
Toward Understanding the Mechanisms of Malignant Peripheral Nerve Sheath Tumor Development


Abstract
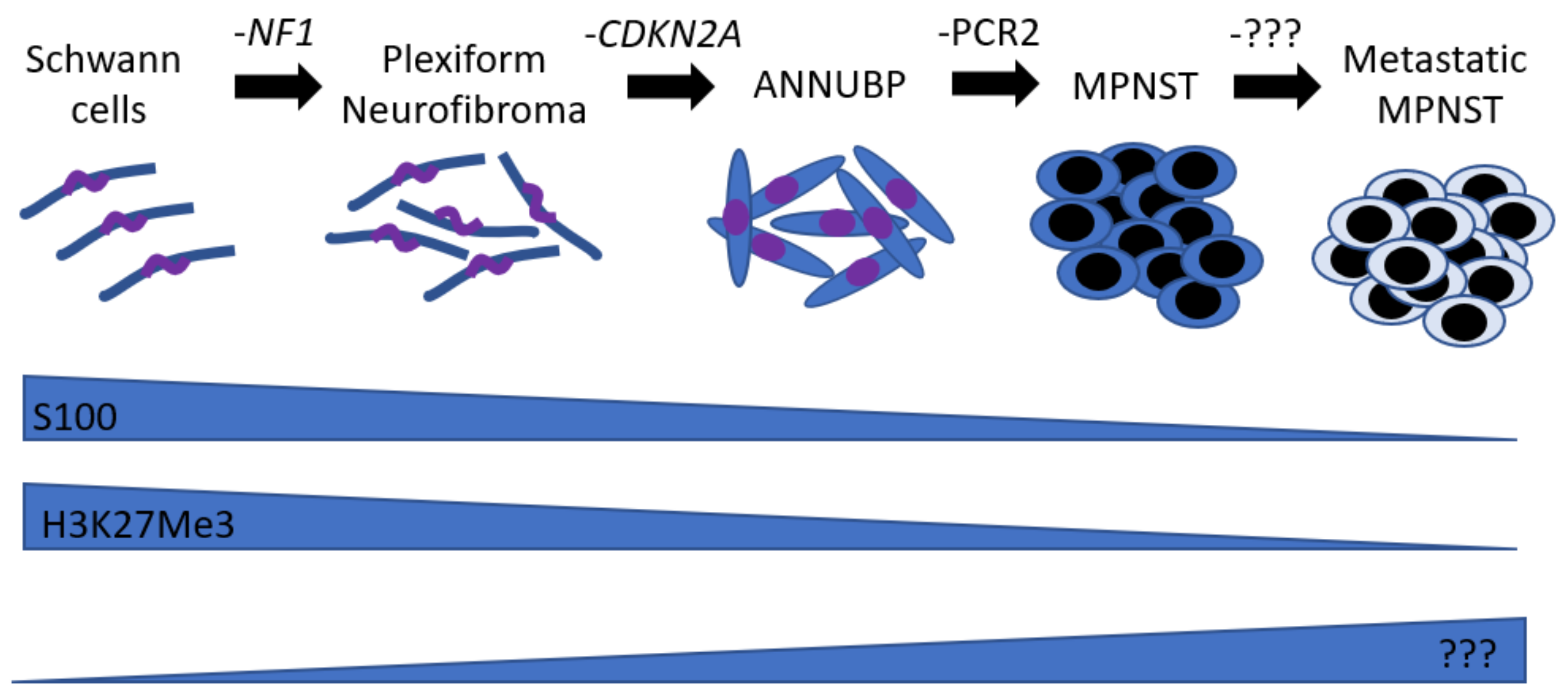
:1. Natural History
2. Mouse Models
3. Tumor Microenvironment
4. Signaling Pathways
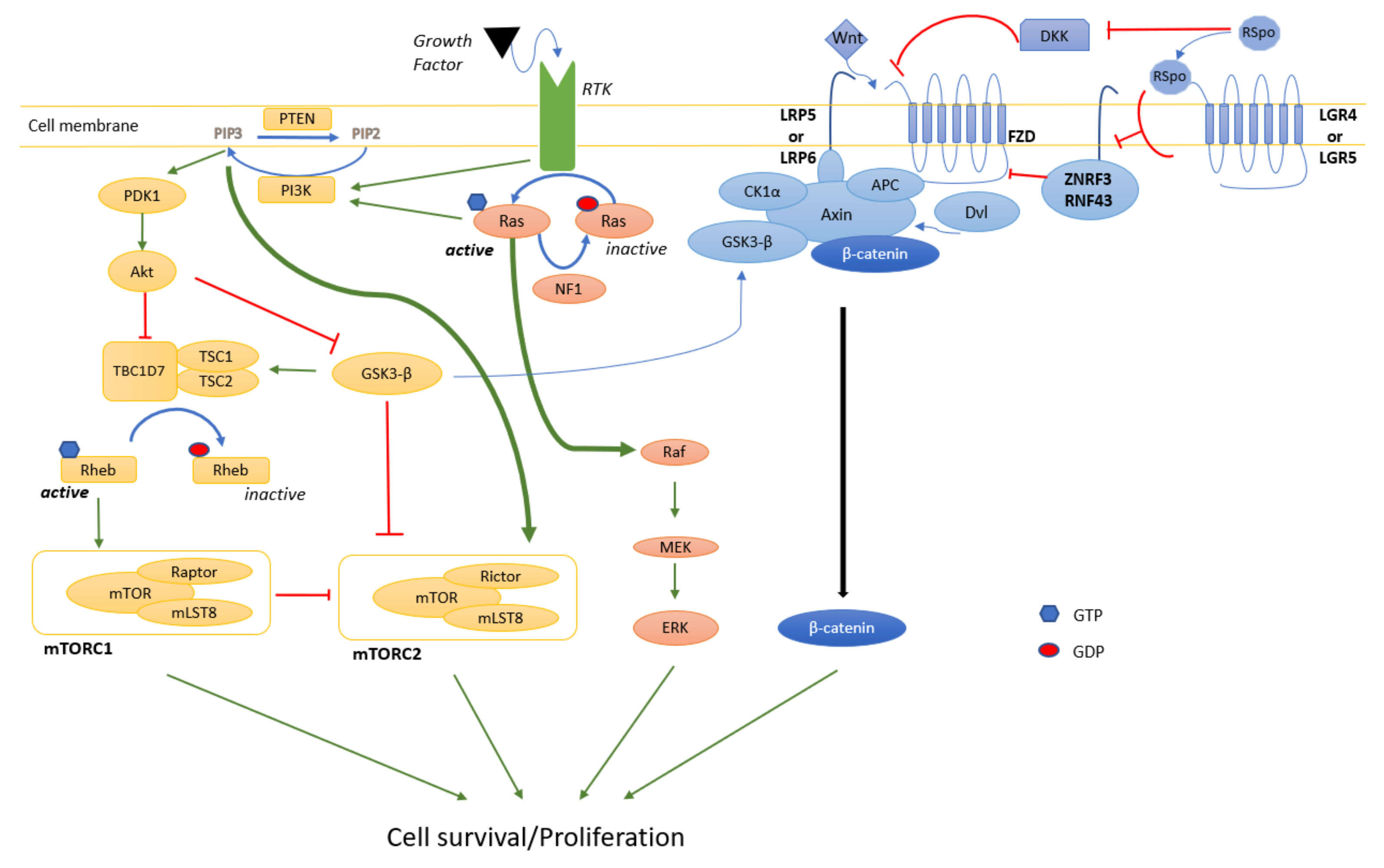
4.1. MAPK
4.2. PI3K/Akt/mTOR
4.3. Wnt
5. Metastasis
6. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cai, Z.; Tang, X.; Liang, H.; Yang, R.; Yan, T.; Guo, W. Prognosis and risk factors for malignant peripheral nerve sheath tumor: A systematic review and meta-analysis. World J. Surg. Oncol. 2020, 18, 257. [Google Scholar] [CrossRef]
- Reilly, K.M.; Kim, A.; Blakely, J.; Ferner, R.E.; Gutmann, D.H.; Legius, E.; Miettinen, M.M.; Randall, R.L.; Ratner, N.; Jumbe, N.L.; et al. Neurofibromatosis Type 1-Associated MPNST State of the Science: Outlining a Research Agenda for the Future. J. Natl. Cancer Inst. 2017, 109, djx124. [Google Scholar] [CrossRef] [Green Version]
- Kahn, J.; Gillespie, A.; Tsokos, M.; Ondos, J.; Dombi, E.; Camphausen, K.; Widemann, B.C.; Kaushal, A. Radiation therapy in management of sporadic and neurofibromatosis type 1-associated malignant peripheral nerve sheath tumors. Front. Oncol. 2014, 4, 324. [Google Scholar] [CrossRef] [Green Version]
- Pemov, A.; Li, H.; Presley, W.; Wallace, M.R.; Miller, D.T. Genetics of human malignant peripheral nerve sheath tumors. Neurooncol. Adv. 2020, 2, i50–i61. [Google Scholar] [CrossRef]
- Pasmant, E.; Sabbagh, A.; Spurlock, G.; Laurendeau, I.; Grillo, E.; Hamel, M.J.; Martin, L.; Barbarot, S.; Leheup, B.; Rodriguez, D.; et al. NF1 microdeletions in neurofibromatosis type 1: From genotype to phenotype. Hum. Mutat. 2010, 31, E1506–E1518. [Google Scholar] [CrossRef] [Green Version]
- Upadhyaya, M.; Ruggieri, M.; Maynard, J.; Osborn, M.; Hartog, C.; Mudd, S.; Penttinen, M.; Cordeiro, I.; Ponder, M.; Ponder, B.A.; et al. Gross deletions of the neurofibromatosis type 1 (NF1) gene are predominantly of maternal origin and commonly associated with a learning disability, dysmorphic features and developmental delay. Hum. Genet. 1998, 102, 591–597. [Google Scholar] [CrossRef]
- Kehrer-Sawatzki, H.; Kluwe, L.; Sandig, C.; Kohn, M.; Wimmer, K.; Krammer, U.; Peyrl, A.; Jenne, D.E.; Hansmann, I.; Mautner, V.F. High frequency of mosaicism among patients with neurofibromatosis type 1 (NF1) with microdeletions caused by somatic recombination of the JJAZ1 gene. Am. J. Hum. Genet. 2004, 75, 410–423. [Google Scholar] [CrossRef] [Green Version]
- Kehrer-Sawatzki, H.; Mautner, V.F.; Cooper, D.N. Emerging genotype-phenotype relationships in patients with large NF1 deletions. Hum. Genet. 2017, 136, 349–376. [Google Scholar] [CrossRef] [Green Version]
- De Raedt, T.; Brems, H.; Wolkenstein, P.; Vidaud, D.; Pilotti, S.; Perrone, F.; Mautner, V.; Frahm, S.; Sciot, R.; Legius, E. Elevated risk for MPNST in NF1 microdeletion patients. Am. J. Hum. Genet. 2003, 72, 1288–1292. [Google Scholar] [CrossRef] [Green Version]
- Evans, D.G.; Baser, M.E.; McGaughran, J.; Sharif, S.; Howard, E.; Moran, A. Malignant peripheral nerve sheath tumours in neurofibromatosis 1. J. Med. Genet. 2002, 39, 311–314. [Google Scholar] [CrossRef] [Green Version]
- De Raedt, T.; Beert, E.; Pasmant, E.; Luscan, A.; Brems, H.; Ortonne, N.; Helin, K.; Hornick, J.L.; Mautner, V.; Kehrer-Sawatzki, H.; et al. PRC2 loss amplifies Ras-driven transcription and confers sensitivity to BRD4-based therapies. Nature 2014, 514, 247–251. [Google Scholar] [CrossRef]
- Lee, W.; Teckie, S.; Wiesner, T.; Ran, L.; Prieto Granada, C.N.; Lin, M.; Zhu, S.; Cao, Z.; Liang, Y.; Sboner, A.; et al. PRC2 is recurrently inactivated through EED or SUZ12 loss in malignant peripheral nerve sheath tumors. Nat. Genet. 2014, 46, 1227–1232. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Wang, Y.; Jones, S.; Sausen, M.; McMahon, K.; Sharma, R.; Wang, Q.; Belzberg, A.J.; Chaichana, K.; Gallia, G.L.; et al. Somatic mutations of SUZ12 in malignant peripheral nerve sheath tumors. Nat. Genet. 2014, 46, 1170–1172. [Google Scholar] [CrossRef] [Green Version]
- Ma, K.H.; Duong, P.; Moran, J.J.; Junaidi, N.; Svaren, J. Polycomb repression regulates Schwann cell proliferation and axon regeneration after nerve injury. Glia 2018, 66, 2487–2502. [Google Scholar] [CrossRef]
- Verdijk, R.M.; den Bakker, M.A.; Dubbink, H.J.; Hop, W.C.; Dinjens, W.N.; Kros, J.M. TP53 mutation analysis of malignant peripheral nerve sheath tumors. J. Neuropathol. Exp. Neurol. 2010, 69, 16–26. [Google Scholar] [CrossRef] [Green Version]
- Gregorian, C.; Nakashima, J.; Dry, S.M.; Nghiemphu, P.L.; Smith, K.B.; Ao, Y.; Dang, J.; Lawson, G.; Mellinghoff, I.K.; Mischel, P.S.; et al. PTEN dosage is essential for neurofibroma development and malignant transformation. Proc. Natl. Acad. Sci. USA 2009, 106, 19479–19484. [Google Scholar] [CrossRef] [Green Version]
- Bradtmoller, M.; Hartmann, C.; Zietsch, J.; Jaschke, S.; Mautner, V.F.; Kurtz, A.; Park, S.J.; Baier, M.; Harder, A.; Reuss, D.; et al. Impaired Pten expression in human malignant peripheral nerve sheath tumours. PLoS ONE 2012, 7, e47595. [Google Scholar] [CrossRef]
- Brohl, A.S.; Kahen, E.; Yoder, S.J.; Teer, J.K.; Reed, D.R. The genomic landscape of malignant peripheral nerve sheath tumors: Diverse drivers of Ras pathway activation. Sci. Rep. 2017, 7, 14992. [Google Scholar] [CrossRef] [Green Version]
- Ferrari, L.; Scuvera, G.; Tucci, A.; Bianchessi, D.; Rusconi, F.; Menni, F.; Battaglioli, E.; Milani, D.; Riva, P. Identification of an atypical microdeletion generating the RNF135-SUZ12 chimeric gene and causing a position effect in an NF1 patient with overgrowth. Hum Genet. 2017, 136, 1329–1339. [Google Scholar] [CrossRef]
- Farid, M.; Demicco, E.G.; Garcia, R.; Ahn, L.; Merola, P.R.; Cioffi, A.; Maki, R.G. Malignant peripheral nerve sheath tumors. Oncologist 2014, 19, 193–201. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Xu, G.; Liu, Z.; Duan, J.; Lin, Y.; Zhu, J.; Baklaushev, V.P.; Chekhonin, V.P.; Peltzer, K.; Wang, G.; et al. Incidence and prognosis of distant metastasis in malignant peripheral nerve sheath tumors. Acta Neurochir. 2021, 163, 521–529. [Google Scholar] [CrossRef]
- Prudner, B.C.; Ball, T.; Rathore, R.; Hirbe, A.C. Diagnosis and management of malignant peripheral nerve sheath tumors: Current practice and future perspectives. Neurooncol. Adv. 2020, 2 (Suppl. 1), i40–i49. [Google Scholar] [CrossRef]
- Mills, A.M.; Karamchandani, J.R.; Vogel, H.; Longacre, T.A. Endocervical fibroblastic malignant peripheral nerve sheath tumor (neurofibrosarcoma): Report of a novel entity possibly related to endocervical CD34 fibrocytes. Am. J. Surg. Pathol. 2011, 35, 404–412. [Google Scholar] [CrossRef]
- Houreih, M.A.; Eyden, B.; Deolekar, M.; Banerjee, S. A case of fibroblastic low-grade malignant peripheral nerve sheath tumor—A true neurofibrosarcoma. Ultrastruct. Pathol. 2007, 31, 347–356. [Google Scholar] [CrossRef]
- Yaga, U.S.; Shivakumar, R.; Kumar, M.A.; Sathyaprakash. Malignant peripheral nerve sheath tumor: A rarity. Indian J. Dent. 2015, 6, 53–56. [Google Scholar] [CrossRef] [Green Version]
- Cichowski, K.; Shih, T.S.; Schmitt, E.; Santiago, S.; Reilly, K.; McLaughlin, M.E.; Bronson, R.T.; Jacks, T. Mouse models of tumor development in neurofibromatosis type 1. Science 1999, 286, 2172–2176. [Google Scholar] [CrossRef]
- Vogel, K.S.; Klesse, L.J.; Velasco-Miguel, S.; Meyers, K.; Rushing, E.J.; Parada, L.F. Mouse tumor model for neurofibromatosis type 1. Science 1999, 286, 2176–2179. [Google Scholar] [CrossRef]
- Hirbe, A.C.; Dahiya, S.; Friedmann-Morvinski, D.; Verma, I.M.; Clapp, D.W.; Gutmann, D.H. Spatially- and temporally-controlled postnatal p53 knockdown cooperates with embryonic Schwann cell precursor Nf1 gene loss to promote malignant peripheral nerve sheath tumor formation. Oncotarget 2016, 7, 7403–7414. [Google Scholar] [CrossRef] [Green Version]
- Ramkissoon, A.; Chaney, K.E.; Milewski, D.; Williams, K.B.; Williams, R.L.; Choi, K.; Miller, A.; Kalin, T.V.; Pressey, J.G.; Szabo, S.; et al. Targeted Inhibition of the Dual Specificity Phosphatases DUSP1 and DUSP6 Suppress MPNST Growth via JNK. Clin. Cancer Res. 2019, 25, 4117–4127. [Google Scholar] [CrossRef] [Green Version]
- Castellsague, J.; Gel, B.; Fernandez-Rodriguez, J.; Llatjos, R.; Blanco, I.; Benavente, Y.; Perez-Sidelnikova, D.; Garcia-Del Muro, J.; Vinals, J.M.; Vidal, A.; et al. Comprehensive establishment and characterization of orthoxenograft mouse models of malignant peripheral nerve sheath tumors for personalized medicine. EMBO Mol. Med. 2015, 7, 608–627. [Google Scholar] [CrossRef]
- Perrin, G.Q.; Li, H.; Fishbein, L.; Thomson, S.A.; Hwang, M.S.; Scarborough, M.T.; Yachnis, A.T.; Wallace, M.R.; Mareci, T.H.; Muir, D. An orthotopic xenograft model of intraneural NF1 MPNST suggests a potential association between steroid hormones and tumor cell proliferation. Lab. Investig. 2007, 87, 1092–1102. [Google Scholar] [CrossRef] [Green Version]
- Brosseau, J.P.; Liao, C.P.; Wang, Y.; Ramani, V.; Vandergriff, T.; Lee, M.; Patel, A.; Ariizumi, K.; Le, L.Q. NF1 heterozygosity fosters de novo tumorigenesis but impairs malignant transformation. Nat. Commun. 2018, 9, 5014. [Google Scholar] [CrossRef]
- DeClue, J.E.; Heffelfinger, S.; Benvenuto, G.; Ling, B.; Li, S.; Rui, W.; Vass, W.C.; Viskochil, D.; Ratner, N. Epidermal growth factor receptor expression in neurofibromatosis type 1-related tumors and NF1 animal models. J. Clin. Investig. 2000, 105, 1233–1241. [Google Scholar] [CrossRef] [Green Version]
- Holtkamp, N.; Malzer, E.; Zietsch, J.; Okuducu, A.F.; Mucha, J.; Mawrin, C.; Mautner, V.F.; Schildhaus, H.U.; von Deimling, A. EGFR and erbB2 in malignant peripheral nerve sheath tumors and implications for targeted therapy. Neuro Oncol. 2008, 10, 946–957. [Google Scholar] [CrossRef] [Green Version]
- Stonecypher, M.S.; Byer, S.J.; Grizzle, W.E.; Carroll, S.L. Activation of the neuregulin-1/ErbB signaling pathway promotes the proliferation of neoplastic Schwann cells in human malignant peripheral nerve sheath tumors. Oncogene 2005, 24, 5589–5605. [Google Scholar] [CrossRef] [Green Version]
- Huijbregts, R.P.; Roth, K.A.; Schmidt, R.E.; Carroll, S.L. Hypertrophic neuropathies and malignant peripheral nerve sheath tumors in transgenic mice overexpressing glial growth factor beta3 in myelinating Schwann cells. J. Neurosci. 2003, 23, 7269–7280. [Google Scholar] [CrossRef] [Green Version]
- Ling, B.C.; Wu, J.; Miller, S.J.; Monk, K.R.; Shamekh, R.; Rizvi, T.A.; Decourten-Myers, G.; Vogel, K.S.; DeClue, J.E.; Ratner, N. Role for the epidermal growth factor receptor in neurofibromatosis-related peripheral nerve tumorigenesis. Cancer Cell 2005, 7, 65–75. [Google Scholar] [CrossRef] [Green Version]
- Kazmi, S.J.; Byer, S.J.; Eckert, J.M.; Turk, A.N.; Huijbregts, R.P.; Brossier, N.M.; Grizzle, W.E.; Mikhail, F.M.; Roth, K.A.; Carroll, S.L. Transgenic mice overexpressing neuregulin-1 model neurofibroma-malignant peripheral nerve sheath tumor progression and implicate specific chromosomal copy number variations in tumorigenesis. Am. J. Pathol. 2013, 182, 646–667. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Williams, J.P.; Rizvi, T.A.; Kordich, J.J.; Witte, D.; Meijer, D.; Stemmer-Rachamimov, A.O.; Cancelas, J.A.; Ratner, N. Plexiform and dermal neurofibromas and pigmentation are caused by Nf1 loss in desert hedgehog-expressing cells. Cancer Cell 2008, 13, 105–116. [Google Scholar] [CrossRef] [Green Version]
- Wu, J.; Patmore, D.M.; Jousma, E.; Eaves, D.W.; Breving, K.; Patel, A.V.; Schwartz, E.B.; Fuchs, J.R.; Cripe, T.P.; Stemmer-Rachamimov, A.O.; et al. EGFR-STAT3 signaling promotes formation of malignant peripheral nerve sheath tumors. Oncogene 2014, 33, 173–180. [Google Scholar] [CrossRef] [Green Version]
- Keng, V.W.; Rahrmann, E.P.; Watson, A.L.; Tschida, B.R.; Moertel, C.L.; Jessen, W.J.; Rizvi, T.A.; Collins, M.H.; Ratner, N.; Largaespada, D.A. PTEN and NF1 inactivation in Schwann cells produces a severe phenotype in the peripheral nervous system that promotes the development and malignant progression of peripheral nerve sheath tumors. Cancer Res. 2012, 72, 3405–3413. [Google Scholar] [CrossRef] [Green Version]
- Chaney, K.E.; Perrino, M.R.; Kershner, L.J.; Patel, A.V.; Wu, J.; Choi, K.; Rizvi, T.A.; Dombi, E.; Szabo, S.; Largaespada, D.A.; et al. Cdkn2a Loss in a Model of Neurofibroma Demonstrates Stepwise Tumor Progression to Atypical Neurofibroma and MPNST. Cancer Res. 2020, 80, 4720–4730. [Google Scholar] [CrossRef]
- Rhodes, S.D.; He, Y.; Smith, A.; Jiang, L.; Lu, Q.; Mund, J.; Li, X.; Bessler, W.; Qian, S.; Dyer, W.; et al. Cdkn2a (Arf) loss drives NF1-associated atypical neurofibroma and malignant transformation. Hum. Mol. Genet. 2019. [Google Scholar] [CrossRef]
- Dodd, R.D.; Lee, C.L.; Overton, T.; Huang, W.; Eward, W.C.; Luo, L.; Ma, Y.; Ingram, D.R.; Torres, K.E.; Cardona, D.M.; et al. NF1(+/-) Hematopoietic Cells Accelerate Malignant Peripheral Nerve Sheath Tumor Development without Altering Chemotherapy Response. Cancer Res. 2017, 77, 4486–4497. [Google Scholar] [CrossRef] [Green Version]
- Miettinen, M.M.; Antonescu, C.R.; Fletcher, C.D.M.; Kim, A.; Lazar, A.J.; Quezado, M.M.; Reilly, K.M.; Stemmer-Rachamimov, A.; Stewart, D.R.; Viskochil, D.; et al. Histopathologic evaluation of atypical neurofibromatous tumors and their transformation into malignant peripheral nerve sheath tumor in patients with neurofibromatosis 1-a consensus overview. Hum. Pathol. 2017, 67, 1–10. [Google Scholar] [CrossRef]
- Cleven, A.H.; Al Sannaa, G.A.; Briaire-de Bruijn, I.; Ingram, D.R.; van de Rijn, M.; Rubin, B.P.; de Vries, M.W.; Watson, K.L.; Torres, K.E.; Wang, W.L.; et al. Loss of H3K27 tri-methylation is a diagnostic marker for malignant peripheral nerve sheath tumors and an indicator for an inferior survival. Mod. Pathol. 2016, 29, 1113. [Google Scholar] [CrossRef] [Green Version]
- Otsuka, H.; Kohashi, K.; Yoshimoto, M.; Ishihara, S.; Toda, Y.; Yamada, Y.; Yamamoto, H.; Nakashima, Y.; Oda, Y. Immunohistochemical evaluation of H3K27 trimethylation in malignant peripheral nerve sheath tumors. Pathol. Res. Pract. 2018, 214, 417–425. [Google Scholar] [CrossRef]
- Prieto-Granada, C.N.; Wiesner, T.; Messina, J.L.; Jungbluth, A.A.; Chi, P.; Antonescu, C.R. Loss of H3K27me3 Expression Is a Highly Sensitive Marker for Sporadic and Radiation-induced MPNST. Am. J. Surg. Pathol. 2016, 40, 479–489. [Google Scholar] [CrossRef] [Green Version]
- Jessen, K.R.; Mirsky, R. The origin and development of glial cells in peripheral nerves. Nat. Rev. Neurosci. 2005, 6, 671–682. [Google Scholar] [CrossRef]
- Brosseau, J.P.; Sathe, A.A.; Wang, Y.; Nguyen, T.; Glass, D.A., 2nd; Xing, C.; Le, L.Q. Human cutaneous neurofibroma matrisome revealed by single-cell RNA sequencing. Acta Neuropathol. Commun. 2021, 9, 11. [Google Scholar] [CrossRef]
- Parrinello, S.; Lloyd, A.C. Neurofibroma development in NF1--insights into tumour initiation. Trends Cell Biol. 2009, 19, 395–403. [Google Scholar] [CrossRef]
- Joseph, N.M.; Mukouyama, Y.S.; Mosher, J.T.; Jaegle, M.; Crone, S.A.; Dormand, E.L.; Lee, K.F.; Meijer, D.; Anderson, D.J.; Morrison, S.J. Neural crest stem cells undergo multilineage differentiation in developing peripheral nerves to generate endoneurial fibroblasts in addition to Schwann cells. Development 2004, 131, 5599–5612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serra, E.; Rosenbaum, T.; Winner, U.; Aledo, R.; Ars, E.; Estivill, X.; Lenard, H.G.; Lazaro, C. Schwann cells harbor the somatic NF1 mutation in neurofibromas: Evidence of two different Schwann cell subpopulations. Hum. Mol. Genet. 2000, 9, 3055–3064. [Google Scholar] [CrossRef] [Green Version]
- Perry, A.; Roth, K.A.; Banerjee, R.; Fuller, C.E.; Gutmann, D.H. NF1 deletions in S-100 protein-positive and negative cells of sporadic and neurofibromatosis 1 (NF1)-associated plexiform neurofibromas and malignant peripheral nerve sheath tumors. Am. J. Pathol. 2001, 159, 57–61. [Google Scholar] [CrossRef] [Green Version]
- Kluwe, L.; Friedrich, R.; Mautner, V.F. Loss of NF1 allele in Schwann cells but not in fibroblasts derived from an NF1-associated neurofibroma. Genes Chromosom. Cancer 1999, 24, 283–285. [Google Scholar] [CrossRef]
- Khalifa, M.A.; Montgomery, E.A.; Ismiil, N.; Azumi, N. What are the CD34+ cells in benign peripheral nerve sheath tumors? Double immunostaining study of CD34 and S-100 protein. Am. J. Clin. Pathol. 2000, 114, 123–126. [Google Scholar] [CrossRef] [Green Version]
- Hirose, T.; Tani, T.; Shimada, T.; Ishizawa, K.; Shimada, S.; Sano, T. Immunohistochemical demonstration of EMA/Glut1-positive perineurial cells and CD34-positive fibroblastic cells in peripheral nerve sheath tumors. Mod. Pathol. 2003, 16, 293–298. [Google Scholar] [CrossRef]
- Wei, C.J.; Gu, Y.H.; Wang, W.; Ren, J.Y.; Cui, X.W.; Lian, X.; Liu, J.; Wang, H.J.; Gu, B.; Li, Q.F.; et al. A narrative review of the role of fibroblasts in the growth and development of neurogenic tumors. Ann. Transl. Med. 2020, 8, 1462. [Google Scholar] [CrossRef]
- Richard, L.; Vedrenne, N.; Vallat, J.M.; Funalot, B. Characterization of Endoneurial Fibroblast-like Cells from Human and Rat Peripheral Nerves. J. Histochem. Cytochem. 2014, 62, 424–435. [Google Scholar] [CrossRef] [Green Version]
- Gesundheit, B.; Parkin, P.; Greenberg, M.; Baruchel, S.; Senger, C.; Kapelushnik, J.; Smith, C.; Klement, G.L. The role of angiogenesis in the transformation of plexiform neurofibroma into malignant peripheral nerve sheath tumors in children with neurofibromatosis type 1. J. Pediatr. Hematol. Oncol. 2010, 32, 548–553. [Google Scholar] [CrossRef] [Green Version]
- Ozerdem, U. Targeting neovascular pericytes in neurofibromatosis type 1. Angiogenesis 2004, 7, 307–311. [Google Scholar] [CrossRef] [Green Version]
- Maki, R.G.; D’Adamo, D.R.; Keohan, M.L.; Saulle, M.; Schuetze, S.M.; Undevia, S.D.; Livingston, M.B.; Cooney, M.M.; Hensley, M.L.; Mita, M.M.; et al. Phase II study of sorafenib in patients with metastatic or recurrent sarcomas. J. Clin. Oncol. 2009, 27, 3133–3140. [Google Scholar] [CrossRef] [Green Version]
- Widemann, B.C.; Meyer, C.F.; Cote, G.M.; Chugh, R.; Milhem, M.M.; Tine, B.A.V.; Kim, A.; Turpin, B.; Dombi, E.; Jayaprakash, N.; et al. SARC016: Phase II study of everolimus in combination with bevacizumab in sporadic and neurofibromatosis type 1 (NF1) related refractory malignant peripheral nerve sheath tumors (MPNST). J. Clin. Oncol. 2016, 34, 11053. [Google Scholar] [CrossRef]
- Fisher, M.J.; Shih, C.S.; Rhodes, S.D.; Armstrong, A.E.; Wolters, P.L.; Dombi, E.; Zhang, C.; Angus, S.P.; Johnson, G.L.; Packer, R.J.; et al. Cabozantinib for neurofibromatosis type 1-related plexiform neurofibromas: A phase 2 trial. Nat. Med. 2021, 27, 165–173. [Google Scholar] [CrossRef]
- Yang, F.C.; Ingram, D.A.; Chen, S.; Hingtgen, C.M.; Ratner, N.; Monk, K.R.; Clegg, T.; White, H.; Mead, L.; Wenning, M.J.; et al. Neurofibromin-deficient Schwann cells secrete a potent migratory stimulus for Nf1+/- mast cells. J. Clin. Investig. 2003, 112, 1851–1861. [Google Scholar] [CrossRef] [Green Version]
- Liao, C.P.; Booker, R.C.; Brosseau, J.P.; Chen, Z.; Mo, J.; Tchegnon, E.; Wang, Y.; Clapp, D.W.; Le, L.Q. Contributions of inflammation and tumor microenvironment to neurofibroma tumorigenesis. J. Clin. Investig. 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, F.C.; Ingram, D.A.; Chen, S.; Zhu, Y.; Yuan, J.; Li, X.; Yang, X.; Knowles, S.; Horn, W.; Li, Y.; et al. Nf1-dependent tumors require a microenvironment containing Nf1+/-- and c-kit-dependent bone marrow. Cell 2008, 135, 437–448. [Google Scholar] [CrossRef] [Green Version]
- Friedrich, R.E.; Naber, U.; Glatzel, M.; Hagel, C. Vessel and Mast Cell Densities in Sporadic and Syndrome-associated Peripheral Nerve Sheath Tumors. Anticancer Res. 2015, 35, 4713–4722. [Google Scholar]
- Vasconcelos, R.A.T.; Guimaraes Coscarelli, P.; Vieira, T.M.; Noguera, W.S.; Rapozo, D.C.M.; Acioly, M.A. Prognostic significance of mast cell and microvascular densities in malignant peripheral nerve sheath tumor with and without neurofibromatosis type 1. Cancer Med. 2019, 8, 972–981. [Google Scholar] [CrossRef] [Green Version]
- Prada, C.E.; Jousma, E.; Rizvi, T.A.; Wu, J.; Dunn, R.S.; Mayes, D.A.; Cancelas, J.A.; Dombi, E.; Kim, M.O.; West, B.L.; et al. Neurofibroma-associated macrophages play roles in tumor growth and response to pharmacological inhibition. Acta Neuropathol. 2013, 125, 159–168. [Google Scholar] [CrossRef]
- Choi, K.; Komurov, K.; Fletcher, J.S.; Jousma, E.; Cancelas, J.A.; Wu, J.; Ratner, N. An inflammatory gene signature distinguishes neurofibroma Schwann cells and macrophages from cells in the normal peripheral nervous system. Sci. Rep. 2017, 7, 43315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haworth, K.B.; Arnold, M.A.; Pierson, C.R.; Choi, K.; Yeager, N.D.; Ratner, N.; Roberts, R.D.; Finlay, J.L.; Cripe, T.P. Immune profiling of NF1-associated tumors reveals histologic subtype distinctions and heterogeneity: Implications for immunotherapy. Oncotarget 2017, 8, 82037–82048. [Google Scholar] [CrossRef] [Green Version]
- Shurell, E.; Singh, A.S.; Crompton, J.G.; Jensen, S.; Li, Y.; Dry, S.; Nelson, S.; Chmielowski, B.; Bernthal, N.; Federman, N.; et al. Characterizing the immune microenvironment of malignant peripheral nerve sheath tumor by PD-L1 expression and presence of CD8+ tumor infiltrating lymphocytes. Oncotarget 2016, 7, 64300–64308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Davis, L.E.; Nicholls, L.A.; Babiker, H.M.; Liau, J.; Mahadevan, D. PD-1 Inhibition Achieves a Complete Metabolic Response in a Patient with Malignant Peripheral Nerve Sheath Tumor. Cancer Immunol. Res. 2019, 7, 1396–1400. [Google Scholar] [CrossRef]
- Brosseau, J.P.; Le, L.Q. Heterozygous Tumor Suppressor Microenvironment in Cancer Development. Trends Cancer 2019, 5, 541–546. [Google Scholar] [CrossRef]
- Brosseau, J.P.; Liao, C.P.; Le, L.Q. Translating current basic research into future therapies for neurofibromatosis type 1. Br. J. Cancer 2020, 123, 178–186. [Google Scholar] [CrossRef]
- Hirbe, A.C.; Kaushal, M.; Sharma, M.K.; Dahiya, S.; Pekmezci, M.; Perry, A.; Gutmann, D.H. Clinical genomic profiling identifies TYK2 mutation and overexpression in patients with neurofibromatosis type 1-associated malignant peripheral nerve sheath tumors. Cancer 2017, 123, 1194–1201. [Google Scholar] [CrossRef] [Green Version]
- Castellano, E.; Downward, J. RAS Interaction with PI3K: More Than Just Another Effector Pathway. Genes Cancer 2011, 2, 261–274. [Google Scholar] [CrossRef] [Green Version]
- Simanshu, D.K.; Nissley, D.V.; McCormick, F. RAS Proteins and Their Regulators in Human Disease. Cell 2017, 170, 17–33. [Google Scholar] [CrossRef] [Green Version]
- Brossier, N.M.; Prechtl, A.M.; Longo, J.F.; Barnes, S.; Wilson, L.S.; Byer, S.J.; Brosius, S.N.; Carroll, S.L. Classic Ras Proteins Promote Proliferation and Survival via Distinct Phosphoproteome Alterations in Neurofibromin-Null Malignant Peripheral Nerve Sheath Tumor Cells. J. Neuropathol. Exp. Neurol. 2015, 74, 568–586. [Google Scholar] [CrossRef] [Green Version]
- Kaplan, H.G. Vemurafenib treatment of BRAF V600E-mutated malignant peripheral nerve sheath tumor. J. Natl. Compr. Cancer Netw. 2013, 11, 1466–1470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ambrosini, G.; Cheema, H.S.; Seelman, S.; Teed, A.; Sambol, E.B.; Singer, S.; Schwartz, G.K. Sorafenib inhibits growth and mitogen-activated protein kinase signaling in malignant peripheral nerve sheath cells. Mol. Cancer Ther. 2008, 7, 890–896. [Google Scholar] [CrossRef] [Green Version]
- Zou, C.; Smith, K.D.; Liu, J.; Lahat, G.; Myers, S.; Wang, W.L.; Zhang, W.; McCutcheon, I.E.; Slopis, J.M.; Lazar, A.J.; et al. Clinical, pathological, and molecular variables predictive of malignant peripheral nerve sheath tumor outcome. Ann. Surg. 2009, 249, 1014–1022. [Google Scholar] [CrossRef]
- Endo, M.; Yamamoto, H.; Setsu, N.; Kohashi, K.; Takahashi, Y.; Ishii, T.; Iida, K.; Matsumoto, Y.; Hakozaki, M.; Aoki, M.; et al. Prognostic significance of AKT/mTOR and MAPK pathways and antitumor effect of mTOR inhibitor in NF1-related and sporadic malignant peripheral nerve sheath tumors. Clin. Cancer Res. 2013, 19, 450–461. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dombi, E.; Baldwin, A.; Marcus, L.J.; Fisher, M.J.; Weiss, B.; Kim, A.; Whitcomb, P.; Martin, S.; Aschbacher-Smith, L.E.; Rizvi, T.A.; et al. Activity of Selumetinib in Neurofibromatosis Type 1-Related Plexiform Neurofibromas. N. Engl. J. Med. 2016, 375, 2550–2560. [Google Scholar] [CrossRef]
- Nagabushan, S.; Lau, L.M.S.; Barahona, P.; Wong, M.; Sherstyuk, A.; Marshall, G.M.; Tyrrell, V.; Wegner, E.A.; Ekert, P.G.; Cowley, M.J.; et al. Efficacy of MEK inhibition in a recurrent malignant peripheral nerve sheath tumor. NPJ Precis. Oncol. 2021, 5, 9. [Google Scholar] [CrossRef]
- Kim, J.; Guan, K.L. mTOR as a central hub of nutrient signalling and cell growth. Nat. Cell Biol. 2019, 21, 63–71. [Google Scholar] [CrossRef]
- Johansson, G.; Mahller, Y.Y.; Collins, M.H.; Kim, M.O.; Nobukuni, T.; Perentesis, J.; Cripe, T.P.; Lane, H.A.; Kozma, S.C.; Thomas, G.; et al. Effective in vivo targeting of the mammalian target of rapamycin pathway in malignant peripheral nerve sheath tumors. Mol. Cancer Ther. 2008, 7, 1237–1245. [Google Scholar] [CrossRef] [Green Version]
- Varin, J.; Poulain, L.; Hivelin, M.; Nusbaum, P.; Hubas, A.; Laurendeau, I.; Lantieri, L.; Wolkenstein, P.; Vidaud, M.; Pasmant, E.; et al. Dual mTORC1/2 inhibition induces anti-proliferative effect in NF1-associated plexiform neurofibroma and malignant peripheral nerve sheath tumor cells. Oncotarget 2016, 7, 35753–35767. [Google Scholar] [CrossRef] [Green Version]
- Sharma, M.; Castro-Piedras, I.; Simmons, G.E., Jr.; Pruitt, K. Dishevelled: A masterful conductor of complex Wnt signals. Cell Signal. 2018, 47, 52–64. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.; Lee, J.K.; Ahn, S.H.; Lee, J.; Nam, D.H. WNT signaling in glioblastoma and therapeutic opportunities. Lab. Investig. 2016, 96, 137–150. [Google Scholar] [CrossRef] [Green Version]
- Walker, J.A.; Upadhyaya, M. Emerging therapeutic targets for neurofibromatosis type 1. Expert Opin. Ther. Targets 2018, 22, 419–437. [Google Scholar] [CrossRef] [PubMed]
- Watson, A.L.; Rahrmann, E.P.; Moriarity, B.S.; Choi, K.; Conboy, C.B.; Greeley, A.D.; Halfond, A.L.; Anderson, L.K.; Wahl, B.R.; Keng, V.W.; et al. Canonical Wnt/beta-catenin signaling drives human schwann cell transformation, progression, and tumor maintenance. Cancer Discov. 2013, 3, 674–689. [Google Scholar] [CrossRef] [Green Version]
- Zhan, T.; Rindtorff, N.; Boutros, M. Wnt signaling in cancer. Oncogene 2017, 36, 1461–1473. [Google Scholar] [CrossRef] [PubMed]
- Ghadimi, M.P.; Young, E.D.; Belousov, R.; Zhang, Y.; Lopez, G.; Lusby, K.; Kivlin, C.; Demicco, E.G.; Creighton, C.J.; Lazar, A.J.; et al. Survivin is a viable target for the treatment of malignant peripheral nerve sheath tumors. Clin. Cancer Res. 2012, 18, 2545–2557. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mo, W.; Chen, J.; Patel, A.; Zhang, L.; Chau, V.; Li, Y.; Cho, W.; Lim, K.; Xu, J.; Lazar, A.J.; et al. CXCR4/CXCL12 mediate autocrine cell- cycle progression in NF1-associated malignant peripheral nerve sheath tumors. Cell 2013, 152, 1077–1090. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karpinsky, G.; Krawczyk, M.A.; Izycka-Swieszewska, E.; Fatyga, A.; Budka, A.; Balwierz, W.; Sobol, G.; Zalewska-Szewczyk, B.; Rychlowska-Pruszynska, M.; Klepacka, T.; et al. Tumor expression of survivin, p53, cyclin D1, osteopontin and fibronectin in predicting the response to neo-adjuvant chemotherapy in children with advanced malignant peripheral nerve sheath tumor. J. Cancer Res. Clin. Oncol. 2018, 144, 519–529. [Google Scholar] [CrossRef] [Green Version]
- Song, D.H.; Sussman, D.J.; Seldin, D.C. Endogenous protein kinase CK2 participates in Wnt signaling in mammary epithelial cells. J. Biol. Chem. 2000, 275, 23790–23797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kendall, J.J.; Chaney, K.E.; Patel, A.V.; Rizvi, T.A.; Largaespada, D.A.; Ratner, N. CK2 blockade causes MPNST cell apoptosis and promotes degradation of beta-catenin. Oncotarget 2016, 7, 53191–53203. [Google Scholar] [CrossRef] [Green Version]
- Kim, K.A.; Wagle, M.; Tran, K.; Zhan, X.; Dixon, M.A.; Liu, S.; Gros, D.; Korver, W.; Yonkovich, S.; Tomasevic, N.; et al. R-Spondin family members regulate the Wnt pathway by a common mechanism. Mol. Biol. Cell 2008, 19, 2588–2596. [Google Scholar] [CrossRef] [Green Version]
- Binnerts, M.E.; Kim, K.A.; Bright, J.M.; Patel, S.M.; Tran, K.; Zhou, M.; Leung, J.M.; Liu, Y.; Lomas, W.E., 3rd; Dixon, M.; et al. R-Spondin1 regulates Wnt signaling by inhibiting internalization of LRP6. Proc. Natl. Acad. Sci. USA 2007, 104, 14700–14705. [Google Scholar] [CrossRef] [Green Version]
- Carmon, K.S.; Gong, X.; Lin, Q.; Thomas, A.; Liu, Q. R-spondins function as ligands of the orphan receptors LGR4 and LGR5 to regulate Wnt/beta-catenin signaling. Proc. Natl. Acad. Sci. USA 2011, 108, 11452–11457. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luscan, A.; Shackleford, G.; Masliah-Planchon, J.; Laurendeau, I.; Ortonne, N.; Varin, J.; Lallemand, F.; Leroy, K.; Dumaine, V.; Hivelin, M.; et al. The activation of the WNT signaling pathway is a Hallmark in neurofibromatosis type 1 tumorigenesis. Clin. Cancer Res. 2014, 20, 358–371. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, P.; Chu, J.; Wu, Y.; Sun, L.; Lv, X.; Zhu, Y.; Li, J.; Guo, Q.; Gong, C.; Liu, B.; et al. NBAT1 suppresses breast cancer metastasis by regulating DKK1 via PRC2. Oncotarget 2015, 6, 32410–32425. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Velez-Reyes, G.L.; Koes, N.; Ryu, J.H.; Kaufmann, G.; Berner, M.; Weg, M.T.; Wolf, N.K.; Rathe, S.K.; Ratner, N.; Moriarity, B.S.; et al. Transposon Mutagenesis-Guided CRISPR/Cas9 Screening Strongly Implicates Dysregulation of Hippo/YAP Signaling in Malignant Peripheral Nerve Sheath Tumor Development. Cancers 2021, 13, 1584. [Google Scholar] [CrossRef]
- Kumar, R.; Gururaj, A.E.; Barnes, C.J. p21-activated kinases in cancer. Nat. Rev. Cancer 2006, 6, 459–471. [Google Scholar] [CrossRef] [PubMed]
- Semenova, G.; Stepanova, D.S.; Dubyk, C.; Handorf, E.; Deyev, S.M.; Lazar, A.J.; Chernoff, J. Targeting group I p21-activated kinases to control malignant peripheral nerve sheath tumor growth and metastasis. Oncogene 2017, 36, 5421–5431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paz, A.; Haklai, R.; Elad-Sfadia, G.; Ballan, E.; Kloog, Y. Galectin-1 binds oncogenic H-Ras to mediate Ras membrane anchorage and cell transformation. Oncogene 2001, 20, 7486–7493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih, T.C.; Fan, Y.; Kiss, S.; Li, X.; Deng, X.N.; Liu, R.; Chen, X.J.; Carney, R.; Chen, A.; Ghosh, P.M.; et al. Galectin-1 inhibition induces cell apoptosis through dual suppression of CXCR4 and Ras pathways in human malignant peripheral nerve sheath tumors. Neuro Oncol. 2019, 21, 1389–1400. [Google Scholar] [CrossRef] [PubMed]
- Palomo-Irigoyen, M.; Perez-Andres, E.; Iruarrizaga-Lejarreta, M.; Barreira-Manrique, A.; Tamayo-Caro, M.; Vila-Vecilla, L.; Moreno-Cugnon, L.; Beitia, N.; Medrano, D.; Fernandez-Ramos, D.; et al. HuR/ELAVL1 drives malignant peripheral nerve sheath tumor growth and metastasis. J. Clin. Investig. 2020, 130, 3848–3864. [Google Scholar] [CrossRef] [Green Version]
- Yang, K.; Du, J.; Shi, D.; Ji, F.; Ji, Y.; Pan, J.; Lv, F.; Zhang, Y.; Zhang, J. Knockdown of MSI2 inhibits metastasis by interacting with caveolin-1 and inhibiting its ubiquitylation in human NF1-MPNST cells. Cell Death Dis. 2020, 11, 489. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
| Mouse Model | Penetrance | Histological Criteria 1 | Precursor Lesion? | Reference |
|---|---|---|---|---|
| cisNf1+/−p53+/− | 30% |
| Not observed | [26] |
| cisNf1+/−p53+/− | N/A |
| Not observed | [27] |
| Gfap-Cre Pten f/+ LSL KrasG12D | 100% |
| Neurofibroma | [16] |
| Dhh-CreNf1f/fPtenf/f | N/A |
| N/A | [41] |
| P0-GGFb3 | 74% |
| Neurofibroma | [38] |
| Dhh-CreNf1f/f CNPase-hEGFR | 33% |
| Neurofibroma | [40] |
| Gfap-CreNf1f/f OR Gfap-CreNf1f/− OR Postn-CreNf1f/f +pTOMOshp53 | 60–70% |
| Hyperplasia | [28] |
| Nf1f/fArff/f OR Nf1f/−Arff/f | N/A |
| Not observed | [44] |
| Plp-creERT2 Nf1f/f | 10% |
| Neurofibroma | [32] |
| Postn-Cre Nf1f/fArff/f OR Postn-Cre Nf1f/fArff/+ | 100% |
| Neurofibroma and ANNUBP | [43] |
| Dhh-Cre Nf1f/fArf−/− OR Dhh-Cre Nf1f/fArf+/− | 40% |
| Neurofibroma and ANNUBP | [42] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mohamad, T.; Plante, C.; Brosseau, J.-P. Toward Understanding the Mechanisms of Malignant Peripheral Nerve Sheath Tumor Development. Int. J. Mol. Sci. 2021, 22, 8620. https://doi.org/10.3390/ijms22168620
Mohamad T, Plante C, Brosseau J-P. Toward Understanding the Mechanisms of Malignant Peripheral Nerve Sheath Tumor Development. International Journal of Molecular Sciences. 2021; 22(16):8620. https://doi.org/10.3390/ijms22168620
Chicago/Turabian StyleMohamad, Teddy, Camille Plante, and Jean-Philippe Brosseau. 2021. "Toward Understanding the Mechanisms of Malignant Peripheral Nerve Sheath Tumor Development" International Journal of Molecular Sciences 22, no. 16: 8620. https://doi.org/10.3390/ijms22168620
APA StyleMohamad, T., Plante, C., & Brosseau, J.-P. (2021). Toward Understanding the Mechanisms of Malignant Peripheral Nerve Sheath Tumor Development. International Journal of Molecular Sciences, 22(16), 8620. https://doi.org/10.3390/ijms22168620