
Highlighting Immune System and Stress in Major Depressive Disorder, Parkinson’s, and Alzheimer’s Diseases, with a Connection with Serotonin



{kind=link}
{kind=link}
{kind=link}
Abstract
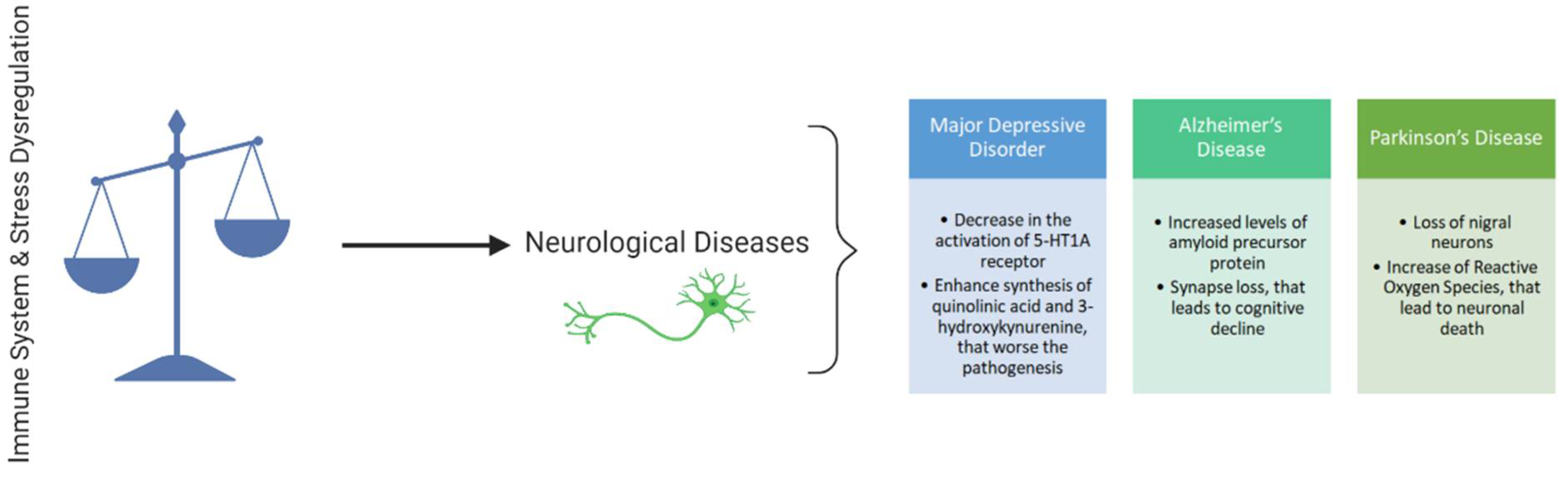
:1. Introduction
2. Stress Influence
2.1. In Major Depressive Disorder
2.2. In Alzheimer’s Disease
2.3. In Parkinson’s Disease
3. Immune System Involvement
3.1. In Major Depressive Disorder
3.2. In Alzheimer’s Disease
3.3. In Parkinson’s Disease
4. An Interplay between Stress and Immune System
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mora, F.; Segovia, G.; del Arco, A.; de Blas, M.; Garrido, P. Stress, neurotransmitters, corticosterone and body–brain integration. Brain Res. 2012, 1476, 71–85. [Google Scholar] [CrossRef] [PubMed]
- Wong, M.L.; Lewis, M.; Licinio, J. 2.2 Translational Research in Endocrinology and Neuroimmunology Applied to Depression. In Biomedical Chemistry; De Gruyter Open: Berlin, Germany, 2015; pp. 119–131. ISBN 9783110468755. [Google Scholar]
- Vaz, R.P.; Cardoso, A.; Serrão, P.; Pereira, P.A.; Madeira, M.D. Chronic stress leads to long-lasting deficits in olfactory-guided behaviors, and to neuroplastic changes in the nucleus of the lateral olfactory tract. Horm. Behav. 2018, 98, 130–144. [Google Scholar] [CrossRef] [PubMed]
- Hemmerle, A.M.; Herman, J.P.; Seroogy, K.B. Stress, depression and Parkinson’s disease. Exp. Neurol. 2012, 233, 79–86. [Google Scholar] [CrossRef] [Green Version]
- Den Buuse, M.; Hale, M.W. Serotonin in stress. In Stress: Physiology, Biochemistry, and Pathology Handbook of Stress Series, Volume 3; Elsevier: Amsterdam, The Netherlands, 2019; pp. 115–123. ISBN 9780128131466. [Google Scholar]
- Bethea, C.L.; Centeno, M.L.; Cameron, J.L. Neurobiology of stress-induced reproductive dysfunction in female macaques. Mol. Neurobiol. 2008, 38, 199–230. [Google Scholar] [CrossRef] [Green Version]
- Houwing, D.J.; Buwalda, B.; Van Der Zee, E.A.; De Boer, S.F.; Olivier, J.D.A. The serotonin transporter and early life stress: Translational perspectives. Front. Cell. Neurosci. 2017, 11, 117. [Google Scholar] [CrossRef] [Green Version]
- Leonard, B.E.; Myint, A. Changes in the immune system in depression and dementia: Causal or coincidental effects? Dialogues Clin. Neurosci. 2006, 8, 163–174. [Google Scholar]
- Tufekci, K.U.; Meuwissen, R.; Genc, S.; Genc, K. Inflammation in parkinson’s disease. In Advances in Protein Chemistry and Structural Biology; Academic Press Inc.: Cambridge, MA, USA, 2012; Volume 88, pp. 69–132. [Google Scholar]
- Su, Y.A.; Lin, J.Y.; Liu, Q.; Lv, X.Z.; Wang, G.; Wei, J.; Zhu, G.; Chen, Q.L.; Tian, H.J.; Zhang, K.R.; et al. Associations among serum markers of inflammation, life stress and suicide risk in patients with major depressive disorder. J. Psychiatr. Res. 2020, 129, 53–60. [Google Scholar] [CrossRef]
- Cowen, P.J.; Browning, M. What has serotonin to do with depression? World Psychiatry 2015, 14, 158–160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- England, M.J.; Sim, L.J. Depression in Parents, Parenting, and Children: Opportunities to Improve Identification, Treatment, and Prevention; National Academies Press: Washington, DC, USA, 2009; ISBN 0309121787. [Google Scholar]
- Yang, L.; Zhao, Y.; Wang, Y.; Liu, L.; Zhang, X.; Li, B.; Cui, R. The effects of psychological stress on depression. Curr. Neuropharmacol. 2015, 13, 494–504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sapolsky, R.; Krey, L.; McEwen, B. Prolonged glucocorticoid exposure reduces hippocampal neuron number: Implications for aging. J. Neurosci. 1985, 5, 1222–1227. [Google Scholar] [CrossRef] [Green Version]
- Tian, L.; Hui, C.W.; Bisht, K.; Tan, Y.; Sharma, K.; Chen, S.; Zhang, X.; Tremblay, M.-E. Microglia under psychosocial stressors along the aging trajectory: Consequences on neuronal circuits, behavior, and brain diseases. Prog. Neuropsychopharmacol. Biol. Psychiatry 2017, 79, 27–39. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, T.; Kimoto, T.; Tanabe, N.; Hattori, T.A.; Yasumatsu, N.; Kawato, S. Corticosterone acutely prolonged N-methyl-d-aspartate receptor-mediated Ca2+ elevation in cultured rat hippocampal neurons. J. Neurochem. 2002, 83, 1441–1451. [Google Scholar] [CrossRef] [Green Version]
- Tata, D.A.; Marciano, V.A.; Anderson, B.J. Synapse loss from chronically elevated glucocorticoids: Relationship to neuropil volume and cell number in hippocampal area CA3. J. Comp. Neurol. 2006, 498, 363–374. [Google Scholar] [CrossRef] [PubMed]
- Liston, C.; Gan, W.-B. Glucocorticoids are critical regulators of dendritic spine development and plasticity in vivo. Proc. Natl. Acad. Sci. USA 2011, 108, 16074–16079. [Google Scholar] [CrossRef] [Green Version]
- Belleau, E.L.; Treadway, M.T.; Pizzagalli, D.A. The impact of stress and major depressive disorder on hippocampal and medial prefrontal cortex morphology. Biol. Psychiatry 2019, 85, 443–453. [Google Scholar] [CrossRef] [PubMed]
- Leonard, B.E. The concept of depression as a dysfunction of the immune system. In Depression: From Psychopathology to Pharmacotherapy; S. Karger AG: Bazel, Switzerland, 2010; Volume 27, pp. 53–71. ISBN 9783805596060. [Google Scholar]
- Czéh, B.; Michaelis, T.; Watanabe, T.; Frahm, J.; De Biurrun, G.; Van Kampen, M.; Bartolomucci, A.; Fuchs, E. Stress-induced changes in cerebral metabolites, hippocampal volume, and cell proliferation are prevented by antidepressant treatment with tianeptine. Proc. Natl. Acad. Sci. USA 2001, 98, 12796–12801. [Google Scholar] [CrossRef] [Green Version]
- Jimenez-Fernandez, S.; Gurpegui, M.; Diaz-Atienza, F.; Perez-Costillas, L.; Gerstenberg, M.; Correll, C.U. Oxidative stress and antioxidant parameters in patients with major depressive disorder compared to healthy controls before and after antidepressant treatment: Results from a meta-analysis. J. Clin. Psychiatry 2015, 76, 1658–1667. [Google Scholar] [CrossRef]
- Bustamante, A.C.; Aiello, A.E.; Galea, S.; Ratanatharathorn, A.; Noronha, C.; Wildman, D.E.; Uddin, M. Glucocorticoid receptor DNA methylation, childhood maltreatment and major depression. J. Affect. Disord. 2016, 206, 181–188. [Google Scholar] [CrossRef] [Green Version]
- Radtke, K.M.; Schauer, M.; Gunter, H.M.; Ruf-Leuschner, M.; Sill, J.; Meyer, A.; Elbert, T. Epigenetic modifications of the glucocorticoid receptor gene are associated with the vulnerability to psychopathology in childhood maltreatment. Transl. Psychiatry 2015, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swartz, J.R.; Hariri, A.R.; Williamson, D.E. An epigenetic mechanism links socioeconomic status to changes in depression-related brain function in high-risk adolescents. Mol. Psychiatry 2017, 22, 209–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Booij, L.; Szyf, M.; Carballedo, A.; Frey, E.M.; Morris, D.; Dymov, S.; Vaisheva, F.; Ly, V.; Fahey, C.; Meaney, J.; et al. DNA methylation of the serotonin transporter gene in peripheral cells and stress-related changes in hippocampal volume: A study in depressed patients and healthy controls. PLoS ONE 2015, 10. [Google Scholar] [CrossRef] [PubMed]
- Song, Y.; Miyaki, K.; Suzuki, T.; Sasaki, Y.; Tsutsumi, A.; Kawakami, N.; Shimazu, A.; Takahashi, M.; Inoue, A.; Kan, C.; et al. Altered DNA methylation status of human brain derived neurotrophis factor gene could be useful as biomarker of depression. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2014, 165, 357–364. [Google Scholar] [CrossRef] [Green Version]
- Weder, N.; Zhang, H.; Jensen, K.; Yang, B.Z.; Simen, A.; Jackowski, A.; Lipschitz, D.; Douglas-Palumberi, H.; Ge, M.; Perepletchikova, F.; et al. Child abuse, depression, and methylation in genes involved with stress, neural plasticity, and brain circuitry. J. Am. Acad. Child Adolesc. Psychiatry 2014, 53. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sadeh, N.; Spielberg, J.M.; Logue, M.W.; Wolf, E.J.; Smith, A.K.; Lusk, J.; Hayes, J.P.; Sperbeck, E.; Milberg, W.P.; Mcglinchey, R.E.; et al. SKA2 methylation is associated with decreased prefrontal cortical thickness and greater PTSD severity among trauma-exposed veterans. Mol. Psychiatry 2016, 21, 357–363. [Google Scholar] [CrossRef] [Green Version]
- Raap, D.K.; Van de Kar, L.D. Selective serotonin reuptake inhibitors and neuroendocrine function. Life Sci. 1999, 65, 1217–1235. [Google Scholar] [CrossRef]
- Mikkelsen, J.D.; Hay-Schmidt, A.; Kiss, A. Serotonergic stimulation of the rat hypothalamo-pituitary-adrenal axis: Interaction between 5-HT1A and 5-HT2A receptors. In Proceedings of the Annals of the New York Academy of Sciences; New York Academy of Sciences: New York, NY, USA, 2004; Volume 1018, pp. 65–70. [Google Scholar]
- Jensen, J.B.; Jessop, D.S.; Harbuz, M.S.; Mørk, A.; Sánchez, C.; Mikkelsen, J.D. Acute and long-term treatments with the selective serotonin reuptake inhibitor citalopram modulate the HPA axis activity at different levels in male rats. J. Neuroendocrinol. 1999, 11, 465–471. [Google Scholar] [CrossRef]
- Rothman, S.M.; Mattson, M.P. Adverse stress, hippocampal networks, and Alzheimer’s disease. NeuroMolecular Med. 2010, 12, 56–70. [Google Scholar] [CrossRef] [Green Version]
- Song, H.; Sieurin, J.; Wirdefeldt, K.; Pedersen, N.L.; Almqvist, C.; Larsson, H.; Valdimarsdóttir, U.A.; Fang, F. Association of Stress-Related Disorders with Subsequent Neurodegenerative Diseases. JAMA Neurol. 2020, 77, 700–709. [Google Scholar] [CrossRef] [PubMed]
- Justice, N.J. The relationship between stress and Alzheimer’s disease. Neurobiol. Stress 2018, 8, 127–133. [Google Scholar] [CrossRef] [PubMed]
- Aznar, S.; Knudsen, G.M. Depression and alzheimer’s disease: Is stress the initiating factor in a common neuropathological cascade? J. Alzheimer’s Dis. 2011, 23, 177–193. [Google Scholar] [CrossRef]
- Swaab, D.F.; Bao, A.M.; Lucassen, P.J. The stress system in the human brain in depression and neurodegeneration. Ageing Res. Rev. 2005, 4, 141–194. [Google Scholar] [CrossRef] [PubMed]
- Giubilei, F.; Patacchioli, F.R.; Antonini, G.; Sepe Monti, M.; Tisei, P.; Bastianello, S.; Monnazzi, P.; Angelucci, L. Altered circadian cortisol secretion in Alzheimer’s disease: Clinical and neuroradiological aspects. J. Neurosci. Res. 2001, 66, 262–265. [Google Scholar] [CrossRef] [PubMed]
- Duksal, F.; Kilic, I.; Tufan, A.C.; Akdogan, I. Effects of different corticosteroids on the brain weight and hippocampal neuronal loss in rats. Brain Res. 2009, 1250, 75–80. [Google Scholar] [CrossRef] [PubMed]
- Aisa, B.; Tordera, R.; Lasheras, B.; Del Río, J.; Ramírez, M.J. Cognitive impairment associated to HPA axis hyperactivity after maternal separation in rats. Psychoneuroendocrinology 2007, 32, 256–266. [Google Scholar] [CrossRef]
- Ouanes, S.; Popp, J. High Cortisol and the Risk of Dementia and Alzheimer’s Disease: A Review of the Literature. Front. Aging Neurosci. 2019, 11. [Google Scholar] [CrossRef]
- Conrad, C.D. Chronic stress-induced hippocampal vulnerability: The glucocorticoid vulnerability hypothesis. Rev. Neurosci. 2008, 19, 395–411. [Google Scholar] [CrossRef] [Green Version]
- Videbech, P.; Ravnkilde, B. Hippocampal volume and depression: A meta-analysis of MRI studies. Am. J. Psychiatry 2004, 161, 1957–1966. [Google Scholar] [CrossRef]
- Lucassen, P.J.; Müller, M.B.; Holsboer, F.; Bauer, J.; Holtrop, A.; Wouda, J.; Hoogendijk, W.J.G.; De Kloet, E.R.; Swaab, D.F. Hippocampal apoptosis in major depression is a minor event and absent from subareas at risk for glucocorticoid overexposure. Am. J. Pathol. 2001, 158, 453–468. [Google Scholar] [CrossRef] [Green Version]
- Müller, M.B.; Lucassen, P.J.; Yassouridis, A.; Hoogendijk, W.J.G.; Holsboer, F.; Swaab, D.F. Neither major depression nor glucocorticoid treatment affects the cellular integrity of the human hippocampus. Eur. J. Neurosci. 2001, 14, 1603–1612. [Google Scholar] [CrossRef] [PubMed]
- Green, K.N.; Billings, L.M.; Roozendaal, B.; McGaugh, J.L.; LaFerla, F.M. Glucocorticoids increase amyloid-β and tau pathology in a mouse model of Alzheimer’s disease. J. Neurosci. 2006, 26, 9047–9056. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sotiropoulos, I.; Catania, C.; Pinto, L.G.; Silva, R.; Pollerberg, G.E.; Takashima, A.; Sousa, N.; Almeida, O.F.X. Stress acts cumulatively to precipitate Alzheimer’s disease-like tau pathology and cognitive deficits. J. Neurosci. 2011, 31, 7840–7847. [Google Scholar] [CrossRef] [Green Version]
- Cai, Z.; Qiao, P.F.; Wan, C.Q.; Cai, M.; Zhou, N.K.; Li, Q. Role of blood-brain barrier in Alzheimer’s disease. J. Alzheimer’s Dis. 2018, 63, 1223–1234. [Google Scholar] [CrossRef]
- Kang, J.-E.; Cirrito, J.R.; Dong, H.; Csernansky, J.G.; Holtzman, D.M. Acute stress increases interstitial fluid amyloid-β via corticotropin-releasing factor and neuronal activity. Proc. Natl. Acad. Sci. USA 2007, 104, 10673–10678. [Google Scholar] [CrossRef] [Green Version]
- Jeong, Y.H.; Park, C.H.; Yoo, J.; Shin, K.Y.; Ahn, S.M.; Kim, H.S.; Suh, Y.H. Chronic stress accelerates learning and memory impairments and increases amyloid deposition in APPV717I-CT100 transgenic mice, an Alzheimer’s disease model. FASEB J. 2006, 20, 729–731. [Google Scholar] [CrossRef] [Green Version]
- Mdawar, B.; Ghossoub, E.; Khoury, R. Selective serotonin reuptake inhibitors and Alzheimer’s disease. Neural Regen. Res. 2020, 15, 41–46. [Google Scholar]
- Geldenhuys, W.J.; Van Der Schyf, C.J. Role of serotonin in Alzheimers disease: A new therapeutic target? CNS Drugs 2011, 25, 765–781. [Google Scholar] [CrossRef] [PubMed]
- Ramirez, M.J.; Lai, M.K.P.; Tordera, R.M.; Francis, P.T. Serotonergic therapies for cognitive symptoms in alzheimer’s disease: Rationale and current status. Drugs 2014, 74, 729–736. [Google Scholar] [CrossRef]
- Cumbo, E.; Cumbo, S.; Torregrossa, S.; Migliore, D. Treatment effects of vortioxetine on cognitive functions in mild Alzheimer’s disease patients with depressive symptoms: A 12 month, open-label, observational study. J. Prev. Alzheimer Dis. 2019, 6, 192–197. [Google Scholar] [CrossRef]
- Djamshidian, A.; Lees, A.J. Can stress trigger Parkinson’s disease? J. Neurol. Neurosurg. Psychiatry 2014, 85, 879–882. [Google Scholar] [CrossRef] [PubMed]
- Giladi, N.; Hausdorff, J.M. The role of mental function in the pathogenesis of freezing of gait in Parkinson’s disease. J. Neurol. Sci. 2006, 248, 173–176. [Google Scholar] [CrossRef] [PubMed]
- Austin, K.W.; Ameringer, S.W.; Cloud, L.J. An integrated review of psychological stress in Parkinson’s disease: Biological mechanisms and symptom and health outcomes. Parkinsons. Dis. 2016, 2016. [Google Scholar] [CrossRef]
- Lauretti, E.; Di Meco, A.; Merali, S.; Praticò, D. Chronic behavioral stress exaggerates motor deficit and neuroinflammation in the MPTP mouse model of Parkinson’s disease. Transl. Psychiatry 2016, 6. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dallé, E.; Daniels, W.M.U.; Mabandla, M.V. Long-term treatment with fluvoxamine decreases nonmotor symptoms and dopamine depletion in a postnatal stress rat model of Parkinson’s disease. Oxid. Med. Cell. Longev. 2020, 2020. [Google Scholar] [CrossRef]
- He, K.-J.; Zhang, Y.-T.; Wei, S.-Z.; Jiang, S.-M.; Xu, L.; Ren, C.; Wang, F. Impact of maternal separation on dopamine system and its association with Parkinson’s disease. NeuroMol. Med. 2020, 22, 335–340. [Google Scholar] [CrossRef]
- Wihan, J.; Grosch, J.; Kalinichenko, L.S.; Müller, C.P.; Winkler, J.; Kohl, Z. Layer-specific axonal degeneration of serotonergic fibers in the prefrontal cortex of aged A53T α-synuclein–expressing mice. Neurobiol. Aging 2019, 80, 29–37. [Google Scholar] [CrossRef] [PubMed]
- Fox, S.H. Pimavanserin as treatment for Parkinson’s disease psychosis. Lancet 2014, 383, 494–496. [Google Scholar] [CrossRef]
- Wilson, H.; Dervenoulas, G.; Pagano, G.; Koros, C.; Yousaf, T.; Picillo, M.; Politis, M. Serotonergic pathology and disease burden in the premotor and motor phase of A53T α-synuclein parkinsonism: A cross-sectional study. Lancet. Neurol. 2019, 18, 748–759. [Google Scholar] [CrossRef] [Green Version]
- Glavaski-Joksimovic, A.; Virag, T.; Chang, Q.A.; West, N.C.; Mangatu, T.A.; McGrogan, M.P.; Bohn, M.C. Reversal of dopaminergic degeneration in a parkinsonian rat following micrografting of human bone marrow-derived neural progenitors. Cell Transplant. 2009, 18, 804–814. [Google Scholar] [CrossRef] [PubMed]
- Leonard, B.E. The concept of depression as a dysfunction of the immune system. Curr. Immunol. Rev. 2010, 6, 205–212. [Google Scholar] [CrossRef] [Green Version]
- Kenis, G.; Maes, M. Effects of antidepressants on the production of cytokines. Int. J. Neuropsychopharmacol. 2002, 5, 401–412. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sluzewska, A.; Rybakowski, J.; Bosmans, E.; Sobieska, M.; Berghmans, R.; Maes, M.; Wiktorowicz, K. Indicators of immune activation in major depression. Psychiatry Res. 1996, 64, 161–167. [Google Scholar] [CrossRef]
- Zunszain, P.A.; Anacker, C.; Cattaneo, A.; Carvalho, L.A.; Pariante, C.M. Glucocorticoids, cytokines and brain abnormalities in depression. Prog. Neuro-Psychopharmacol. Biol. Psychiatry 2011, 35, 722–729. [Google Scholar] [CrossRef] [Green Version]
- Raison, C.L.; Miller, A.H. Is depression an inflammatory disorder? Curr. Psychiatry Rep. 2011, 13, 467–475. [Google Scholar] [CrossRef] [Green Version]
- Fourrier, C.; Sampson, E.; Hori, H.; Schubert, K.O.; Clark, S.; Mills, N.T.; Baune, B.T. Exploratory study of association between blood immune markers and cognitive symptom severity in major depressive disorder: Stratification by body mass index status. Brain. Behav. Immun. 2020, 88, 242–251. [Google Scholar] [CrossRef] [PubMed]
- Das, R.; Emon, M.P.Z.; Shahriar, M.; Nahar, Z.; Islam, S.M.A.; Bhuiyan, M.A.; Islam, S.N.; Islam, M.R. Higher levels of serum IL-1β and TNF-α are associated with an increased probability of major depressive disorder. Psychiatry Res. 2021, 295, 113568. [Google Scholar] [CrossRef] [PubMed]
- Köhler, C.A.; Freitas, T.H.; Maes, M.; de Andrade, N.Q.; Liu, C.S.; Fernandes, B.S.; Stubbs, B.; Solmi, M.; Veronese, N.; Herrmann, N.; et al. Peripheral cytokine and chemokine alterations in depression: A meta-analysis of 82 studies. Acta Psychiatr. Scand. 2017, 135, 373–387. [Google Scholar] [CrossRef]
- Hepgul, N.; Cattaneo, A.; Zunszain, P.A.; Pariante, C.M. Depression pathogenesis and treatment: What can we learn from blood mRNA expression? BMC Med. 2013, 11, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Stein, D.J.; Vasconcelos, M.F.; Albrechet-Souza, L.; Ceresér, K.M.M.; de Almeida, R.M.M. Microglial over-activation by social defeat stress contributes to anxiety- and depressive-like behaviors. Front. Behav. Neurosci. 2017, 11, 207. [Google Scholar] [CrossRef] [Green Version]
- Wohleb, E.S.; McKim, D.B.; Sheridan, J.F.; Godbout, J.P. Monocyte trafficking to the brain with stress and inflammation: A novel axis of immune-to-brain communication that influences mood and behavior. Front. Neurosci. 2015, 8, 447. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.H.; Raison, C.L. The role of inflammation in depression: From evolutionary imperative to modern treatment target. Nat. Rev. Immunol. 2016, 16, 22–34. [Google Scholar] [CrossRef] [Green Version]
- Ascoli, B.M.; Géa, L.P.; Colombo, R.; Barbé-Tuana, F.M.; Kapczinski, F.; Rosa, A.R. The role of macrophage polarization on bipolar disorder: Identifying new therapeutic targets. Aust. N. Z. J. Psychiatry 2016, 50, 618–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramirez, K.; Shea, D.T.; McKim, D.B.; Reader, B.F.; Sheridan, J.F. Imipramine attenuates neuroinflammatory signaling and reverses stress-induced social avoidance. Brain. Behav. Immun. 2015, 46, 212–220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feng, X.; Zhao, Y.; Yang, T.; Song, M.; Wang, C.; Yao, Y.; Fan, H. Glucocorticoid-driven NLRP3 inflammasome activation in hippocampal microglia mediates chronic stress-induced depressive-like behaviors. Front. Mol. Neurosci. 2019, 12, 210. [Google Scholar] [CrossRef] [PubMed]
- Holmes, S.E.; Hinz, R.; Conen, S.; Gregory, C.J.; Matthews, J.C.; Anton-Rodriguez, J.M.; Talbot, P.S. Elevated translocator protein in anterior cingulate in major depression and a role for inflammation in suicidal thinking: A positron emission tomography study. Biol. Psychiatry 2018, 83, 61–69. [Google Scholar] [CrossRef] [Green Version]
- Torres-Platas, S.G.; Cruceanu, C.; Chen, G.G.; Turecki, G.; Mechawar, N. Evidence for increased microglial priming and macrophage recruitment in the dorsal anterior cingulate white matter of depressed suicides. Brain. Behav. Immun. 2014, 42, 50–59. [Google Scholar] [CrossRef] [PubMed]
- Amodeo, G.; Allegra Trusso, M.; Fagiolini, A. Depression and inflammation: Disentangling a clear yet complex and multifaceted link. Neuropsychiatry 2018, 07, 448–457. [Google Scholar] [CrossRef]
- Medina-Rodriguez, E.M.; Lowell, J.A.; Worthen, R.J.; Syed, S.A.; Beurel, E. Involvement of innate and adaptive immune systems alterations in the pathophysiology and treatment of depression. Front. Neurosci. 2018, 12, 547. [Google Scholar] [CrossRef] [Green Version]
- Li, M.X.; Zheng, H.L.; Luo, Y.; He, J.G.; Wang, W.; Han, J.; Zhang, L.; Wang, X.; Ni, L.; Zhou, H.Y.; et al. Gene deficiency and pharmacological inhibition of caspase-1 confers resilience to chronic social defeat stress via regulating the stability of surface AMPARs. Mol. Psychiatry 2018, 23, 556–568. [Google Scholar] [CrossRef] [Green Version]
- Franklin, T.C.; Wohleb, E.S.; Zhang, Y.; Fogaça, M.; Hare, B.; Duman, R.S. Persistent increase in microglial RAGE contributes to chronic stress-induced priming of depressive-like behavior. Biol. Psychiatry 2018, 83, 50–60. [Google Scholar] [CrossRef]
- Rogers, G.B.; Keating, D.J.; Young, R.L.; Wong, M.L.; Licinio, J.; Wesselingh, S. From gut dysbiosis to altered brain function and mental illness: Mechanisms and pathways. Mol. Psychiatry 2016, 21, 738–748. [Google Scholar] [CrossRef] [Green Version]
- Valles-Colomer, M.; Falony, G.; Darzi, Y.; Tigchelaar, E.F.; Wang, J.; Tito, R.Y.; Schiweck, C.; Kurilshikov, A.; Joossens, M.; Wijmenga, C.; et al. The neuroactive potential of the human gut microbiota in quality of life and depression. Nat. Microbiol. 2019, 4, 623–632. [Google Scholar] [CrossRef] [PubMed]
- Köhler, O.; Benros, M.E.; Nordentoft, M.; Farkouh, M.E.; Iyengar, R.L.; Mors, O.; Krogh, J. Effect of anti-inflammatory treatment on depression, depressive symptoms, and adverse effects: A systematic review and meta-analysis of randomized clinical trials. JAMA Psychiatry 2014, 71, 1381–1391. [Google Scholar] [CrossRef]
- Felger, J.C.; Li, Z.; Haroon, E.; Woolwine, B.J.; Jung, M.Y.; Hu, X.; Miller, A.H. Inflammation is associated with decreased functional connectivity within corticostriatal reward circuitry in depression. Mol. Psychiatry 2015, 21, 1358–1365. [Google Scholar] [CrossRef] [Green Version]
- Lee, C.H.; Giuliani, F. The role of inflammation in depression and fatigue. Front. Immunol. 2019, 10, 1696. [Google Scholar] [CrossRef] [Green Version]
- Beurel, E.; Toups, M.; Nemeroff, C.B. The bidirectional relationship of depression and inflammation: Double trouble. Neuron 2020, 107, 234–256. [Google Scholar] [CrossRef]
- Wu, H.; Denna, T.H.; Storkersen, J.N.; Gerriets, V.A. Beyond a neurotransmitter: The role of serotonin in inflammation and immunity. Pharmacol. Res. 2019, 140, 100–114. [Google Scholar] [CrossRef]
- Vollmar, P.; Nessler, S.; Kalluri, S.R.; Hartung, H.P.; Hemmer, B. The antidepressant venlafaxine ameliorates murine experimental autoimmune encephalomyelitis by suppression of pro-inflammatory cytokines. Int. J. Neuropsychopharmacol. 2009, 12, 525–536. [Google Scholar] [CrossRef] [Green Version]
- Gaur, A.; Boehme, S.A.; Chalmers, D.; Crowe, P.D.; Pahuja, A.; Ling, N.; Brocke, S.; Steinman, L.; Conlon, P.J. Amelioration of relapsing experimental autoimmune encephalomyelitis with altered myelin basic protein peptides involves different cellular mechanisms. J. Neuroimmunol. 1997, 74, 149–158. [Google Scholar] [CrossRef]
- Chen, Y.; Jiang, T.; Chen, P.; Ouyang, J.; Xu, G.; Zeng, Z.; Sun, Y. Emerging tendency towards autoimmune process in major depressive patients: A novel insight from Th17 cells. Psychiatry Res. 2011, 188, 224–230. [Google Scholar] [CrossRef] [PubMed]
- Grosse, L.; Carvalho, L.A.; Birkenhager, T.K.; Hoogendijk, W.J.; Kushner, S.A.; Drexhage, H.A.; Bergink, V. Circulating cytotoxic T cells and natural killer cells as potential predictors for antidepressant response in melancholic depression. Restoration of T regulatory cell populations after antidepressant therapy. Psychopharmacology 2016, 233, 1679–1688. [Google Scholar] [CrossRef] [PubMed]
- Grosse, L.; Hoogenboezem, T.; Ambrée, O.; Bellingrath, S.; Jörgens, S.; de Wit, H.J.; Wijkhuijs, A.M.; Arolt, V.; Drexhage, H.A. Deficiencies of the T and natural killer cell system in major depressive disorder. T regulatory cell defects are associated with inflammatory monocyte activation. Brain. Behav. Immun. 2016, 54, 38–44. [Google Scholar] [CrossRef]
- Di Rosso, M.E.; Palumbo, M.L.; Genaro, A.M. Immunomodulatory effects of fluoxetine: A new potential pharmacological action for a classic antidepressant drug? Pharmacol. Res. 2016, 109, 101–107. [Google Scholar] [CrossRef]
- Mp, G.-P.; Rochina, J.; La, D.; Je, R.; Pilar, M.; Pardo, G. Role of stress, immune system and well-being in patients with Alzheimer’s disease. J. Neurol. Neurosci. 2017, 8, 171. [Google Scholar] [CrossRef] [Green Version]
- Serrano-Pozo, A.; Mielke, M.L.; Gómez-Isla, T.; Betensky, R.A.; Growdon, J.H.; Frosch, M.P.; Hyman, B.T. Reactive glia not only associates with plaques but also parallels tangles in Alzheimer’s disease. Am. J. Pathol. 2011, 179, 1373–1384. [Google Scholar] [CrossRef]
- Wang, W.Y.; Tan, M.S.; Yu, J.T.; Tan, L. Role of pro-inflammatory cytokines released from microglia in Alzheimer’s disease. Ann. Transl. Med. 2015, 3, 136. [Google Scholar] [PubMed]
- Heneka, M.T.; Kummer, M.P.; Stutz, A.; Delekate, A.; Schwartz, S.; Vieira-Saecker, A.; Griep, A.; Axt, D.; Remus, A.; Tzeng, T.C.; et al. NLRP3 is activated in Alzheimer’s disease and contributes to pathology in APP/PS1 mice. Nature 2013, 493, 674–678. [Google Scholar] [CrossRef] [PubMed]
- Kinney, J.W.; Bemiller, S.M.; Murtishaw, A.S.; Leisgang, A.M.; Salazar, A.M.; Lamb, B.T. Inflammation as a central mechanism in Alzheimer’s disease. Alzheimer Dement. Transl. Res. Clin. Interv. 2018, 4, 575–590. [Google Scholar] [CrossRef] [PubMed]
- Morgan, B.P. Complement in the pathogenesis of Alzheimer’s disease. Semin. Immunopathol. 2018, 40, 113–124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, Q.; Chowdhury, S.; Ma, R.; Le, K.X.; Hong, S.; Caldarone, B.J.; Stevens, B.; Lemere, C.A. Complement C3 deficiency protects against neurodegeneration in aged plaque-rich APP/PS1 mice. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saresella, M.; Calabrese, E.; Marventano, I.; Piancone, F.; Gatti, A.; Alberoni, M.; Nemni, R.; Clerici, M. Increased activity of Th-17 and Th-9 lymphocytes and a skewing of the post-thymic differentiation pathway are seen in Alzheimer’s disease. Brain. Behav. Immun. 2011, 25, 539–547. [Google Scholar] [CrossRef]
- Smith, J.A.; Das, A.; Ray, S.K.; Banik, N.L. Role of pro-inflammatory cytokines released from microglia in neurodegenerative diseases. Brain Res. Bull. 2012, 87, 10–20. [Google Scholar] [CrossRef] [PubMed]
- Cribbs, D.H.; Berchtold, N.C.; Perreau, V.; Coleman, P.D.; Rogers, J.; Tenner, A.J.; Cotman, C.W. Extensive innate immune gene activation accompanies brain aging, increasing vulnerability to cognitive decline and neurodegeneration: A microarray study. J. Neuroinflammation 2012, 9, 1–18. [Google Scholar] [CrossRef] [Green Version]
- Ojala, J.; Alafuzoff, I.; Herukka, S.K.; van Groen, T.; Tanila, H.; Pirttilä, T. Expression of interleukin-18 is increased in the brains of Alzheimer’s disease patients. Neurobiol. Aging 2009, 30, 198–209. [Google Scholar] [CrossRef] [PubMed]
- Salani, F.; Sterbini, V.; Sacchinelli, E.; Garramone, M.; Bossù, P. Is innate memory a double-edge sword in Alzheimer’s disease? A reappraisal of new concepts and old data. Front. Immunol. 2019, 10, 1768. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ke, K.F.; Liu, Z.; Qiu, Y.H.; Peng, Y.P. Th17 cell-mediated neuroinflammation is involved in neurodegeneration of Aβ1-42-induced Alzheimer’s disease model rats. PLoS ONE 2013, 8, 75786. [Google Scholar] [CrossRef]
- Cristiano, C.; Volpicelli, F.; Lippiello, P.; Buono, B.; Raucci, F.; Piccolo, M.; Iqbal, A.J.; Irace, C.; Miniaci, M.C.; Perrone Capano, C.; et al. Neutralization of IL-17 rescues amyloid-β-induced neuroinflammation and memory impairment. Br. J. Pharmacol. 2019, 176, 3544–3557. [Google Scholar] [CrossRef]
- Leyns, C.E.G.; Ulrich, J.D.; Finn, M.B.; Stewart, F.R.; Koscal, L.J.; Serrano, J.R.; Robinson, G.O.; Anderson, E.; Colonna, M.; Holtzman, D.M. TREM2 deficiency attenuates neuroinflammation and protects against neurodegeneration in a mouse model of tauopathy. Proc. Natl. Acad. Sci. USA 2017, 114, 11524–11529. [Google Scholar] [CrossRef] [Green Version]
- Elsworthy, R.J.; Aldred, S. Depression in Alzheimer’s disease: An alternative role for selective serotonin reuptake inhibitors? J. Alzheimer’s Dis. 2019, 69, 651–661. [Google Scholar] [CrossRef]
- Caraci, F.; Copani, A.; Nicoletti, F.; Drago, F. Depression and Alzheimer’s disease: Neurobiological links and common pharmacological targets. Eur. J. Pharmacol. 2010, 626, 64–71. [Google Scholar] [CrossRef]
- Metaxas, A.; Anzalone, M.; Vaitheeswaran, R.; Petersen, S.; Landau, A.M.; Finsen, B. Neuroinflammation and amyloid-beta 40 are associated with reduced serotonin transporter (SERT) activity in a transgenic model of familial Alzheimer’s disease. Alzheimer Res. Ther. 2019, 11, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.B.; Blakely, R.D.; Hewlett, W.A. The proinflammatory cytokines interleukin-1beta and tumor necrosis factor-alpha activate serotonin transporters. Neuropsychopharmacology 2006, 31, 2121–2131. [Google Scholar] [CrossRef] [Green Version]
- Kong, E.; Sucic, S.; Monje, F.J.; Savalli, G.; Diao, W.; Khan, D.; Ronovsky, M.; Cabatic, M.; Koban, F.; Freissmuth, M.; et al. STAT3 controls IL6-dependent regulation of serotonin transporter function and depression-like behavior. Sci. Rep. 2015, 5. [Google Scholar] [CrossRef] [Green Version]
- Ledo, J.H.; Azevedo, E.P.; Beckman, D.; Ribeiro, F.C.; Santos, L.E.; Razolli, D.S.; Kincheski, G.C.; Melo, H.M.; Bellio, M.; Teixeira, A.L.; et al. Cross talk between brain innate immunity and serotonin signaling underlies depressive-like behavior induced by Alzheimer’s amyloid-β oligomers in mice. J. Neurosci. 2016, 36, 12106–12116. [Google Scholar] [CrossRef] [PubMed]
- Cirrito, J.R.; Disabato, B.M.; Restivo, J.L.; Verges, D.K.; Goebel, W.D.; Sathyan, A.; Hayreh, D.; D’Angelo, G.; Benzinger, T.; Yoon, H.; et al. Serotonin signaling is associated with lower amyloid-β levels and plaques in transgenic mice and humans. Proc. Natl. Acad. Sci. USA 2011, 108, 14968–14973. [Google Scholar] [CrossRef] [Green Version]
- Anderson, K.M.; Augusto, D.G.; Dandekar, R.; Shams, H.; Zhao, C.; Yusufali, T.; Montero-Martín, G.; Marin, W.M.; Nemat-Gorgani, N.; Creary, L.E.; et al. Killer cell immunoglobulin-like receptor variants are associated with protection from symptoms associated with more severe course in PD. J. Immunol. 2020, 205, 1323–1330. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Guajardo, V.; Febbraro, F.; Kirik, D.; Romero-Ramos, M. Microglia acquire distinct activation profiles depending on the degree of α-synuclein neuropathology in a rAAV based model of Parkinson’s disease. PLoS ONE 2010, 5. [Google Scholar] [CrossRef]
- Cheng, J.; Liao, Y.; Dong, Y.; Hu, H.; Yang, N.; Kong, X.; Li, S.; Li, X.; Guo, J.; Qin, L.; et al. Microglial autophagy defect causes parkinson disease-like symptoms by accelerating inflammasome activation in mice. Autophagy 2020, 16, 2193–2205. [Google Scholar] [CrossRef]
- Reish, H.E.A.; Standaert, D.G. Role of α-synuclein in inducing innate and adaptive immunity in Parkinson disease. J. Parkinsons. Dis. 2015, 5, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Brochard, V.; Combadière, B.; Prigent, A.; Laouar, Y.; Perrin, A.; Beray-Berthat, V.; Bonduelle, O.; Alvarez-Fischer, D.; Callebert, J.; Launay, J.M.; et al. Infiltration of CD4+ lymphocytes into the brain contributes to neurodegeneration in a mouse model of Parkinson disease. J. Clin. Invest. 2009, 119, 182–192. [Google Scholar] [CrossRef]
- Baba, Y.; Kuroiwa, A.; Uitti, R.J.; Wszolek, Z.K.; Yamada, T. Alterations of T-lymphocyte populations in Parkinson disease. Park. Relat. Disord. 2005, 11, 493–498. [Google Scholar] [CrossRef] [PubMed]
- Mogi, M.; Harada, M.; Kondo, T.; Riederer, P.; Inagaki, H.; Minami, M.; Nagatsu, T. Interleukin-1β, interleukin-6, epidermal growth factor and transforming growth factor-α are elevated in the brain from parkinsonian patients. Neurosci. Lett. 1994, 180, 147–150. [Google Scholar] [CrossRef]
- Kraft, A.D.; Jean Harry, G. Features of microglia and neuroinflammation relevant to environmental exposure and neurotoxicity. Int. J. Environ. Res. Public Health 2011, 8, 2980–3018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Da Chagas, L.S.; Sandre, P.C.; Ribeiro, N.C.A.R.E.; Marcondes, H.; Silva, P.O.; Savino, W.; Serfaty, C.A. Environmental signals on microglial function during brain development, neuroplasticity, and disease. Int. J. Mol. Sci. 2020, 21, 2111. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacMahon Copas, A.N.; McComish, S.F.; Fletcher, J.M.; Caldwell, M.A. The pathogenesis of Parkinson’s disease: A complex interplay between astrocytes, microglia, and T lymphocytes? Front. Neurol. 2021, 12, 771. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Münch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef]
- Nash, Y.; Schmukler, E.; Trudler, D.; Pinkas-Kramarski, R.; Frenkel, D. DJ-1 deficiency impairs autophagy and reduces alpha-synuclein phagocytosis by microglia. J. Neurochem. 2017, 143, 584–594. [Google Scholar] [CrossRef] [Green Version]
- Mackie, P.; Lebowitz, J.; Saadatpour, L.; Nickoloff, E.; Gaskill, P.; Khoshbouei, H. The dopamine transporter: An unrecognized nexus for dysfunctional peripheral immunity and signaling in Parkinson’s Disease. Brain. Behav. Immun. 2018, 70, 21–35. [Google Scholar] [CrossRef]
- Kotagal, V.; Spino, C.; Bohnen, N.I.; Koeppe, R.; Albin, R.L. Serotonin, β-amyloid, and cognition in Parkinson disease. Ann. Neurol. 2018, 83, 994–1002. [Google Scholar] [CrossRef]
- Maydych, V. The interplay between stress, inflammation, and emotional attention: Relevance for depression. Front. Neurosci. 2019, 13, 384. [Google Scholar] [CrossRef]
- Kempuraj, D.; Ahmed, M.E.; Selvakumar, G.P.; Thangavel, R.; Raikwar, S.P.; Zaheer, S.A.; Iyer, S.S.; Burton, C.; James, D.; Zaheer, A. Psychological stress–induced immune response and risk of Alzheimer’s disease in veterans from operation enduring freedom and operation iraqi freedom. Clin. Ther. 2020, 42, 974–982. [Google Scholar] [CrossRef]
- Segerstrom, S.C.; Miller, G.E. Psychological stress and the human immune system: A meta-analytic study of 30 years of inquiry. Psychol. Bull. 2004, 130, 601–630. [Google Scholar] [CrossRef] [Green Version]
- Wohleb, E.S.; Hanke, M.L.; Corona, A.W.; Powell, N.D.; La’Tonia, M.S.; Bailey, M.T.; Sheridan, J.F. β-Adrenergic receptor antagonism prevents anxiety-like behavior and microglial reactivity induced by repeated social defeat. J. Neurosci. 2011, 31, 6277–6288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dantzer, R. Cytokine, sickness behavior, and depression. Immunol. Allergy Clin. North Am. 2009, 29, 247–264. [Google Scholar] [CrossRef] [Green Version]
- Niklasson, F.; Ågren, H. Brain energy metabolism and blood-brain barrier permeability in depressive patients: Analyses of creatine, creatinine, urate, and albumin in CSF and blood. Biol. Psychiatry 1984, 19, 1183–1206. [Google Scholar] [PubMed]
- Beurel, E.; Harrington, L.E.; Jope, R.S. Inflammatory T helper 17 cells promote depression-like behavior in mice. Biol. Psychiatry 2013, 73, 622–630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beurel, E.; Lowell, J.A.; Jope, R.S. Distinct characteristics of hippocampal pathogenic TH17 cells in a mouse model of depression. Brain. Behav. Immun. 2018, 73, 180–191. [Google Scholar] [CrossRef]
- Milior, G.; Lecours, C.; Samson, L.; Bisht, K.; Poggini, S.; Pagani, F.; Maggi, L. Fractalkine receptor deficiency impairs microglial and neuronal responsiveness to chronic stress. Brain. Behav. Immun. 2016, 55, 114–125. [Google Scholar] [CrossRef]
- Tong, L.; Gong, Y.; Wang, P.; Hu, W.; Wang, J.; Chen, Z.; Huang, C. Microglia loss contributes to the development of major depression induced by different types of chronic stresses. Neurochem. Res. 2017, 42, 2698–2711. [Google Scholar] [CrossRef]
- Zhao, Q.; Xie, X.; Fan, Y.; Zhang, J.; Jiang, W.; Wu, X.; Yan, S.; Chen, Y.; Peng, C.; You, Z. Phenotypic dysregulation of microglial activation in young offspring rats with maternal sleep deprivation-induced cognitive impairment. Sci. Rep. 2015, 5, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diz-Chaves, Y.; Pernía, O.; Carrero, P.; Garcia-Segura, L.M. Prenatal stress causes alterations in the morphology of microglia and the inflammatory response of the hippocampus of adult female mice. J. Neuroinflamm. 2012, 9, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Ong, L.K.; Zhao, Z.; Kluge, M.; TeBay, C.; Zalewska, K.; Dickson, P.W.; Walker, F.R. Reconsidering the role of glial cells in chronic stress-induced dopaminergic neurons loss within the substantia nigra? Friend or foe? Brain. Behav. Immun. 2017, 60, 117–125. [Google Scholar] [CrossRef] [PubMed]
- Iwata, M.; Ota, K.T.; Li, X.Y.; Sakaue, F.; Li, N.; Dutheil, S.; Duman, R.S. Psychological stress activates the inflammasome via release of adenosine triphosphate and stimulation of the purinergic type 2X7 receptor. Biol. Psychiatry 2016, 80, 12–22. [Google Scholar] [CrossRef] [PubMed]
- McQuillin, A.; Bass, N.J.; Choudhury, K.; Puri, V.; Kosmin, M.; Lawrence, J.; Gurling, H.M.D. Case-control studies show that a non-conservative amino-acid change from a glutamine to arginine in the P2RX7 purinergic receptor protein is associated with both bipolar- and unipolar-affective disorders. Mol. Psychiatry 2009, 14, 614–620. [Google Scholar] [CrossRef] [PubMed]
- Kreisel, T.; Frank, M.G.; Licht, T.; Reshef, R.; Ben-Menachem-Zidon, O.; Baratta, M.V.; Yirmiya, R. Dynamic microglial alterations underlie stress-induced depressive-like behavior and suppressed neurogenesis. Mol. Psychiatry 2014, 19, 699–709. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bae, Y.S.; Shin, E.C.; Bae, Y.S.; Van Eden, W. Editorial: Stress and immunity. Front. Immunol. 2019, 10, 245. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Correia, A.S.; Cardoso, A.; Vale, N. Highlighting Immune System and Stress in Major Depressive Disorder, Parkinson’s, and Alzheimer’s Diseases, with a Connection with Serotonin. Int. J. Mol. Sci. 2021, 22, 8525. https://doi.org/10.3390/ijms22168525
Correia AS, Cardoso A, Vale N. Highlighting Immune System and Stress in Major Depressive Disorder, Parkinson’s, and Alzheimer’s Diseases, with a Connection with Serotonin. International Journal of Molecular Sciences. 2021; 22(16):8525. https://doi.org/10.3390/ijms22168525
Chicago/Turabian StyleCorreia, Ana Salomé, Armando Cardoso, and Nuno Vale. 2021. "Highlighting Immune System and Stress in Major Depressive Disorder, Parkinson’s, and Alzheimer’s Diseases, with a Connection with Serotonin" International Journal of Molecular Sciences 22, no. 16: 8525. https://doi.org/10.3390/ijms22168525
APA StyleCorreia, A. S., Cardoso, A., & Vale, N. (2021). Highlighting Immune System and Stress in Major Depressive Disorder, Parkinson’s, and Alzheimer’s Diseases, with a Connection with Serotonin. International Journal of Molecular Sciences, 22(16), 8525. https://doi.org/10.3390/ijms22168525