
Proteomics of Mouse Heart Ventricles Reveals Mitochondria and Metabolism as Major Targets of a Post-Infarction Short-Acting GLP1Ra-Therapy

 , , ,
, , ,
Abstract
:1. Introduction
2. Results
2.1. Mitochondrial Proteins Have a Central Role in the Course of Cardiac Remodeling
2.2. Oxidative Phosphorylation and Multiple Cellular Metabolic Pathways Are Regulated in the First Week of Post-Infarction Remodeling
2.3. Cross-Comparative Analysis Identifies Mitochondrial Compartments as the Main Cellular Components of DEPs
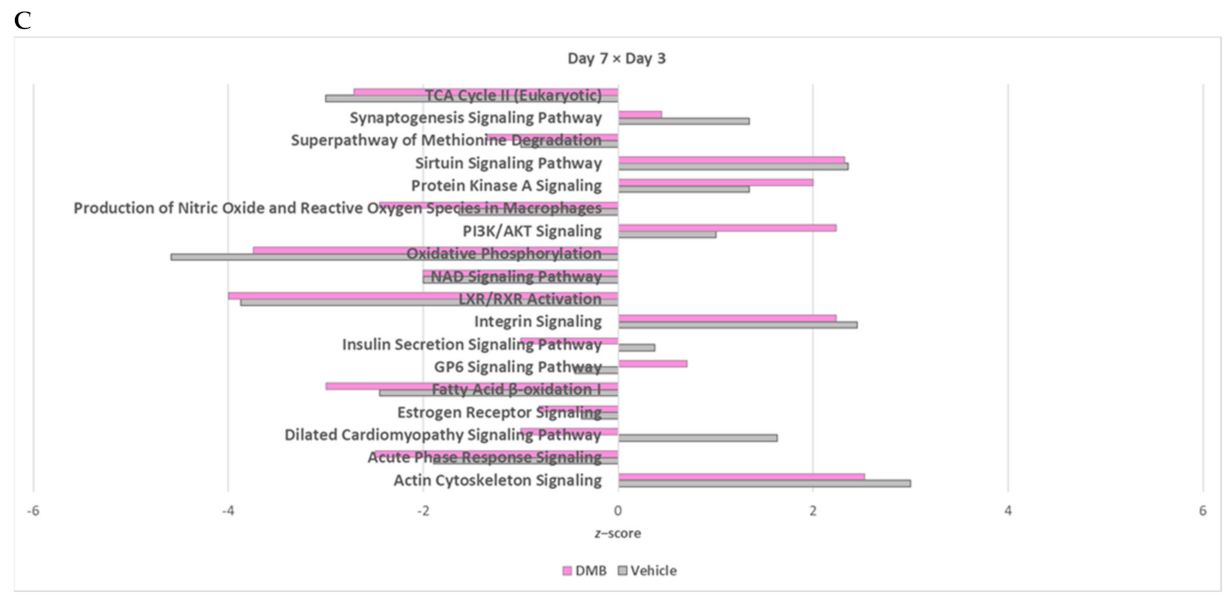
2.4. The Cross-Comparative Analysis Reveals a Predominant Role of DMB in Regulating the Redox Signaling, ATP Production and Overal Metabolism during Cardiac Ischemia
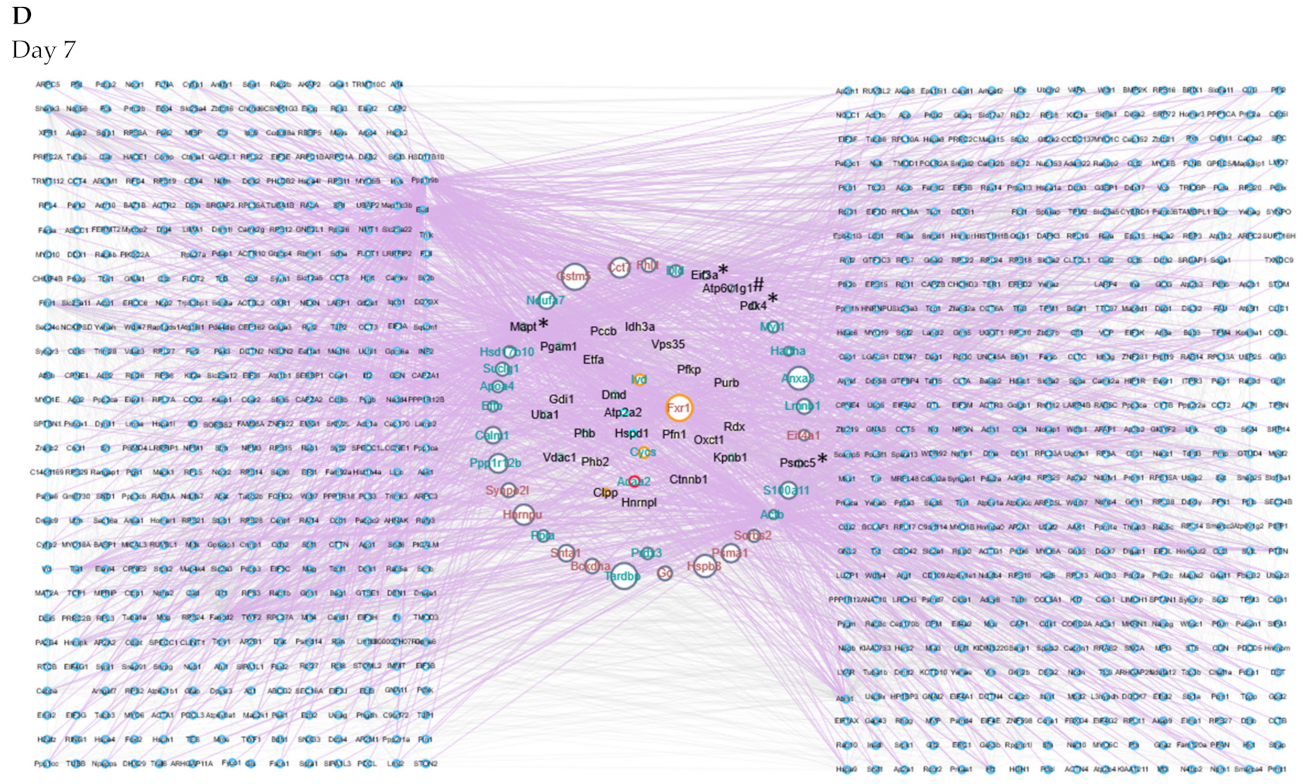
2.5. Protein–Protein Interaction Networks Reveal Dynamic Changes in Protein Expression in Early Remodeling
3. Discussion
4. Materials and Methods
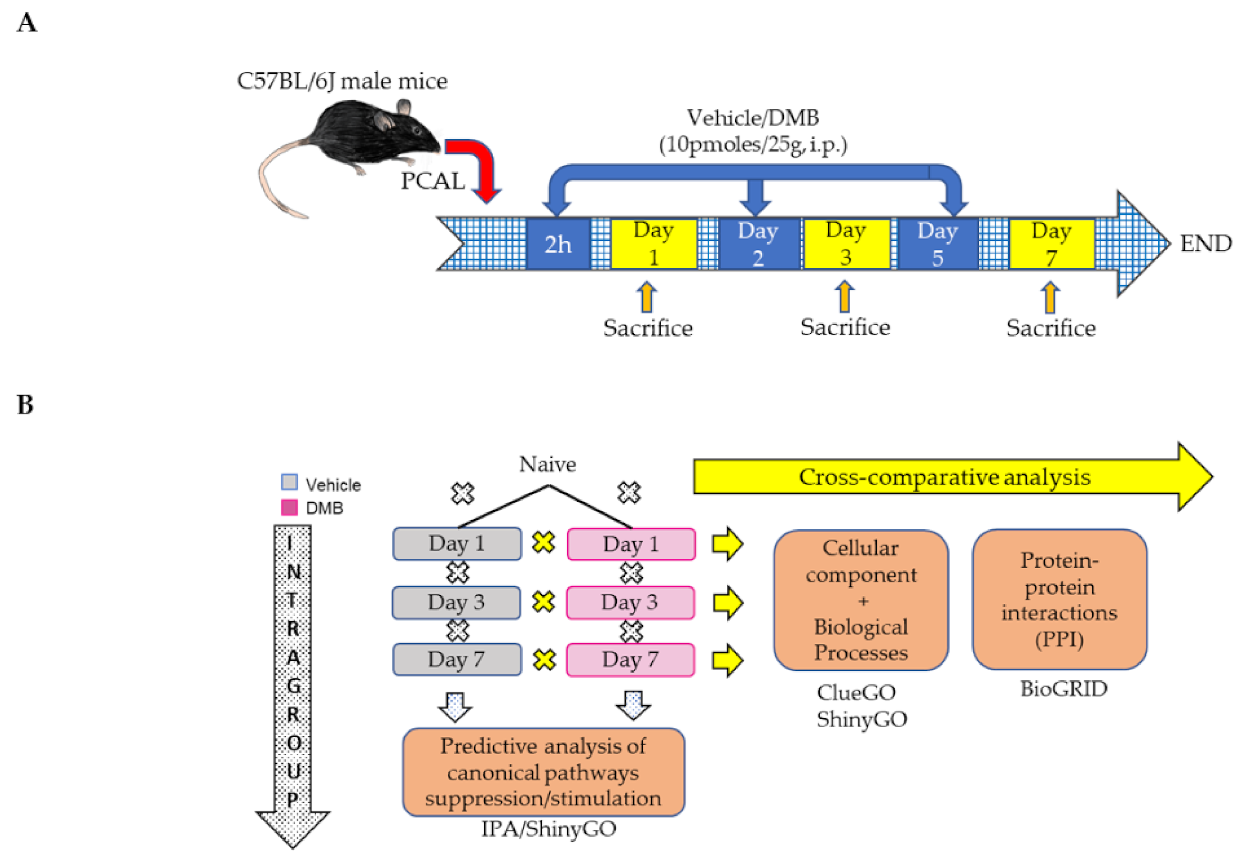
4.1. Permanent Coronary Artery Ligation (PCAL) and DMB Treatment
4.2. Proteomic Analysis
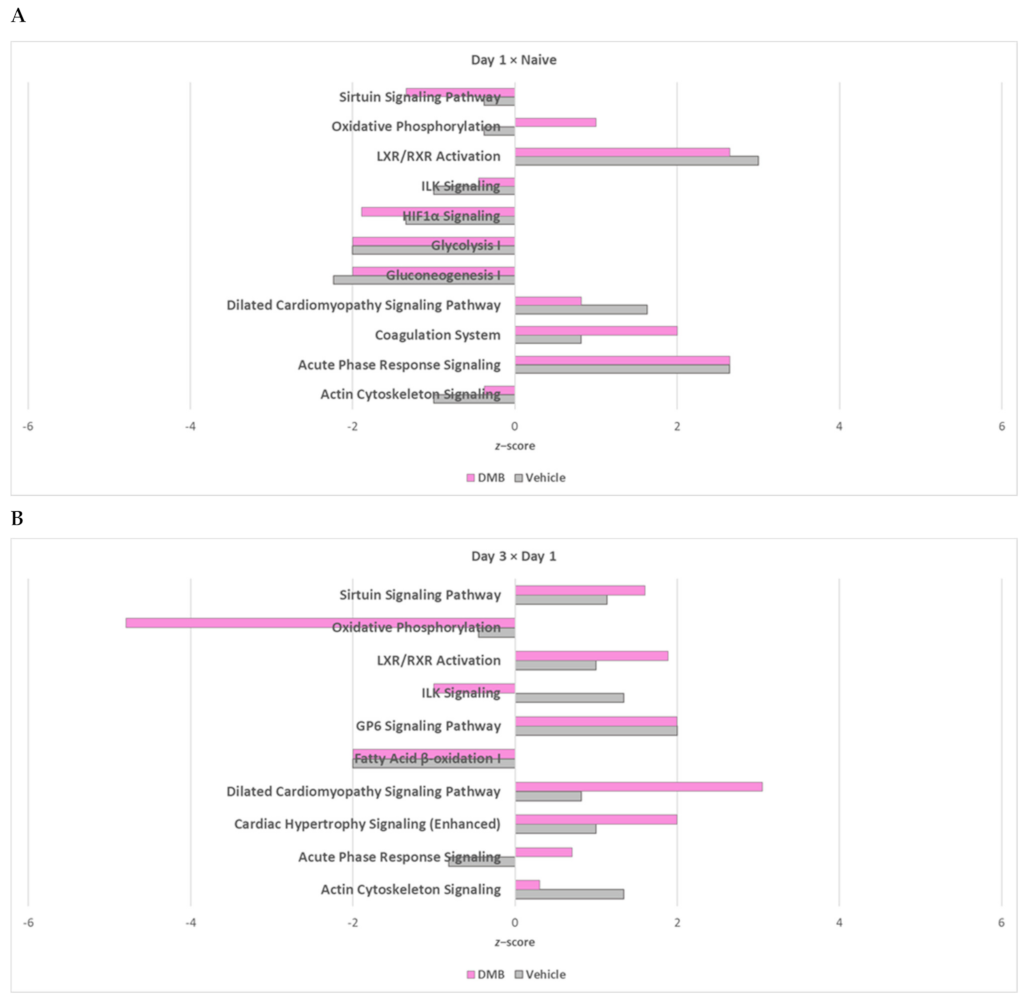
4.3. Bioinformatic and Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Galli, A.; Lombardi, F. Postinfarct left ventricular remodelling: A prevailing cause of heart failure. Cardiol. Res. Pract. 2016, 2016, 2579832. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Virani, S.S.; Alonso, A.; Benjamin, E.J.; Bittencourt, M.S.; Callaway, C.W.; Carson, A.P.; Chamberlain, A.M.; Chang, A.R.; Cheng, S.; Delling, F.N.; et al. Heart disease and stroke statistics–2020 update: A report from the american heart association. Circulation 2020, 141, e139–e596. [Google Scholar] [CrossRef] [PubMed]
- McMurray, J.J.; Packer, M.; Desai, A.S.; Gong, J.; Lefkowitz, M.P.; Rizkala, A.R.; Rouleau, J.L.; Shi, V.C.; Solomon, S.D.; Swedberg, K.; et al. Angiotensin-neprilysin inhibition versus enalapril in heart failure. N. Engl. J. Med. 2014, 371, 993–1004. [Google Scholar] [CrossRef] [Green Version]
- Dai, W.; Hale, S.; Kloner, R.A. Delayed therapeutic hypothermia protects against the myocardial no-reflow phenomenon independently of myocardial infarct size in a rat ischemia/reperfusion model. Int. J. Cardiol. 2017, 236, 400–404. [Google Scholar] [CrossRef]
- Hernandez, A.F.; Green, J.B.; Janmohamed, S.; D’Agostino, R.B.; Granger, C.B.; Jones, N.P.; Leiter, L.A.; Rosenberg, A.E.; Sigmon, K.N.; Somerville, M.C.; et al. Albiglutide and cardiovascular outcomes in patients with type 2 diabetes and cardiovascular disease (harmony outcomes): A double-blind, randomised placebo-controlled trial. Lancet 2018, 392, 1519–1529. [Google Scholar] [CrossRef] [Green Version]
- Marso, S.P.; Daniels, G.H.; Brown-Frandsen, K.; Kristensen, P.; Mann, J.F.; Nauck, M.A.; Nissen, S.E.; Pocock, S.; Poulter, N.R.; Ravn, L.S.; et al. Liraglutide and cardiovascular outcomes in type 2 diabetes. N. Engl. J. Med. 2016, 375, 311–322. [Google Scholar] [CrossRef] [Green Version]
- Marso, S.P.; Bain, S.C.; Consoli, A.; Eliaschewitz, F.G.; Jódar, E.; Leiter, L.A.; Lingvay, I.; Rosenstock, J.; Seufert, J.; Warren, M.L.; et al. Semaglutide and cardiovascular outcomes in patients with type 2 diabetes. N. Engl. J. Med. 2016, 375, 1834–1844. [Google Scholar] [CrossRef] [Green Version]
- Gottlieb, R.A.; Piplani, H.; Sin, J.; Sawaged, S.; Hamid, S.M.; Taylor, D.J.; de Freitas Germano, J. At the heart of mitochondrial quality control: Many roads to the top. Cell. Mol. Life Sci. 2021, 78, 3791–3801. [Google Scholar] [CrossRef] [PubMed]
- Andres, A.M.; Tucker, K.C.; Thomas, A.; Taylor, D.J.; Sengstock, D.; Jahania, S.M.; Dabir, R.; Pourpirali, S.; Brown, J.A.; Westbrook, D.G.; et al. Mitophagy and mitochondrial biogenesis in atrial tissue of patients undergoing heart surgery with cardiopulmonary bypass. JCI Insight 2017, 2, e89303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bravo-San Pedro, J.M.; Kroemer, G.; Galluzzi, L. Autophagy and mitophagy in cardiovascular disease. Circ. Res. 2017, 120, 1812–1824. [Google Scholar] [CrossRef]
- Qiao, H.; Ren, H.; Du, H.; Zhang, M.; Xiong, X.; Lv, R. Liraglutide repairs the infarcted heart: The role of the sirt1/parkin/mitophagy pathway. Mol. Med. Rep. 2018, 17, 3722–3734. [Google Scholar] [CrossRef] [Green Version]
- Kyhl, K.; Lonborg, J.; Hartmann, B.; Kissow, H.; Poulsen, S.S.; Ali, H.E.; Kjaer, A.; Dela, F.; Engstrom, T.; Treiman, M. Lack of effect of prolonged treatment with liraglutide on cardiac remodeling in rats after acute myocardial infarction. Peptides 2017, 93, 1–12. [Google Scholar] [CrossRef]
- Germano, J.F.; Huang, C.; Sin, J.; Song, Y.; Tucker, K.C.; Taylor, D.J.R.; Saadaeijahromi, H.; Stotland, A.; Piplani, H.; Gottlieb, R.A.; et al. Intermittent use of a short-course glucagon-like peptide-1 receptor agonist therapy limits adverse cardiac remodeling via parkin-dependent mitochondrial turnover. Sci. Rep. 2020, 10, 8284. [Google Scholar] [CrossRef]
- Doenst, T.; Nguyen, T.D.; Abel, E.D. Cardiac metabolism in heart failure: Implications beyond atp production. Circ. Res. 2013, 113, 709–724. [Google Scholar] [CrossRef] [Green Version]
- Burgoyne, J.R.; Mongue-Din, H.; Eaton, P.; Shah, A.M. Redox signaling in cardiac physiology and pathology. Circ. Res. 2012, 111, 1091–1106. [Google Scholar] [CrossRef] [PubMed]
- Zorov, D.B.; Juhaszova, M.; Sollott, S.J. Mitochondrial reactive oxygen species (ros) and ros-induced ros release. Physiol. Rev. 2014, 94, 909–950. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Münzel, T.; Camici, G.G.; Maack, C.; Bonetti, N.R.; Fuster, V.; Kovacic, J.C. Impact of oxidative stress on the heart and vasculature: Part 2 of a 3-part series. J. Am. Coll. Cardiol. 2017, 70, 212–229. [Google Scholar] [CrossRef]
- Tanigaki, K.; Sundgren, N.; Khera, A.; Vongpatanasin, W.; Mineo, C.; Shaul, P.W. Fcγ receptors and ligands and cardiovascular disease. Circ. Res. 2015, 116, 368–384. [Google Scholar] [CrossRef] [Green Version]
- Swirski, F.K.; Nahrendorf, M. Leukocyte behavior in atherosclerosis, myocardial infarction, and heart failure. Science 2013, 339, 161–166. [Google Scholar] [CrossRef] [Green Version]
- Kearney, C.J.; Martin, S.J. An inflammatory perspective on necroptosis. Mol. Cell 2017, 65, 965–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Frieler, R.A.; Nadimpalli, S.; Boland, L.K.; Xie, A.; Kooistra, L.J.; Song, J.; Chung, Y.; Cho, K.W.; Lumeng, C.N.; Wang, M.M.; et al. Depletion of macrophages in cd11b diphtheria toxin receptor mice induces brain inflammation and enhances inflammatory signaling during traumatic brain injury. Brain Res. 2015, 1624, 103–112. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al Batran, R.; Almutairi, M.; Ussher, J.R. Glucagon-like peptide-1 receptor mediated control of cardiac energy metabolism. Peptides 2018, 100, 94–100. [Google Scholar] [CrossRef]
- Bajic, V.P.; van Neste, C.; Obradovic, M.; Zafirovic, S.; Radak, D.; Bajic, V.B.; Essack, M.; Isenovic, E.R. Glutathione “redox homeostasis” and its relation to cardiovascular disease. Oxid. Med. Cell Longev. 2019, 2019, 5028181. [Google Scholar] [CrossRef] [Green Version]
- Mariani, J.A.; Smolic, A.; Preovolos, A.; Byrne, M.J.; Power, J.M.; Kaye, D.M. Augmentation of left ventricular mechanics by recirculation-mediated aav2/1-serca2a gene delivery in experimental heart failure. Eur. J. Heart Fail. 2011, 13, 247–253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hayward, C.; Banner, N.R.; Morley-Smith, A.; Lyon, A.R.; Harding, S.E. The current and future landscape of serca gene therapy for heart failure: A clinical perspective. Hum. Gene Ther. 2015, 26, 293–304. [Google Scholar] [CrossRef] [PubMed]
- Chu, M.; Novak, S.M.; Cover, C.; Wang, A.A.; Chinyere, I.R.; Juneman, E.B.; Zarnescu, D.C.; Wong, P.K.; Gregorio, C.C. Increased cardiac arrhythmogenesis associated with gap junction remodeling with upregulation of rna-binding protein fxr1. Circulation 2018, 137, 605–618. [Google Scholar] [CrossRef]
- Betrie, A.H.; Ayton, S.; Bush, A.I.; Angus, J.A.; Lei, P.; Wright, C.E. Evidence of a cardiovascular function for microtubule-associated protein tau. J. Alzheimer’s Dis. 2017, 56, 849–860. [Google Scholar] [CrossRef] [PubMed]
- Sumpter, R.; Sirasanagandla, S.; Fernández, Á.; Wei, Y.; Dong, X.; Franco, L.; Zou, Z.; Marchal, C.; Lee, M.Y.; Clapp, D.W.; et al. Fanconi anemia proteins function in mitophagy and immunity. Cell 2016, 165, 867–881. [Google Scholar] [CrossRef] [Green Version]
- Stark, C.; Breitkreutz, B.J.; Reguly, T.; Boucher, L.; Breitkreutz, A.; Tyers, M. Biogrid: A general repository for interaction datasets. Nucleic Acids Res. 2006, 34, D535–D539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohl, M.; Wiese, S.; Warscheid, B. Cytoscape: Software for visualization and analysis of biological networks. Methods Mol. Biol. 2011, 696, 291–303. [Google Scholar]
- Assenov, Y.; Ramírez, F.; Schelhorn, S.E.; Lengauer, T.; Albrecht, M. Computing topological parameters of biological networks. Bioinformatics 2008, 24, 282–284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- He, X.; Zhang, J. Why do hubs tend to be essential in protein networks? PLoS Genet. 2006, 2, e88. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, A.; Cinti, C.; Capobianco, E. Multitype network-guided target controllability in phenotypically characterized osteosarcoma: Role of tumor microenvironment. Front. Immunol. 2017, 8, 918. [Google Scholar] [CrossRef] [Green Version]
- Ge, S.X.; Jung, D.; Yao, R. Shinygo: A graphical gene-set enrichment tool for animals and plants. Bioinformatics 2020, 36, 2628–2629. [Google Scholar] [CrossRef]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pagès, F.; Trajanoski, Z.; Galon, J. ClueGO: A cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Vehicle | DMB | |||
|---|---|---|---|---|
| Pathway | z-Score | Pathway | z-Score | |
| Day 1 vs. Naive | TCA Cycle II (Eukaryotic) | −2 | Estrogen Receptor Signaling | 1.342 |
| NAD signaling pathway | −1.633 | GP6 Signaling Pathway | 0.447 | |
| Production of Nitric Oxide and Reactive Oxygen Species in Macrophages | 0.447 | |||
| RhoA Signaling | 1.633 | |||
| RhoGDI Signaling | −1.000 | |||
| Signaling by Rho Family GTPases | 2.000 | |||
| Unfolded protein response | 2.000 | |||
| Xenobiotic Metabolism AHR Signaling Pathway | −2.236 | |||
| Xenobiotic Metabolism CAR Signaling Pathway | 0.378 | |||
| Xenobiotic Metabolism PXR Signaling Pathway | −1.633 | |||
| Day 3 vs. Day 1 | PFKFB4 Signaling Pathway | −1.000 | Apelin Cardiomyocyte Signaling Pathway | −0.447 |
| RhoA signaling | 2.000 | Aspartate Degradation II | −2.000 | |
| Calcium signaling | 0.477 | BAG2 Signaling Pathway | 1.342 | |
| Cardiac Hypertrophy Signaling | −0.447 | |||
| Cdc42 Signaling | −1.000 | |||
| Coagulation System | 0.447 | |||
| Death Receptor Signaling | −0.447 | |||
| ERK/MAPK Signaling | 2.000 | |||
| Estrogen Receptor Signaling | −2.309 | |||
| Gluconeogenesis I | −2.000 | |||
| Hepatic Fibrosis Signaling Pathway | −1.633 | |||
| HER-2 Signaling in Breast Cancer | 2.000 | |||
| Insulin Secretion Signaling Pathway | −0.447 | |||
| Integrin Signaling | 1.134 | |||
| NAD Signaling Pathway | −2.449 | |||
| Necroptosis Signaling Pathway | −2.000 | |||
| Nitric Oxide Signaling in the Cardiovascular System | 1.000 | |||
| PI3K/AKT Signaling | 1.000 | |||
| PPARα/RXRα Activation | −2.236 | |||
| Production of Nitric Oxide and Reactive Oxygen Species in Macrophages | 1.000 | |||
| Regulation of Actin-based Motility by Rho | −1.000 | |||
| RhoGDI Signaling | 1.342 | |||
| Role of PKR in Interferon Induction and Antiviral Response | −1.342 | |||
| Semaphorin Neuronal Repulsive Signaling Pathway | −2.236 | |||
| Senescence Pathway | −1.633 | |||
| Signaling by Rho Family GTPases | −0.447 | |||
| Superpathway of Methionine Degradation | −2.236 | |||
| TCA Cycle II (Eukaryotic) | −3.317 | |||
| Unfolded protein response | 1.633 | |||
| Isoleucine Degradation I | −2.000 | |||
| Day 7 vs. Day 3 | Adrenomedullin signaling pathway | 1.000 | 14-3-3-mediated Signaling | 2.000 |
| Apelin Adipocyte Signaling Pathway | −2.000 | Acetyl-CoA Biosynthesis I (Pyruvate Dehydrogenase Complex) | −2.000 | |
| BAG2 Signaling Pathway | 1.000 | Complement System | 0.816 | |
| Breast Cancer Regulation by Stathmin1 | −0.477 | Gluconeogenesis I | 1.342 | |
| Cardiac Hypertrophy Signaling | 1.000 | Glutaryl-CoA Degradation | −2.236 | |
| Cardiac Hypertrophy Signaling (Enhanced) | 1.342 | Glycolysis I | 2.000 | |
| Coagulation System | −1.342 | Hepatic Fibrosis Signaling Pathway | 1.342 | |
| Coronavirus Pathogenesis Pathway | −2.000 | ILK Signaling | 1.732 | |
| Death Receptor Signaling | 2.000 | Inhibition of ARE-Mediated mRNA Degradation Pathway | 2.236 | |
| Endocannabinoid Neuronal Synapse Pathway | 1.000 | Intrinsic Prothrombin Activation Pathway | −0.447 | |
| Fcγ Receptor-mediated Phagocytosis in Macrophages and Monocytes | 2.000 | Isoleucine Degradation I | −2.236 | |
| HIF1α Signaling | 1.000 | Ketogenesis | −1.342 | |
| Leukocyte Extravasation Signaling | 2.000 | Ketolysis | −1.000 | |
| Necroptosis Signaling Pathway | 0.816 | MSP-RON Signaling in Cancer Cells Pathway | 2.236 | |
| Opioid Signaling Pathway | 1.000 | Necroptosis Signaling Pathway | −0.447 | |
| PPARα/RXRα Activation | −2.121 | Senescence Pathway | −1.000 | |
| RhoA Signaling | 2.236 | Tryptophan Degradation III (Eukaryotic) | −2.236 | |
| RhoGDI Signaling | −2.000 | Valine Degradation I | −2.646 | |
| Role of NFAT in Cardiac Hypertrophy | 2.000 | |||
| Signaling by Rho Family GTPases | 2.449 | |||
| Unfolded protein response | 2.000 | |||
| Xenobiotic Metabolism CAR Signaling Pathway | 1.000 | |||
| Xenobiotic Metabolism PXR Signaling Pathway | −1.633 | |||
| Vehicle | DMB | ||||
|---|---|---|---|---|---|
| Day 1 | Day 3 | Day 7 | Day 1 | Day 3 | Day 7 |
| COFA1, COX8B, CTNB1, DCTN2, DUS3, EIF3B, EZRI, FAHD2, HNRPF, IMB1, KCC2D, KINH, LACE1, MARE2, MGDP1, MIC26, NDUC2, PDC6I, PSA7, RAN, RMD1, RS5, SAM50, SEPT2, STX3, TALDO, UBE2N, UBP5, 6PGD, APOC1, ARF4, C4BPA, CFAB, CHIL3, CMC2, CO4B, CRIP1, EGFR, FETUB, GPX41, HRG, ICAL, ITIH3, KLH41, LMOD2, LUM, MYH11, NEDD4, PRS10, PRS6A, RD23B, RS20, RS3, SYDC, TCPH, THTR, TOM1, UN45B, AN32A, CBR1, DHDH, FAHD1, GLRX1, HDHD2, HOT, NUCG, OXND1, PGS2, PIMT, PSA3, SIR5, SPEG, VP26A, CBR4, CLPP, DDX5, GPD1L, KV5AA, LGUL, MAOX, MIME, PYC, VPS29 | CLPP, COX2, NP1L1, TSP1, CX7A2, XDH, FXR1, FBLI1 | NUCG, GLRX3, AKAP9, CAND1, G3BP1, PRELP, VATG1, MYL4, MPPB | MLRA, MSRB2, PGFS, PSA6, NQO2, LYZ2 | PDK4, LGMN, PMGE, RADI, CX6A1, COX8B, VATA, MTPN, RS3, TCPE, IC1, PSME2, PSMD1, PCBP2, SAM50, UN45B, DDX1, CPT1B, PLST, NAMPT, CAP2, IPYR, MECR, PFKAP, PSD13 | ITIH3, CLPP, YBOX3, TAU, ARC1B, EIF3A, FBN1, NDUB1, PRS8, SYRC, PDK4 |
| Day | Total Number of Nodes (Seed + Interacting Partners) | Number of Interactions | Unique Proteins and DEPs in Network (Seed Proteins) | High-Degree Proteins (Unique for That Day) |
|---|---|---|---|---|
| Day 1 | 198 | 612 | 43 | Fancd2 |
| Atp2a2 | ||||
| Eed | ||||
| Day 3 | 768 | 3314 | 81 | Fxr1 |
| Fancd2 | ||||
| Actb | ||||
| Day 7 | 922 | 4317 | 39 | Mapt |
| Actb | ||||
| Fancd2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Freitas Germano, J.; Sharma, A.; Stastna, M.; Huang, C.; Aniag, M.; Aceves, A.; Van Eyk, J.E.; Mentzer, R.M., Jr.; Piplani, H.; Andres, A.M.; et al. Proteomics of Mouse Heart Ventricles Reveals Mitochondria and Metabolism as Major Targets of a Post-Infarction Short-Acting GLP1Ra-Therapy. Int. J. Mol. Sci. 2021, 22, 8711. https://doi.org/10.3390/ijms22168711
de Freitas Germano J, Sharma A, Stastna M, Huang C, Aniag M, Aceves A, Van Eyk JE, Mentzer RM Jr., Piplani H, Andres AM, et al. Proteomics of Mouse Heart Ventricles Reveals Mitochondria and Metabolism as Major Targets of a Post-Infarction Short-Acting GLP1Ra-Therapy. International Journal of Molecular Sciences. 2021; 22(16):8711. https://doi.org/10.3390/ijms22168711
Chicago/Turabian Stylede Freitas Germano, Juliana, Ankush Sharma, Miroslava Stastna, Chengqun Huang, Marianne Aniag, Angie Aceves, Jennifer E. Van Eyk, Robert M. Mentzer, Jr., Honit Piplani, Allen M. Andres, and et al. 2021. "Proteomics of Mouse Heart Ventricles Reveals Mitochondria and Metabolism as Major Targets of a Post-Infarction Short-Acting GLP1Ra-Therapy" International Journal of Molecular Sciences 22, no. 16: 8711. https://doi.org/10.3390/ijms22168711
APA Stylede Freitas Germano, J., Sharma, A., Stastna, M., Huang, C., Aniag, M., Aceves, A., Van Eyk, J. E., Mentzer, R. M., Jr., Piplani, H., Andres, A. M., & Gottlieb, R. A. (2021). Proteomics of Mouse Heart Ventricles Reveals Mitochondria and Metabolism as Major Targets of a Post-Infarction Short-Acting GLP1Ra-Therapy. International Journal of Molecular Sciences, 22(16), 8711. https://doi.org/10.3390/ijms22168711