkan, L.; Vaňourková, Z.; Táborský, M.; Nishiyama, A.; Hwang, S.H.; Hammock, B.D.; Sadowski, J.; et al. Effects of angiotensin-converting enzyme blockade, alone or combined with blockade of soluble epoxide hydrolase, on the course of congestive heart failure and occurrence of renal dysfunction in Ren-2 transgenic hypertensive rats with aorto-caval fistula. Physiol. Res. 2018, 67, 401–415. [Google Scholar] [CrossRef]
- Dube, P.; Weber, K.T. Congestive heart failure: Pathophysiologic consequences of neurohormonal activation and the potential for recovery: Part I. Am. J. Med. Sci. 2011, 342, 348–351. [Google Scholar] [CrossRef] [PubMed]
- Packer, M.; McMurray, J.J.V. Importance of endogenous compensatory vasoactive peptides in broadening the effects of inhibitors of the renin-angiotensin system for the treatment of heart failure. Lancet 2017, 389, 1831–1840. [Google Scholar] [CrossRef]
- Hartupee, J.; Mann, D.L. Neurohormonal activation in heart failure with reduced ejection fraction. Nat. Rev. Cardiol. 2017, 14, 30–38. [Google Scholar] [CrossRef] [Green Version]
- Castrop, H.; Hocherl, K.; Kurtz, A.; Schweda, F.; Todorov, V.; Wagner, C. Physiology of kidney renin. Physiol. Rev. 2010, 90, 607–673. [Google Scholar] [CrossRef] [PubMed]
- Sparks, M.A.; Crowley, S.D.; Gurley, S.B.; Mirotsou, M.; Coffman, T.M. Clasical renin-angiotensin system in kidney physiology. Compr. Physiol. 2014, 4, 1201–1228. [Google Scholar]
- Ocaranza, M.P.; Riquelme, J.A.; Garcia, L.; Jalil, J.E.; Chiong, M.; Santos, R.A.S.; Lavandero, S. Counter-regulatory renin-angiotensin system in cardiovascular disease. Nat. Rev. Cardiol. 2020, 17, 116–129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davenport, A.P.; Hyndman, K.A.; Dhaun, N.; Southan, C.; Kohan, D.E.; Pollock, J.S.; Pollock, D.M.; Webb, D.J.; Maguire, J.J. Endothelin. Pharmacol. Rev. 2016, 68, 357–418. [Google Scholar] [CrossRef] [Green Version]
- Vaneckova, I.; Kramer, H.J.; Bäcker, A.; Schejbalová, S.; Vernerová, Z.; Eis, V.; Opočenský, M.; Dvořák, P.; Červenka, L. Early-onset endothelin receptor blockade in hypertensive heterozygous Ren-2 rats. Vasc. Pharmacol. 2006, 45, 163–170. [Google Scholar] [CrossRef] [PubMed]
- Vernerová, Z.; Kramer, H.J.; Bäcker, A.; Červenka, L.; Opočenský, M.; Husková, Z.; Vaňourková, Z.; Eis, V.; Čertíková Chábová, V.; Tesař, V.; et al. Late-onset endothelin receptor blockade in hypertensive heterozygous Ren-2 transgenic rats. Vasc. Pharmacol. 2008, 48, 165–173. [Google Scholar] [CrossRef]
- Sedláková, L.; Čertíková Chábová, V.; Doleželová, Š.; Škaroupková, P.; Kopkan, L.; Husková, Z.; Červenková, L.; Kikerlová, S.; Vaněčková, I.; Sadowski, J.; et al. Renin-angiotensin system blockade alone or combined with ETA receptor blockade: Effects on the course of chronic kidney disease in 5/6 nephrectomized Ren-2 transgenic hypertensive rats. Clin. Exp. Hypertens. 2017, 39, 183–195. [Google Scholar] [CrossRef]
- Miyauchi, T.; Sakai, S. Endothelin and the heart in health and diseases. Peptides 2019, 111, 77–88. [Google Scholar] [CrossRef] [PubMed]
- El-Sherbeni, A.A.; Aboutabl, M.E.; Zordoky, B.N.M.; Anwa-Mohamed, A.; El-Kadi, A.O.S. Determination of the dominant arachidonic acid cytochrome P450 monooxygenase in rat heart, lung, kidney and liver: Protein expression and metabolic kinetics. AAPS J. 2013, 15, 112–122. [Google Scholar] [CrossRef] [Green Version]
- Lai, J.; Chen, C. The role of epoxyeicosatrienoic acids in cardiac remodeling. Front. Physiol. 2021, 12, 642470. [Google Scholar] [CrossRef]
- Imig, J.D. Epoxyeicosanoids in Hypertension. Physiol. Res. 2019, 68, 695–704. [Google Scholar] [CrossRef] [PubMed]
- Červenka, L.; Melenovský, V.; Husková, Z.; Škaroupková, P.; Nishiyama, A.; Sadowski, J. Inhibition of soluble epoxide hydrolase counteracts the development of renal dysfunction and progression of congestive heart failure in Ren-2 transgenic hypertensive rats with aorto-caval fistula. Clin. Exp. Pharmacol. Physiol. 2015, 42, 795–807. [Google Scholar] [CrossRef]
- Červenka, L.; Husková, Z.; Kopkan, L.; Kikerlová, S.; Sedláková, L.; Vaňourková, Z.; Alánová, P.; Kolář, F.; Hammock, B.D.; Hwang, S.H.; et al. Two pharmacological epoxyeicosatrienoic acid-enhancing therapies are effectively antihypertensive and reduce the severity of ischemic arrhythmias in rats with angiotensin II-dependent hypertension. J. Hypertens. 2018, 36, 1326–1341. [Google Scholar] [CrossRef]
- Alsaad, A.M.S.; Zordoky, B.N.M.; Tse, M.M.Y.; El-Kadi, A.O.S. Role of cytochrome 450-mediated arachidonic acid metabolites in the pathogenesis of cardiac hypertrophy. Drug Metab. Rev. 2013, 45, 173–195. [Google Scholar] [CrossRef] [PubMed]
- Roman, R.J.; Fan, F. 20-HETE, hypertension and beyond. Hypertension 2018, 72, 12–18. [Google Scholar] [CrossRef]
- DiBona, G.F.; Esler, M. Translation medicine: The antihypertensive effect of renal denervation. Am. J. Physiol. 2010, 298, R245–R253. [Google Scholar]
- Schmieder, R.E. Renal denervation: Where do we stand and what is the relevance to the nephrologist? Nephrol. Dial. Transplant. 2020. [Google Scholar] [CrossRef] [PubMed]
- Jang, H.-S.; Kim, J.; Padanilam, B.J. Renal sympathetic nerve activation via α2-adrenergic receptors in chronic kidney disease progression. Kidney Res. Clin. Pract. 2019, 38, 6–14. [Google Scholar] [CrossRef] [Green Version]
- Insel, P.A.; Snavely, M.D. Catecholamines and the kidney: Receptors and renal function. Annu. Rev. Physiol. 1981, 43, 625–636. [Google Scholar] [CrossRef] [PubMed]
- Karlstaedt, A.; Zhang, X.; Vitrac, H.; Harmancey, R.; Vasauez, H.; Wang, J.H.; Goodell, M.A.; Taegtmeyer, H. Oncometabolite d-2-hydroyglutarated impairs α-ketoglutarate dehydrogenase and contractile function in rodent heart. Proc. Natl. Acad. Sci. USA 2016, 113, 10436–10441. [Google Scholar] [CrossRef] [Green Version]
- Meijers, W.C.; Maglione, M.; Bakker, S.J.L.; Oberhuber, R.; Kieneker, L.M.; de Jong, S.; Haubner, B.J.; Nagengast, W.B.; Lyon, A.R.; van Veldhuisen, D.J.; et al. Heart failure stimulates tumor growth by circulating factors. Circulation 2018, 138, 678–691. [Google Scholar] [CrossRef]
- Giebisch, G.H. A long affair with renal tubules. Annu. Rev. Physiol. 2011, 73, 1–28. [Google Scholar] [CrossRef] [PubMed]
- Ayla, S.; Seckin, I.; Tanriverdi, G.; Cengiz, M.; Eser, M.; Soner, B.C.; Oktem, G. Doxorubicin induced nephrotoxicity: Protective effect of nicotinamide. Int. J. Cell Biol. 2011, 2011, 390238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xiang, C.; Yan, Y.; Zhang, D. Alleviation of the doxorubicin-induced nephrotoxicity by fasudil in vivo and in vitro. J. Pharmacol. Sci. 2021, 145, 6–15. [Google Scholar] [CrossRef]
- Mullins, J.J.; Peters, J.; Ganten, D. Fulminant hypertension in transgenic rats harbouring the mouse Ren-2 gene. Nature 1990, 344, 541–544. [Google Scholar] [CrossRef]
- Jíchová, Š.; Doleželová, Š.; Kopkan, L.; Kompanowska-Jezierska, E.; Sadowski, J.; Červenka, L. Fenofibrate attenuates malignant hypertension by suppression of the renin-angiotensin system: A study in Cyp1a1-Ren-2 transgenic rats. Am. J. Med. Sci. 2016, 352, 618–630. [Google Scholar] [CrossRef]
- Sporková, A.; Čertíková Chábová, V.; Doleželová, Š.; Jíchová, Š.; Vaňourková, Z.; Kompanowska-Jezierska, E.; Sadowski, J.; Maxová, H.; Červenka, L. Fenofibrate attenuates hypertension in Goldblatt hypertensive rats: Role of 20-hydroxyeicosatrienoic acid in the nonclipped kidney. Am. J. Med. Sci. 2017, 353, 568–579. [Google Scholar] [CrossRef] [PubMed]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2−ΔΔCT method. Methods 2001, 25, 402–408. [Google Scholar] [CrossRef]
- Bas, A.; Forsberg, G.; Hammarstrom, S.; Hammarstrom, M.L. Utility of the housekeeping genes 18S rRNA, beta-actin and glyceraldehyde-3-phosphate-dehydrogenase for normalization in real-time quantitative reverse transcriptase-polymerase chain reaction analysis of gene expression in human T lymphocytes. Scand. J. Immunol. 2004, 59, 566–573. [Google Scholar] [CrossRef] [PubMed]
- Bustin, S.A.; Benes, V.; Garson, J.A.; Hellemans, J.; Hugget, J.; Kubista, M.; Mueller, R.; Nolan, T.; Pfaffl, M.W.; Shipley, G.L.; et al. The MIQE Guidelines: Minimum information for publication of quantitative real-time PCR experiments. Clin. Chem. 2009, 55, 611–622. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.-M.; Yan, D.; Liu, Z.-F.; Hu, S.-Z.; Yan, S.-H.; He, X.-W. Density distribution of gene expression profiles and evaluation of using maximal information coefficient to identify differentially expressed genes. PLoS ONE 2019, 14, e0219551. [Google Scholar] [CrossRef] [PubMed] [Green Version]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID Assay | Gene Name | Abbreviation |
|---|---|---|
| Rn00664637_g1 | natriuretic peptid A | Nppa |
| Rn01488781_g1 | myosin, heavy chain 6, cardiac muscle, alpha | Myh6 |
| Rn01488777_g1 | myosin, heavy chain 7, cardiac muscle, beta | Myh7 |
| Rn01426628_g1 | actin, alpha 1, skeletal muscle | Acta1 |
| Rn00568762_m1 | ATPase, Ca++ transporting, cardiac muscle, slow twitch 2 | SERCA |
| Rn01463848_m1 | collagen, type I, alpha 1 | Col1a1 |
| Rn01437681_m1 | collagen, type III, alpha 1 | Col3a1 |
| Rn00824536_s1 | adrenoceptor beta 1 | Adrb1 |
| Hs99999901_s1 | 18S rRNA ribosomal subunit | 18s rRNA |
| Rn00572010_m1 | transforming growth factor, beta 1 | Tgfb1 |
| Rn01434045_m1 | phospholamban | Pln |
| Rn00572711_m1 | Interleukin-6 | IL-6 |
| ID Assay Number | Gene Name | Abbreviation |
|---|---|---|
| Rn00561847_m1 | renin | Ren |
| Rn00561094_m1 | angiotensin I converting enzyme | Ace |
| Rn01416293_m1 | angiotensin I converting enzyme 2 | Ace2 |
| Rn00593114_m1 | angiotensinogen (serpin peptidase inhibitor, clade A, member 8) | Agt |
| Rn02758772_s1 | angiotensin II receptor, type 1a | Agtr1a |
| Rn00562673_s1 | MAS1 proto-oncogene, G protein-coupled receptor | Mas1 |
| Rn00561129_m1 | prepro-Endothelin 1 | Edn1 |
| Rn00585943_m1 | endothelin converting enzyme 1 | Ece1 |
| Rn00561137_m1 | endothelin receptor type A | Ednra |
| Rn00569139_m1 | endothelin receptor type B | Ednrb |
| Hs99999901_s1 | 18S rRNA ribosomal subunit | 18s rRNA |
| Rn00598510_m1 | cytochrome P450, family 4, subfamily a, polypeptide 1 | Cyp4a1 |
| Rn01413752_m1 | cytochrome P450, family 2, subfamily c, polypeptide 23 | Cyp2c23 |
| Rn00567876_m1 | adrenoceptor alpha 1A | Adra1a |
| Rn01471343_m1 | adrenoceptor alpha 1B | Adra1b |
| Rn00562488_s1 | adrenoceptor alpha 2A | Adra2a |
| Rn00593312_s1 | adrenoceptor alpha 2B | Adra2b |
| Rn00593341_s1 | adrenoceptor alpha 2C | Adra2c |
| Rn00824536_s1 | adrenoceptor beta 1 | Adrb1 |
| Rn00560650_s1 | adrenoceptor beta 2, surface | Adrb2 |
| Rn00560677_s1 | angiotensin II receptor, type 2 | Agtr2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jíchová, Š.; Gawryś, O.; Kompanowska-Jezierska, E.; Sadowski, J.; Melenovský, V.; Hošková, L.; Červenka, L.; Kala, P.; Veselka, J.; Čertíková Chábová, V. Kidney Response to Chemotherapy-Induced Heart Failure: mRNA Analysis in Normotensive and Ren-2 Transgenic Hypertensive Rats. Int. J. Mol. Sci. 2021, 22, 8475. https://doi.org/10.3390/ijms22168475
Jíchová Š, Gawryś O, Kompanowska-Jezierska E, Sadowski J, Melenovský V, Hošková L, Červenka L, Kala P, Veselka J, Čertíková Chábová V. Kidney Response to Chemotherapy-Induced Heart Failure: mRNA Analysis in Normotensive and Ren-2 Transgenic Hypertensive Rats. International Journal of Molecular Sciences. 2021; 22(16):8475. https://doi.org/10.3390/ijms22168475
Chicago/Turabian StyleJíchová, Šárka, Olga Gawryś, Elżbieta Kompanowska-Jezierska, Janusz Sadowski, Vojtěch Melenovský, Lenka Hošková, Luděk Červenka, Petr Kala, Josef Veselka, and Věra Čertíková Chábová. 2021. "Kidney Response to Chemotherapy-Induced Heart Failure: mRNA Analysis in Normotensive and Ren-2 Transgenic Hypertensive Rats" International Journal of Molecular Sciences 22, no. 16: 8475. https://doi.org/10.3390/ijms22168475
APA StyleJíchová, Š., Gawryś, O., Kompanowska-Jezierska, E., Sadowski, J., Melenovský, V., Hošková, L., Červenka, L., Kala, P., Veselka, J., & Čertíková Chábová, V. (2021). Kidney Response to Chemotherapy-Induced Heart Failure: mRNA Analysis in Normotensive and Ren-2 Transgenic Hypertensive Rats. International Journal of Molecular Sciences, 22(16), 8475. https://doi.org/10.3390/ijms22168475